

Abstract
We report a novel approach for determining the enzymatic activity within a single suspended cell. Using a steady-state microfluidic delivery device, and timed exposure to the pore-forming agent digitonin, we controlled the plasma membrane permeation of individual NG108-15 cells. Mildly permeabilized cells (~100 pores) were exposed to a series of concentrations of fluorescein diphosphate (FDP), a fluorogenic alkaline phosphatase substrate, with and without levamisole, an alkaline phosphatase inhibitor. We generated quantitative estimates for intracellular enzyme activity, and were able to construct both dose-response and dose-inhibition curves, at the single-cell level, resulting in an apparent Michaelis contant Km of 15.3 μM ± 1.02 (mean ± standard error of the mean (SEM), n=16), and an inhibition constant Ki of 0.59 mM ± 0.07 (mean ± SEM, n=14). Enzymatic activity could be monitored just 40 s after permeabilization, and 5 point dose-inhibition curves could be obtained within 150 s. This rapid approach offers a new methodology for characterizing enzyme activity within single cells.
INTRODUCTION
New genomics, proteomics, and transcriptomics tools, as well as more traditional methods such as in situ hybridization and immunohistochemistry, have provided extensive knowledge on the genetic makeup and biochemical composition of cells1. In many cases we have a fine-grained understanding, down to the level of individual organelles, of their composition, and in some instances, we even know the exact number of key proteins they harbor2. However, exactly what tasks and operations many of these proteins perform, and how fast they accomplish this, i.e. a detailed functional, and kinetic understanding, is presently far from established1,3. Probing enzymatic activity is of central importance in understanding the cellular machinery. Furthermore, it would be optimal to measure the activity of enzymes in situ as they may exist in inactive forms that demand post-translational processing, or they may be inactive for other reasons such as natural inhibitor binding or requiring complex organization to become active4–6. It is today well acknowledged that cell populations exhibit biological “noise”, and that phenotypically identical cells can differ in their enzymatic activity5.
Traditional ways to measure the activity of intracellular enzymes typically use suspensions of lysed cells, purified, or recombinant proteins, and therefore are unable to reveal functional differences between cells, generating an ensemble average response. Furthermore, results obtained in such measurements, may have limited applicability to in vivo situations, as several key physical and chemical parameters of cells are neglected6–10. Methods to measure enzyme activity in single cells have recently been reviewed by Kovarik and Allbritton5. As in bulk measurements, product formation can be detected based on fluorescence or electrochemical properties, with new functional substrates being continually developed5. In order to increase throughput, facilitate substrate application, and follow readout of enzymatic activity within single cells, they can be confined in arrays on microfluidics devices11. Lysed cells can also be confined in arrays of microfabricated wells or droplets, minimizing cell content dilution12–15. Single-cell measurements can furthermore be performed in flowing streams such as in flow cytometry, or in microfluidic devices16–17. Using capillary electrophoresis, it has been possible to separate and measure the activity of up to 5 different enzymes from a single cell5,18. Despite the rapid development of methods to measure enzyme activity within single cells, none have provided full dose-response curves of in situ enzymes, and thus access to meaningful data on apparent binding constants such as Ki, and Km values, which are required for full pharmacological profiling. This is an important shortcoming that cannot be easily overcome, unless a method is found to titrate enzymes in situ with known concentrations of activators and inhibitors.
In this report, we describe a calibrated method for directly titrating intracellular enzymes within single cells and to map their function and performance under different conditions. To demonstrate the method we studied endogenous phosphatase activity in NG108-15 cells, using the weakly fluorescent substrate FDP. FDP is a substrate for phosphatases such as alkaline phosphatase (AP) and protein tyrosine phosphatase (PTP). Although we cannot omit the possibility of measuring an enzyme population including different phosphatases, characterization using different inhibitors (see Supporting Note 11) suggests that the majority of the measured activity comes from AP. AP is an essential intrinsic membrane protein that has a broad hydrolysis and transphosporlation function and is involved in the metabolism of nucleotides19. Our technique utilizes a microfluidic system that generates a large number of discrete solution environments, together with a high precision solution exchange capability (millisecond exchange times)20, to permeabilize the cell plasma membrane and subsequently expose the cell to a sequence of varying concentrations of enzyme substrates and inhibitors. Digitonin was used, which creates pores 8–10 nm in diameter21–22, to permeabilize the cells to allow for controlled delivery of substrate through the membrane. Importantly, by timing the digitonin-exposure, the number of pores formed can be controlled, and a mild permeabilization can be obtained. The microfluidic system we employed has been described elsewhere20,23, and has been demonstrated for both receptor screening20, creation of complex concentration waves24, and for control of the intracellular environment25. Here, we present the application of this microfluidic system to monitor changes in intracellular enzymatic activity, to create dose-response and dose-inhibition curves, and to estimate apparent Km, and Ki values. We demonstrate the comparison of alkaline phosphatase efficacy in two different conditions within 80 seconds of membrane permeabilization, and measurement of 5-point dose inhibition curves within 150 seconds. The protocol here presented, can furthermore be applied to receptor characterization, to investigate the role of modulatory agents in cell signaling, and allows for on-line monitoring of signal pathway regulation.
MATERIALS AND METHODS
Cell Preparations
NG108-15 (mouse neuroblastoma and rat glioma hybrid) cells were cultured as described elsewhere 26. Before each experiment, the cells were washed and detached in extracellular buffer (ECB) buffer containing: 140 mM NaCl, 1 mM CaCl2, 10 mM D-glucose and 10 mM HEPES (pH 7.2). All chemicals used in the cell culturing were obtained from Sigma-Aldrich AB (Stockholm, Sweden).
Microfluidic Platform
Experiments were performed using a microfluidic superfusion device (Dynaflow 16, Cellectricon, Gothenburg, Sweden) which is described in detail elsewhere23–24. The standard glass cover slip of the device was exchanged to a thinner, #1 coverslip of thickness ~130 μm in order to allow the use of high magnification confocal microscopy. The device was placed onto a computer-control motorized scanning stage (Proscan, Prior Scientific, Cambridge, U.K), maneuvered with a custom control program. A description of the cell holding pipette can be found elsewhere25. ECB solutions supplemented with fluorescein, digitonin, FDP (substrate) and levamisole (inhibitor) were loaded into the 16 sample reservoirs. Probenecid (4-(dipropylsulfamoyl)benzoic acid, Sigma-Aldrich AB, Stockholm, Sweden) was added to all solutions with exception of the digitonin solution. Probenecid is an inhibitor of organic-anion transporters located in cell membrane that is commonly used to prevent transport of dyes out from cells or into cell compartments27. Thus, when probenecid was used, a reasonably homogenous fluorescence distribution was observed within the cell, while highly fluorescent accumulated spots were recorded in the cells when probenecid was excluded.
Confocal Laser Scanning Microscopy
All experiments were performed using a confocal laser scanning microscopy system (Leica TCS SP2 RS, Leica Microsystems, Wetzlar, Germany) with a PL APO CS 63× magnification 1.25 NA oil immersion lens. A region of interest (ROI) analysis was performed on the confocal micrographs, enabling fluorescence mean intensities to be extracted. An investigation into using the validity of utilizing confocal micrographs to represent the mean cell intensities was performed for both ROI position and axial displacement, the results of which are shown in Supporting Figure 2.
Data Analysis
Fluorescence intensity curves were extracted from images using the Leica LAS AF Lite software (Leica Microsystems, Wetzlar, Germany). All further analysis and fitting was performed using Matlab 7.6.0 and the Matlab curvefitting toolbox (MathWorks, Inc., Natick, MA).
Permeability and Calibration Tests
As described elsewhere 25, the concentration inside a cell placed in a solution containing membrane permeable specie at time t=0, can be described by
| (1) |
where Qe (mol/m3) is the extracellular concentration and Q0 (mol/m3) is the initial intracellular concentration, P (m/s) is the membrane permeability coefficient for the considered specie, A (m2) is the cell surface area, V (m3) is the volume occupied by the cell in space, and α is the fraction of this volume that is accessible to the considered specie. Permeability was estimated through fitting equation (1) to experimental curves assuming the cell to be spherical and with α set to 1. As discussed in Supporting Note 7 we see indications that α is smaller than 1. Assuming α to be one then gives an overestimation of the membrane permeability coefficient, which we therefore chose to denote an apparent membrane permeability cofficient. To verify that the membrane permeability for fluorescein was not substantially affected by levamisole the permeability of the same permeabilized cells was tested both in the presence and absence of levamisole in separate experiments. Cells were permeabilized at times between 2 and a few seconds, yielding an apparent plasma membrane permeability coefficient ≤ 0.3μm/s. For more details see Supporting Note 7.
Estimates of KM and KI
Dose-response data from FDP titration and dose-inhibition data from levamisole titration of individual cells were analysed using Michaelis-Menten kinetics. To estimate the Michaelis constant Km, dose-response data were fit to the equation
| (2) |
where v (mol/s) is the product formation rate, Vmax (mol/s) is the maximum rate of product formation, and Xi (mol/m3) is the substrate concentration.
To estimate the inhibition constant Ki from individual cells, single-cell dose-inhibition data were fit using the equation
| (3) |
where Z (mol/m3) is the concentration of inhibitor and B is a constant adjustment for offset. The substrate concentration was set to 20 μM or 150 μM depending on which was used in the experiment, while Km vas set to 15.3 μM (the average value obtained from the FDP dose-response experiments).
RESULTS
Experimental Outline
To demonstrate the method we studied endogenous phosphatase activity in NG108-15 cells, using the weakly fluorescent substrate FDP. AP hydrolyzes FDP, yielding the strongly fluorescent product fluorescein in a two-step process, which makes it possible to follow the enzymatic activity using fluorescence detection28. The method utilizes a microfluidic device that can be used to create a patterned laminar flow, with discrete solution environments including a known concentration of digitonin (pore-forming agent), fluorescein (permeabilization marker), wash solutions (buffer), and different concentrations of substrate (FDP) and inhibitor (levamisole) (Supporting Figure 1). The cell to be characterized is held by a pipette and positioned in the patterned flow. The microfluidic device is placed on a computer controlled scanning stage such that it can be translated relative to the cell. Using this set-up the cell can be exposed to different solutions with high precision in timing and with millisecond switching times20,23. In rapid sequence the cell is 1; Placed at its starting point outside a channel containing buffer solution. 2; Permeabilized by exposure to 10 μM digitonin for a duration of 2 to a few seconds, followed by 20–30 s of waiting time in buffer solution, to allow the plasma membrane to stabilize. 3; Titrated with different concentrations of substrate (FDP) or inhibitor (levamisole). 4; Subjected to a test of the membrane permeability and a calibration of the dependence between intracellular product concentration and fluorescence intensity. This is done through exposing the cell to a solution containing fluorescein for (typically) 30 s while measuring the dynamics of the fluorescein diffusion over the cell membrane as well as the end-point fluorescence intensity. During the entire cause of each experiment the intracellular concentration of product is measured continuously together with the background signal directly adjacent to the cell using confocal microscopy. Figure 1 shows a schematic of the basic experimental procedure used to measure single-cell enzyme activity, while a detailed scheme can be found in Supporting Figure 1.
Figure 1.
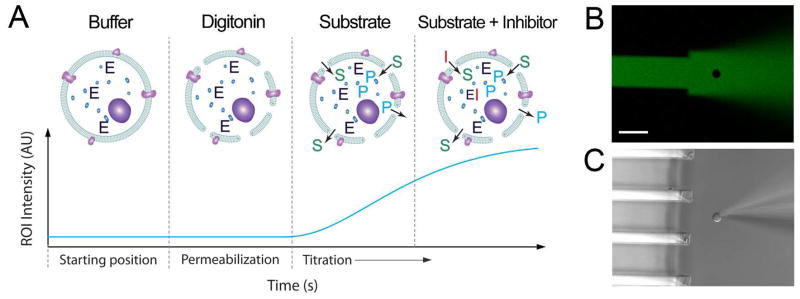
Experimental scheme for measuring single cell enzymatic activity. (A) Depicts a representation of the analysis stages with the corresponding signal-response curve below. Each stage utilizes one or several distinct chemical environments. The cell is in sequence: 1. Moved to the starting position which is in front of a buffer containing channel; 2. Permeabilized through exposure to digitonin; 3. Titrated through exposure to varying concentrations of enzymatic substrate (S) and inhibitor (I). This enables enzyme (E) kinetics to be extracted, by monitoring the formation of fluorescent product (P). (B) An example cell positioning image in fluorescence, showing the orientation and arrangement of microfluidic channels and the holding pipette, with a corresponding DIC image in (C). Channels exiting a microfluidic device into an open volume allow for the formation of laminar flow streams. The cell (NG108-15) which is held by a glass micropipette can be translated through different solution environments at a fixed distance from the channel array. In (B) and (C) the cell has not yet been porated and is held in a solution environment containing 10 μM fluorescein and ECB buffer, pH 7.4. The fluorescein solution is excluded from the cell, seen as an absence of fluorescence within its cross section, due to the plasma membrane integrity. The scale bar represents 50 μm.
Permeabilization of the Plasma Membrane
The kinetically controlled permeabilization of cell membranes using digitonin is a key feature of the experimental protocol. By translating the cell through a region of a digitonin solution with variable speed and residence time, we can choose between creating very few pores (estimated to be fewer than ~100, see Supporting Note 5) using exposure times of approximately 1 s or less (where the cells will self-heal on the timescale of minutes), few pores (estimated to be on the order of 100) using exposure times of 2 s to a few seconds (the cells do not manage to self-heal), or to heavily porate the cell membrane, using exposure times on the order of several tens of seconds.
Single Cell Titration
An example of a single cell titration with varying concentrations of the fluorogenic substrate FDP is shown in Figure 2. Fluorescence and differential interference contrast (DIC) images are presented for testing the permeability of the cell membrane and intracellular product buildup (Figure 2A–B).
Figure 2.

Titration of a single NG108-15 cell using varying concentrations of the alkaline phosphatase substrate FDP. (A,B) Fluorescence and DIC images from two different time points. Fluorescence images are overlaid with ROIs for measuring both cellular response and background fluorescence intensity. In (A) the cell is exposed to a fluorescein solution prior to analysis, indicating the viability of the membrane. In (B), the cell is exposed to 300 μM FDP and the cell exhibits its maximum intracellular product concentration during the experiment. The scale bar in (B) represents 10 μm and is applicable to both (A, B). (C) The compiled analysis of the single cell titration experiment. The red curve, extracted from the red ROI in (A) and (B), represents the fluorescence intensity within the cell, being a composite of both substrate solution and product. The blue curve represents the background fluorescence intensity measured outside the cell, as observed from the mean signal within the ROI shown in blue in (A) and (B). Step changes in the background intensity are due to the extracellular solution exchange, as each hydrodynamically confined region has a distinct concentration of the weakly fluorescent FDP. These concentrations were 20, 80, 100, and 300 μM for the sections denoted by I, II, III and IV, respectively.
Upon translation of the permeabilized cell into a solution that contains substrate, substrate diffuses through the created pores, unless being too large to pass through the pores. Inside the cell, the substrate is processed by enzymes and product is formed with a rate v, which leads to a build-up of product inside the cell. Diffusive transport of product molecules out of the cell will occur at a rate proportional to the product concentration gradient over the permeabilized membrane. As the product concentration within the cell increases, the diffusion out of the cell increases as well, resulting in a gradual decrease in the rate of product build up. Eventually, the intracellular product concentration is sufficiently high that the loss of product through diffusion out of the cell equals the enzymatic production of new product molecules; the concentration of product inside the cell then has reached a steady-state value, hereafter referred to as a plateau value. This plateau value is proportional to v (see Supporting Note 1). Any change in v manifests as a change in the rate of product build-up, and will result in a concomitant change in the intracellular steady-state product concentration.
FDP Dose-response Experiments and the Estimation of Enzyme Activity From Fluorescence Plateau Values
The permeabilized NG108-15 cells were titrated with increasing concentrations of FDP while measuring the changes of intracellular fluorescein (product) concentration using confocal microscopy. In a first set of experiment the concentrations 20, 100, 300, and 500 μM was used, which allowed us to confirm that the enzyme activity had reached a maximum at 300 μM as no increase was seen when increasing the concentration further. In following experiments we therefore changed to the titration concentrations 20, 80, 100, and 300 μM. An example of a dose-response curve is shown in Figure 2C. As the diffusive supply of substrate is proportional to the concentration gradient across the membrane, a high substrate supply can be obtained, despite low membrane permeability, through application of a steep concentration gradient. We take advantage of this, to accelerate the initial substrate delivery to the cytosol, by briefly exposing the cells to a higher substrate concentration than aimed for within the cell (in the experiment shown in Figure 2 the cell was exposed to 40 μM FDP concentration for 20 s before being exposed to 20 μM FDP). Optimal times and concentrations for this enhanced substrate gradient are based upon the extent of permeability, intracellular volume, and desired intracellular end-concentration.
As the intracellular product concentration at steady state (background and baseline subtracted) is directly proportional to the rate of product formation (v) we can estimate relative changes in enzyme activity by comparing the respective plateau values (see Supporting Note 3 for details). Lack of fluorescent species within the buffer channels allows for a region outside the cell to be used as a baseline reference to account for any detector offset.
In Figure 3, single-cell dose-response data from the experiment in Figure 2, and similar experiments is shown. The concentration of substrate within the cell is lower than that being supplied to the exterior, due to intracellular consumption of substrate, establishing a concentration gradient over the membrane enabling refilling of substrate by diffusion. It can be shown that at steady state
Figure 3.
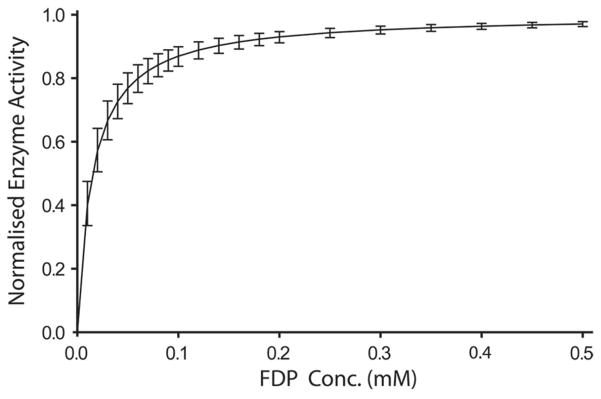
Average dose-response plot constructed from measurements of alkaline phosphatase within individual NG108-15 cells. Cells were titrated with 20, 80, 100 and 300 μM FDP or 20, 100, 300, and 500 μM FDP and the increase in rate of product formation was measured via ROI analysis of confocal microscopy images. In total 16 cells were measured across multiple cell line passages, with multiple dishes from each passage, to obtain a statistically representative sample. The data extracted from each cell was normalized to unity at highest concentration for inter-cell comparisons while Michaelis-Menten kinetics was assumed to allow for extraction of apparent Km values from fits to the individual curves (for details see Materials and Methods). The displayed curve shows the dose response behavior for the mean value of Km from all cells, and the error bars denote the standard deviation between the single-cell dose response curves (Individual data curves shown in Supporting Figure 7).
| (4) |
where Xi is the intracellular substrate concentration, Xe is the bulk extracellular substrate concentration, and Yi is the intracellular product concentration (see Supporting Note 2 for further details). A linear relation between intracellular fluorescein concentration and fluorescence intensity was verified in a separate experiment addressing the dynamic range of the method, as described below. Single point calibrations, assuming a linear dependence, were then performed for each experiment by placing the cell in a known concentration of product (10 μM fluorescein) and measuring the corresponding intracellular intensity. Using this calibration we are able to estimate the intracellular product concentrations in absolute terms. This allows both for calculating the average product formation rate, which was 0.033 μM/s ± 0.0039 (mean ± SEM, n=16) for 20 μM and 0.065 μM/s ± 0.0045 (mean ± SEM, n=16) for 100 μM FDP, and for calculating the true intracellular substrate concentration using equation (4). We obtained the mean intracellular concentrations 12.0 μM, 70.2 μM, 83.1 μM, and 283.2 μM respectively, for the outside concentrations 20 μM, 80 μM, 100 μM, and 300 μM (n=16 for 20 μM, 100 μM and 300 μM; n=7 for 80 μM). Dose-response data from individual cells was fit using Michaelis-Menten kinetics and yielded an average apparent Km of 15.3 μM ± 1.02 (mean ± SEM, n=16). (See Materials and Methods for details).
Determination of the dynamic range and the limits of sensitivity were performed using a systematic introduction of fluorescein into porated cells. The sensitivity of the measurement and the dynamic range were both performed at typical experimentation conditions. The minimum detectable rate was measured to be 0.0047 μM/s, and the maximum measurable rate was determined to be 0.59 μM/s. Under these experimental conditions, the minimum detectable change of rate was calculated to be 0.0010 μM/s. A further description of how the detection range values were extracted can be found in Supporting Figure 10 and Supporting Note 10.
Dose-inhibition Experiments Using Levamisole and Estimation of Enzyme Activities From the Slope of Fluorescence Curves
Single NG108-15 cells were titrated with different concentrations of the alkaline phosphatase inhibitor levamisole19, to obtain quantitative dose-inhibition data. Examples of fluorescence intensity plots can be seen in Figure 4. In the experiment shown in Figure 4A and B, a cell was first porated and its permeability tested, then titrated with 150 μM FDP supplemented with 0, 0.5, 2.5, 10, and 25 mM levamisole. The residence time in each levamisole concentration was 60 s. In order to test the accuracy of the methodology, we varied both the substrate concentration and the exposure times. Figure 4D shows an experiment where 20 μM concentration of FDP was used, in combination with 20 s residence time in each levamisole concentration. The level of poration was tested after the conclusion of the run, by transferring to an external fluorescein solution (Figure 4C).
Figure 4.

Titration of single NG108-15 cells with the alkaline phosphatase inhibitor levamisole. (A–D) display the extracted data from ROIs taken both inside and outside the cell, during exposure to the varying environments. (A,B) The compiled fluorescence curves for a titration experiment. In (A) the level of poration is tested by exposure to a 10 μm solution of fluorescein. In (B), the cell is subsequently translated through channels containing 150 μM FDP supplemented with increasing concentrations of levamisole. The levamisole titration concentrations were 0, 0.5, 2.5, 10, and 25 mM, corresponding to regions I–V (additional details of the experimental scheme are in Supporting Figure 1). Relative changes in rate of product formation when increasing the levamisole concentration from 0 to 0.5 mM, and from 0.5 mM to 2.5 mM, were estimated using equation (5), while changes from 2.5 mM to 10 mM, and 1 mM to 25 mM, were analyzed using the difference in plateau values. Black lines on either side of the red curve indicate the regions of the data used for linear slope fitting, following the protocol detailed in the Supporting Information (Steady-state of Fluorescence Intensities and Fluorescence Intensity Curve Slope Values and Supporting Figure 4). (C,D) A similar experiment as in (A, B) but using 20 μM FDP as substrate concentration, and shorter residence times in each levamisole concentration. The levamisole concentrations and the experimental exposure procedure in (D) are the same as for (B). In (C) the extent of poration is tested at the end of the experiment to check for the level of cell self repair and to ensure that continuous kinetics could be measured.
Changes in enzyme activity can be evaluated by comparing plateau values (as described for Km determination) or alternatively, through considering changes in the rate of intracellular product build up, i.e. changes of the slopes in the curve describing the increase or decrease in intracellular fluorescence. The significant advantage to this approach is that it does not require a steady state to be achieved, expediting the analysis. When the intracellular product concentration is so low that the diffusion of product out from the cell can be neglected in comparison to the considered product formation rates, differences between rates can be accessed through direct comparison of slope values as described by
| (5) |
where vo is the rate of product formation for the first condition, vn is the rate of product formation at a following condition, and S0 and Sn the slopes measured for respective condition. Any diffusion that is not adjusted for, leads to an overestimate of the difference between the product formation rates. In the case of characterization of enzyme inhibition, not accounting for diffusion gives an underestimation of the residual enzymatic activity. When diffusion of product out of the cell is too substantial to be neglected, we can still utilize that the change in slope that takes place when the product formation rate is altered, does represent a difference between the two rates. If the magnitude of this difference is expressed in terms of a reference velocity, the ratio between the two rates can be estimated. Based on this strategy, a general recursion formula for estimating alterations in enzymatic activity from changes in fluorescence curve slope values can, as shown in Supporting Note 4, be derived as
| (6) |
where vo is the product formation rate for the first condition (e.g. when no inhibitor is present) which is also used as the reference rate; vn the rate during the nth following condition (e.g. the nth inhibitor concentration); So the slope of the fluorescence curve representing the reference product formation rate vo,, measured during an interval where diffusion of product out of the cell can be neglected; Sn-1,R the slope representing vn-1 close to the breakpoint Bn where the velocity changes to vn; and Sn,L the slope representing vn close to the breakpoint Bn (see Supporting Figure 4). Equation (6) inherently adjusts for the gradual increase of diffusion of product out from the cell in-between any pair of slopes Sn,L and Sn,R (which both represent the velocity vn, but are used to compare vn to the preceding and succeeding velocities respectively), but not for increase of diffusion in the interval where the slopes are measured. It is therefore important that the slopes can be measured close to the respectively break point.
Average dose inhibition curves based on single cell dose inhibition data (extracted from the curve in Figure 4B and similar curves), where the enzymatic activity was measured for the concentrations; 0, 0.5, 2.5, 10, and 25 mM of levamisole, is presented in Figure 5. Ki values were estimated for each cell and yielded an average apparent Ki of 0.59 mM ± 0.07 (mean ± SEM, n=14) (see Materials and Methods for details).
Figure 5.

Average dose inhibition plots constructed from levamisole inhibition experiments on single cells. Cells were exposed to either 150 or 20 μM FDP and varying levamisole concentrations (0, 0.5, 2.5, 10 and 25 mM). For each FDP concentration 7 cells were monitored (individual data curves shown in Supporting Figure 7). The data for each cell was normalized to the highest measured rate of product formation for that cell and the apparent inhibition constant Ki was extracted from the single-cell dose-inhibition plots (for details see Materials and Methods). The displayed curves are dose inhibition curves constructed using the mean value for Ki from all cells of each FDP concentration, and the error bars denote the standard deviation between the single-cell curves.
There might be experimental situations where diffusion out from the cell cannot be neglected and it is desirable to use equation (6), but where the noise level is too high to accurately resolve Sn,L and Sn,R as two different slopes. Sn,L and Sn,R will then equal each other and most terms in the summation from equation (6) cancel out so that it reduces to equation (5), with Sn=Sn,L=Sn,R. Although it is known that applying equation (5) or (6) in such cases gives an overestimation of changes in enzyme activity, this analysis may still be used.
Figure 4D presents a five-point-dose-inhibition curve recorded in 150 seconds, including a 20 s waiting time for the pores to stabilize before measurement. In this example the noise level is too high, to resolve two different slopes Sn,L and Sn,R during the relatively short residence times used, and equation (6) thus cannot be adequately applied. Instead using equation (5) to compare the product formation rate for the first three conditions, we arrive at the relative enzymatic activities 0.73 and 0.31 for inhibitor concentration 0.5 mM and 2.5 mM compared to no inhibitor present. Further, comparing the plateau heights for 10 mM and 25 mM levamisole, we find that the effect of this step in inhibitor concentration only decreases the enzymatic activity by 13%. Although the change in enzyme activity when increasing the inhibitor concentration from 2.5 mM to 10 mM could not be quantified, and the analyse using equation (5) may substantially underestimate the remaining enzyme activity (in particular for 2.5 mM levamisole), taken together, the analysis of the curve from the dose-inhibition experiment suggests that the Ki for levamisole is close to or higher than 0.5 mM and clearly lower than 10 mM.
DISCUSSION
We developed a microfluidic strategy for in situ titration of enzymes within individual cells. The cells require no pre-incubation with dyes and are virtually undisturbed until the moment of permeabilization, taking place 20–30 seconds before measurement. This approach allows rate determination while circumventing two of the critical issues usually apparent in enzyme studies; substrate depletion and build up of product concentration.
In our presented method, the cell becomes a reaction vessel with controlled permeability, where substrate is continually refreshed through membrane pores and product build up is mediated by diffusion out of the cell. Consequently, measurements can be performed at a steady-state condition for as long as the activity of the enzyme is stable. Steady-state turnover in combination with high temporal control of substrate concentration makes this method ideal for characterizing changes in enzymatic activity, with an additional benefit of being able to monitor delays in activity onset. This approach was applied to enzymatic inhibition using levamisole, an uncompetitive reversible inhibitor for alkaline phosphatase19. While the degree of inhibition for uncompetitive inhibitors is generally dependent on the concentration of substrate, our experimental results are in agreement with our theoretical calculations which predicts similar degrees of inhibition for our experimentally derived Km, at the substrate concentrations used (as described in the Supporting Note 6 and Supporting Figure 5).
Mildly permeabilized cells were found to not reach the same level of intracellular fluorescence as the exterior solution during equilibration with fluorescein, examples of which can be seen in Figure 4A,C. Equilibrium experiments where the fluorescence sampling rate had been reduced, confirmed that this was not an effect of photobleaching the product of the substrate (fluorescein). Instead, this is likely due to maintenance of the intracellular environment, excluding fluorescein molecules from regions of the cell interior, and moreover the intracellular conditions may mediate the fluorescence response, as the fluorophore is known to be e.g. strongly pH sensitive 29. We occasionally observed that further exposure to digitonin for such cells, gave rise to an apparent decrease in permeability concomitant with a higher fluorescence when placed in fluorescein solution. In those cases we believe the additional poration makes the intracellular environment less preserved and increases the fraction of the intracellular volume that is accessible to fluorescein molecules. As we cannot measure what fraction of the cell volume that is accessible to the considered species, we assume α = 1, which corresponds to the entire volume being accessible (see Materials and Methods). The assumption that α = 1 leads to an overestimation of the membrane permeability coefficient P which is why we chose to denote P as an apparent membrane permeability coefficient. When comparing the enzymatic rates for different conditions, the absolute values of α and P have no implications, as they cancel out in the analysis. The only implication to data presented in this article, is a potential overestimation of the numbers of pores in the cell membrane for a given membrane permeability. Further implications for the general analysis of experimental curves are discussed in the Supporting Note 7. The Km values reported in the literature for phosphatases such as AP vary broadly. For AP, values between 1μM and 0.34 mM have been reported, and values from 1 to 8 μM have been reported when FDP is the substrate30–32. The activity of AP is known to differ between isoenzymes and isoforms present in different tissues and species33 and the measurements of purified enzymes demonstrate that the activity can be very sensitive to the environment. The sensitivity of AP to the inhibitor levamisole which was used in our study, has been previously reported to have Ki values in the range 0.05–8.2 mM34–35, variances which have been linked to different isoenzymes33. One of the main arguments for developing the method presented here is the possibility that the behaviour of enzymes differs in a native state, compared to when measured in purified form or in heavily porated cells. In control experiments (see Supporting Figure 8 and 9) we measured the enzymatic cleavage of FDP in NG18-15 cells in bulk using fluorescence spectroscopy, both for different FDP concentrations and for different concentrations of the inhibitor levamisole. In this case we could not fine-tune the permeabilization of the plasma membrane, and it was more heavily permeabilized compared to our single-cell experiments. A difference was noted between the Km and Ki in bulk experiments compared to the experiments performed on single-cells with presumably well-preserved intracellular environment. While the measured Km was lower in bulk experiments (15.3 μM in single-cells versus 0.505 μM in bulk), the Ki was higher (0.59 mM in single-cells versus 5.9 mM in bulk). Experimental details are given in Supporting Note 8 and Supporting Note 9. It is expected that macromolecular crowding generally increases the rate of slow, transition-state limited association reactions while decreases the rate of fast, diffusion-limited association reactions10. Alkaline phosphatase is a highly efficient enzyme that can catalyze reactions close to the diffusion-controlled limit36. It therefore appears reasonable that higher concentrations of FDP are needed to reach the maximum product formation rate in our single-cell experiment compared to in our bulk experiment, and that a higher Km is measured with our method. Levamisole on the other hand is an inhibitor and is not consumed in the enzymatic reaction. Therefore it appears reasonable that the inhibition is more efficient in the single-cell experiment compared to in bulk, resulting in a lower Ki with our method. Our results thus appear to be consistent with general predictions of differences in enzyme behaviour for environmens that deviate from the native environments inside intact cells. It should however be noted that our single-cell experiments uniquely address intracellular enzymes, while our bulk measurements also include enzymes situated extracellularly, so that the enzyme populations in these two cases are not entirely identical.
CONCLUSION
This microfluidic strategy, in combination with our newly established analysis, enables enzymatic kinetics to be determined on single cells, and thereby to map cell population heterogeneity. The described approach targets intracellular enzymes, as products of extracellular enzymes are rapidly removed by the flow. Post-translational modification of enzymes affects activity and cellular distribution, both of which can be addressed using our demonstrated approach. Continued investigation into enzymatic modification in situ will hopefully address some of the beckoning questions governing disease mediated enzyme function.
Supplementary Material
Acknowledgments
This work was supported by funds provided by the Swedish research council VR, the European research council ERC, and NIH R01 GM066018.
References
- 1.Bruggeman FJ, Westerhoff HV. Trends Microbiol. 2007;15:45–50. doi: 10.1016/j.tim.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Mutch SA, Kensel-Hammes P, Gadd JC, Fujimoto BS, Allen RW, Schiro PG, Lorenz RM, Kuyper CL, Kuo JS, Bajjalieh SM, Chiu DT. J Neurosci. 2011;31:1461–1470. doi: 10.1523/JNEUROSCI.3805-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Resendis-Antonio O. Plos One. 2009;4 doi: 10.1371/journal.pone.0004967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boonacker E, Stap J, Koehler A, Van Noorden CJF. Acta Histochem. 2004;106:89–96. doi: 10.1016/j.acthis.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Kovarik ML, Allbritton NL. Trends Biotechnol. 2011;29:222–230. doi: 10.1016/j.tibtech.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrado RJ, Varner JD, DeLisa MP. Curr Opin Biotechnol. 2008;19:492–499. doi: 10.1016/j.copbio.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Lizana L, Bauer B, Orwar O. Proc Natl Acad Sci U S A. 2008;105:4099–4104. doi: 10.1073/pnas.0709932105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minton AP. J Cell Sci. 2006;119:2863–2869. doi: 10.1242/jcs.03063. [DOI] [PubMed] [Google Scholar]
- 9.Schnell S, Turner TE. Prog Biophys Mol Biol. 2004;85:235–260. doi: 10.1016/j.pbiomolbio.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Zhou HX, Rivas GN, Minton AP. Annual Review of Biophysics. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Carlo D, Aghdam N, Lee LP. Anal Chem. 2006;78:4925–4930. doi: 10.1021/ac060541s. [DOI] [PubMed] [Google Scholar]
- 12.Sasuga Y, Iwasawa T, Terada K, Oe Y, Sorimachi H, Ohara O, Harada Y. Anal Chem. 2008;80:9141–9149. doi: 10.1021/ac8016423. [DOI] [PubMed] [Google Scholar]
- 13.Wudl L, Paigen K. Science. 1974;184:992–994. doi: 10.1126/science.184.4140.992. [DOI] [PubMed] [Google Scholar]
- 14.He M, Edgar JS, Jeffries GDM, Lorenz RM, Shelby JP, Chiu DT. Anal Chem. 2005;77:1539–1544. doi: 10.1021/ac0480850. [DOI] [PubMed] [Google Scholar]
- 15.Huebner A, Olguin LF, Bratton D, Whyte G, Huck WTS, de Mello AJ, Edel JB, Abell C, Hollfelder F. Anal Chem. 2008;80:3890–3896. doi: 10.1021/ac800338z. [DOI] [PubMed] [Google Scholar]
- 16.Gao N, Wang W, Zhang X, Jin W, Yin X, Fang Z. Anal Chem. 2006;78:3213–3220. doi: 10.1021/ac060278v. [DOI] [PubMed] [Google Scholar]
- 17.Baret JC, Miller OJ, Taly V, Ryckelynck M, El-Harrak A, Frenz L, Rick C, Samuels ML, Hutchison JB, Agresti JJ, Link DR, Weitz DA, Griffiths AD. Lab Chip. 2009;9:1850–1858. doi: 10.1039/b902504a. [DOI] [PubMed] [Google Scholar]
- 18.Meredith GD, Sims CE, Soughayer JS, Allbritton NL. Nat Biotechnol. 2000;18:309–312. doi: 10.1038/73760. [DOI] [PubMed] [Google Scholar]
- 19.Millan JL. Mammalian Alkaline Phosphatases - From Biology to Applications in Medicine and Biotechnology. Wiley-VCH; 2006. [Google Scholar]
- 20.Sinclair J, Pihl J, Olofsson J, Karlsson M, Jardemark K, Chiu DT, Orwar O. Anal Chem. 2002;74:6133–6138. doi: 10.1021/ac026133f. [DOI] [PubMed] [Google Scholar]
- 21.Dourmashkin RR, Harris RJC, Dougherty RM. Nature. 1962;194:1116–1119. doi: 10.1038/1941116a0. [DOI] [PubMed] [Google Scholar]
- 22.Schulz I. Methods Enzymol. 1990;191:280–300. doi: 10.1016/0076-6879(90)92077-q. [DOI] [PubMed] [Google Scholar]
- 23.Olofsson J, Pihl J, Sinclair J, Sahlin E, Karlsson M, Orwar O. Anal Chem. 2004;76:4968–4976. doi: 10.1021/ac035527j. [DOI] [PubMed] [Google Scholar]
- 24.Olofsson J, Bridle H, Sinclair J, Granfeldt D, Sahlin E, Orwar O. Proc Natl Acad Sci U S A. 2005;102:8097–8102. doi: 10.1073/pnas.0500230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olofsson J, Bridle H, Jesorka A, Isaksson I, Weber S, Orwar O. Anal Chem. 2009;81:1810–1818. doi: 10.1021/ac802081m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryttsen F, Farre C, Brennan C, Weber SG, Nolkrantz K, Jardemark K, Chiu DT, Orwar O. Biophys J. 2000;79:1993–2001. doi: 10.1016/S0006-3495(00)76447-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinberg TH, Newman AS, Swanson JA, Silverstein SC. Journal of Cell Biology. 1987;105:2695–2702. doi: 10.1083/jcb.105.6.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang ZJ, Olson NA, You WM, Haugland RP. J Immunol Methods. 1992;149:261–266. doi: 10.1016/0022-1759(92)90258-u. [DOI] [PubMed] [Google Scholar]
- 29.Martin MM, Lindqvist L. Journal of Luminescence. 1975;10:381–390. [Google Scholar]
- 30.Miller EM, Wheeler AR. Anal Chem. 2008;80:1614–1619. doi: 10.1021/ac702269d. [DOI] [PubMed] [Google Scholar]
- 31.Karlsson A, Sott K, Markstrom M, Davidson M, Konkoli Z, Orwar O. J Phys Chem B. 2005;109:1609–1617. doi: 10.1021/jp0459716. [DOI] [PubMed] [Google Scholar]
- 32.Leach AM, Wheeler AR, Zare RN. Anal Chem. 2003;75:967–972. doi: 10.1021/ac026112l. [DOI] [PubMed] [Google Scholar]
- 33.Van Belle H. Clin Chem. 1976;22:972–976. [PubMed] [Google Scholar]
- 34.Cyboron GW, Wuthier RE. J Biol Chem. 1981;256:7262–7268. [PubMed] [Google Scholar]
- 35.Rezende LA, Ciancaglini P, Pizauro JM, Leone FA. Cell Mol Biol (Noisy-le-grand) 1998;44:293–302. [PubMed] [Google Scholar]
- 36.Simopoulos TT, Jencks WP. Biochemistry. 1994;33:10375–10380. doi: 10.1021/bi00200a018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.