

Abstract
Two functionally important β1-adrenergic receptor (β1AR) polymorphisms have been identified. The R389G polymorphism influences coupling to the Gs-cAMP pathway. R389-β1ARs display enhanced activation of cAMP/PKA; they provide short-term inotropic support but also cause a predisposition to cardiomyopathic decompensation. A second S49G polymorphism is implicated in the evolution of heart failure, but the mechanism remains uncertain. This study shows that position 49 and 389 polymorphisms function in a coordinate manner to influence agonist-dependent cAMP/PKA and ERK responses. cAMP/PKA and ERK responses are more robust in HEK293 cells that heterologously overexpress G49-β1ARs, compared with S49-β1ARs. However, this phenotype is most obvious on a G389-β1AR background; the more robust agonist-dependent cAMP/PKA and ERK responses in R389-β1AR cells effectively obscure the effect of the S49G polymorphism. We also show that isoproterenol (Iso) and carvedilol activate ERK via a similar EGFR-independent mechanism in cells expressing various β1AR haplotypes. However, Iso activates ERK via an Src-independent pathway, but carvedilol-dependent ERK activation requires Src. Since the S49G polymorphism has been linked to changes in β1AR trafficking, we examined whether β1AR polymorphisms influence partitioning to lipid raft membranes. Biochemical fractionation studies show that all four β1AR variants are recovered in buoyant flotillin-enriched membranes; the distinct signaling phenotypes of the different β1AR variants could not be attributed to any gross differences in basal compartmentalization to lipid raft membranes. The allele-specific differences in β1AR signaling phenotypes identified in this study could underlie interindividual differences in responsiveness to β-blocker therapy and clinical outcome in heart failure.
Keywords: adrenergic receptor, protein kinase A, extracellular signal-regulated kinase, lipid raft
β1-adrenergic receptors (β1ARs) are the principle mediators of catecholamine-dependent changes in the force and rate of cardiac contraction. β1ARs adjust cardiac output by activating a Gs-adenylyl cyclase (AC) pathway that increases cAMP, activates protein kinase A (PKA), and phosphorylates substrates involved in excitation-contraction coupling (16). However, heightened β1AR drive in the setting of heart failure (HF) activates signaling pathways that promote cardiomyocyte hypertrophy/apoptosis, interstitial fibrosis, disordered energetics, and contractile dysfunction (8, 16). βAR inhibitors that prevent these maladaptive βAR responses have become standard therapy for HF.
While there is ample evidence that βAR blockers decrease morbidity and mortality in HF, there is considerable interindividual variability in drug responsiveness that is not readily attributable to known clinical or demographic factors. This interindividual variability has been attributed at least in part to two common nonsynonymous single nucleotide polymorphisms (SNPs) at positions 49 and 389 of the human of the β1AR receptor (3). Most studies have focused on the R389G polymorphism at the conformationally sensitive Gs binding domain in the juxtamembrane region of the COOH-terminal cytoplasmic tail. Studies to date indicate that R389-β1ARs display enhanced coupling to the Gs-cAMP pathway and enhanced agonist-dependent desensitization (relative to G389-β1ARs) when overexpressed in cultured fibroblasts (10). Transgenic cardiac-specific R389-β1AR overexpression also leads to enhanced receptor signaling and contractile function in young mice, compared with age-matched G389-β1AR hearts (11). These findings in cell culture and cardiomyocyte-targeted transgenic mouse models are consistent with clinical studies showing that the R389-β1AR (in association with a α2c-AR variant that regulates presynaptic release of norepinephrine) is a risk factor for human HF (23) and that the R389-β1AR allele predicts responsiveness to βAR inhibitor therapy in patients with HF (7, 11).
A second S49G polymorphism in the relatively short NH2 terminus of the β1AR has been implicated as a genetic determinant of βAR inhibitor responses and clinical outcome in HF (1, 9, 17, 25–27). However, the molecular basis for the clinical impact of this polymorphism is less obvious, since the relatively short/featureless βAR NH2 terminus generally is ignored in structure-function studies. While several studies have provided consistent evidence that G49-β1ARs are more susceptible, and S49-β1ARs are relatively resistant, to agonist-dependent downregulation, the available literature on the effects of the S49G polymorphism on β1AR affinity for ligands or β1AR coupling to cAMP is more ambiguous (6, 18, 21).
Despite considerable evidence that R389-β1ARs act as “gain-of-function” variants for the AC/cAMP/PKA pathway (relative to G389-β1ARs), there is only scant information on whether position 389 or 49 polymorphisms influence the magnitude or mechanism for β1AR coupling to effectors that may contribute to cardioprotection, such as ERK (4). Moreover, studies to date that typically focus on the independent effect of a single polymorphic variation in isolation may have only limited relevance to clinical phenotypes that result from the distinct haplotypes that exist in clinical populations. This study identifies allele-specific differences in β1AR coupling to cAMP/PKA and ERK pathways that have implications for the pathogenesis and treatment of clinical HF phenotypes.
MATERIALS AND METHODS
Materials.
Antibodies were from the following sources: β1-AR and flotillin-1 were from Santa Cruz Biotechnology. ERK1/2, p-ERK1/2, and p-PKA substrate antibodies were from Cell Signaling Technology (Danvers, MA). Isoproterenol (Iso), forskolin, and EGF were from Sigma (St. Louis, MO). All other chemicals were reagent grade.
Plasmids.
A plasmid that drives expression of the human S49R389-β1-AR, with an NH2-terminal Flag, was obtained from Addgene. Site-directed mutagenesis of β1-AR-Flag with the QuikChange mutagenesis system (Agilent Technologies) was performed to generate G49R389-β1ARs, S49G389-β1ARs, or G49G389-β1ARs. All constructs were confirmed by sequencing.
Cell culture and transfection.
HEK293 cells were cultured in DMEM containing 10% FCS, and 100 units/ml penicillin-streptomycin. Cells were transfected with plasmids that drive expression of human β1-AR polymorphic variants with the Effectene Transfection reagent (Qiagen) according to the instruction manual. Stable cell lines were generated by selection with G418 (400 μg/ml).
Western blotting.
Immunoblotting was performed on cell extracts according to methods described previously or manufacturer's instructions. Each panel in each figure represents the results of a single gel (exposed for a uniform duration); detection was with enhanced chemiluminescence. All results were replicated in at least three experiments on separate culture preparations.
Preparation of caveolar membranes.
Caveolin-enriched fractions were prepared according to a detergent-free purification scheme described previously (20). All steps were carried out at 4°C. In brief, cells from five 100 mm diameter dishes were washed twice with ice-cold phosphate-buffered saline and then scraped into 0.5 m sodium carbonate, pH 11.0 (0.5 ml/dish). Cells from five dishes were combined for each preparation. The extract was sequentially disrupted by homogenization with a loose-fitting Dounce homogenizer (10 strokes), a Polytron tissue grinder (three 10 s bursts), and a tip sonicator (three 20 s bursts). The homogenate was then adjusted to 40% sucrose by adding an equal volume of 80% sucrose prepared in MES-buffered saline (25 mm MES, pH 6.5, and 0.15 m NaCl), placed on the bottom of an ultracentrifuge tube, overlaid with a 5–30% discontinuous sucrose gradient (3 ml of 5% sucrose and 4 ml of 35% sucrose, both in MES-buffered saline containing 0.25 m sodium carbonate), and centrifuged at 260,000 g for 16–18 h in a SW40 rotor (Beckman Coulter, Palo Alto, CA). After centrifugation, 12 1 ml fractions were collected. A pooled caveolar fraction (fractions 4–5), a pooled fraction 8–12 (which contains the bulk of the cellular material including the cytosol and most of the particulate membrane fraction), and the insoluble pellet (which is solubilized in SDS-PAGE sample buffer) were subjected to SDS-PAGE and immunoblotting.
Measurements of βAR density and cAMP accumulation.
Radioligand binding experiments with [125I]CYP were performed on membranes prepared from each stable cell line according to published methods (24). cAMP measurements also were performed according to standard methods. In brief, cells were cultured in six-well plates, grown to confluence, and then serum deprived for 12 h. Cells were preincubated with 10 mM theophylline for 60 min and then challenged for 5 min with vehicle, Iso (10−6 M), or forskolin (10−5 M) as indicated. Assays were terminated by aspiration of the incubation buffer and addition of 0.5 ml of 100% ice-cold ethanol to each well. Cell lysates were dried in a spin vacuum, and cAMP in the residue was determined with a commercially available enzyme-linked immunosorbent assay kit (R & D Systems, Minneapolis, MN) according to manufacturer's instructions.
Statistics.
Results are shown as means ± SE and were analyzed by Student's t-test followed by Bonferroni adjustment for multiple comparisons with GraphPad Prism software.
RESULTS
The initial studies compared signaling responses induced by activation of individual β1-AR haplotypes transiently overexpressed at similar levels in HEK293 cells. Figure 1 shows that Iso induces a modest increase in ERK phosphorylation in vector-infected HEK293 that contain low levels of endogenous β2 (but not β1) ARs; HEK293 cells also display a modest increase in ERK phosphorylation in response to direct activation of AC by forskolin. β1AR overexpression leads to an increase in Iso-dependent ERK phosphorylation without any associated changes in ERK phosphorylation in response to forskolin or EGF (included as controls in the experiment). Studies with an anti-phospho-PKA substrate antibody (that recognizes protein phosphorylation at the PKA consensus RXXpS motif, used as a surrogate to track agonist-dependent activation of cAMP) show that heterologously overexpressed β1ARs also couple to the cAMP/PKA pathway. Iso induces a modest increase in anti-phospho-PKA substrate immunoreactivity in vector-infected HEK293. The Iso-dependent increase in anti-phospho-PKA substrate immunoreactivity is enhanced by heterologous overexpression of β1-ARs, without any changes in anti-phospho-PKA substrate immunoreactivity in response to treatment with forskolin (included as control in the experiments). Importantly, the magnitude of the Iso-dependent responses differed considerably for the different molecular forms of the β1AR. R389-β1ARs (with either S or G at position 49) displayed robust ERK and PKA substrate phosphorylation responses. Both responses are lower in cells that heterologously overexpress G389-β1ARs. The effect of an S49G polymorphism is most obvious in this context, where a β1AR harboring a position 49 glycine residue induces more robust ERK and PKA substrate phosphorylation responses than the G389-β1AR with a position 49 serine residue.
Fig. 1.
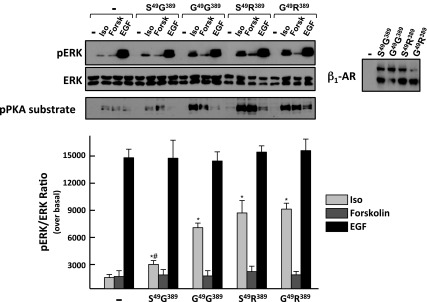
Agonist-dependent ERK and PKA responses in HEK293 cells that heterologously overexpress S49G389, G49G389, S49R389, and G49R389 haplotypes of the β1-adrenergic receptor (β1AR). Cell lysates from cells treated for 5 min with vehicle, isoproterenol (Iso, 1 μM), forskolin (Forsk, 10 μM), or EGF (100 ng/ml) were subjected to immunoblot analysis for pERK and pPKA substrate, as well as ERK protein (to verify equal protein loading) and β1AR (to track transgene expression) according to materials and methods. Results (means ± SE) from 3 separate experiments on separate culture preparations are quantified. *P < 0.05 vs. uninfected. #P < 0.05 vs. S49R389 or G49R389. S49R389 and G49R389 do not differ from each other.
The importance of S49G and R389G polymorphisms was interrogated further in clonal HEK293 cell lines that overexpress similar levels of HA-tagged S49G389-, G49G389-, or S49R389-β1ARs (Bmax values 776 ± 52, 735 ± 33, and 755 ± 41 fmol/mg protein, respectively). We compared signaling in response to activation of S49G389-β1ARs vs. S49R389-β1ARs to resolve the functional importance of the R389G polymorphism. Since the effect of the position 49 polymorphism is most pronounced on a G389-β1AR background, we resolved the functional importance of the S49G polymorphism by comparing signaling responses that result from activation of S49G389-β1ARs vs. G49G389-β1ARs. Figure 2A shows that all three β1AR variants activate the cAMP and ERK pathways. In each case, Iso-dependent ERK phosphorylation is rapid (maximal at 2–5 min) and transient (wanes with 30–60 min of continuous agonist stimulation, Fig. 2B). However, the magnitude of the Iso-dependent responses is influenced by both R389G and S49G polymorphisms. Iso-dependent increases in cAMP accumulation and ERK phosphorylation are higher in S49R389-β1AR cells than in S49G389-β1AR cells. However, a position 389 glycine residue does not necessarily impose a low level of signaling, since G389-β1ARs that harbor a position 49 glycine residue elicit more robust cAMP and ERK phosphorylation responses. Control studies verify that the differences in Iso-dependent activation of cAMP and ERK reflect the distinct signaling phonotypes of the individual heterologously overexpressed β1ARs, since forskolin-dependent cAMP accumulation and EGF-dependent ERK phosphorylation are similar in all three cell lines.
Fig. 2.

S49G389, G49G389, and S49R389 variants of β1-AR recruit the cAMP and ERK pathways. A: HEK293 cell lines that stably expressing similar amounts of S49G389-, G49G389-, or S49R389-β1ARs were treated for the indicated times with Iso (1 μM). Cell lysates were subjected to immunoblot analysis for pERK and ERK protein according to materials and methods. The results are from a single experiment and are representative of data obtained in three separate experiments. B: cAMP and pERK responses were measured in HEK293 cells treated for 5 min with vehicle (Cont), Iso (1 μM), carvedilol (Carv, 1 μM), or forskolin (10 μM) according to materials and methods. The results (means ± SE) represent data from 5–7 (for cAMP) or 3 (for ERK) separate experiments performed on separate culture preparations. *P < 0.05 vs. response to cognate agonist in G49G389 or S49R389 cells.
β1ARs adopt distinct “active” conformations that differ in their ability to activate G protein-dependent vs. G protein-independent (β-arrestin-dependent) responses. Ligands that selectively activate only certain signaling pathways are referred to as “biased” ligands. Carvedilol has been characterized as a β-arrestin-biased ligand for the mouse (S49G389) β1AR; carvedilol acts as an antagonist for the classical β1AR-Gs-cAMP pathway but stabilizes a β1AR conformation that activates the β-arrestin-ERK pathway (5, 12). Figure 2 shows that carvedilol acts as a partial agonist at S49G389-, G49G389-, and S49R389-β1ARs. In each case, carvedilol treatment leads to an increase in ERK phosphorylation that is not associated with a detectable increase in cAMP. Carvedilol-dependent ERK phosphorylation responses are ∼⅓ as robust as the cognate Iso-dependent ERK phosphorylation responses in any given cell line. Control studies established that carvedilol-dependent increases in ERK phosphorylation are maximal (do not increase further at higher carvedilol concentrations) and are specifically mediated by β1ARs (are inhibited by propranolol; data not shown).
Previous studies have attributed β1AR-dependent activation of ERK to a mechanism involving Src and the activation of a matrix metalloproteinase that cleaves HB-EGF and trans-activates EGFRs (12). However, these studies were performed on HEK293 cells that heterologously overexpress the mouse S49G389-β1ARs variant and EGFRs. Studies that rigorously compare the magnitude and mechanism for ERK activation by individual polymorphic variants of the β1AR at more physiologically relevant levels of EGFR expression are illustrated in Figure 3. The results show that AG1478 (an EGFR kinase inhibitor) completely abrogates EGF-dependent ERK phosphorylation, but AG1478 does not block either Iso- or carvedilol-dependent increases in ERK phosphorylation in β1AR cells that do not heterologously overexpress EGFRs. These results indicate that all three β1AR variants couple to an ERK pathway that does not require EGFR activity (i.e., the EGFR transactivation presumably is recruited only at high level of EGFR overexpression). The PKA inhibitor H89 also does not interfere with β1AR-dependent ERK activation (under conditions where H89 pretreatment completely abrogated Iso-dependent increases in phospho-PKA substrate immunoreactivity; data not shown). However, an agonist-dependent difference in the mechanism for ERK activation by Iso and carvedilol was exposed in studies with PP1, an inhibitor of Src kinase activity. Figure 3 shows that carvedilol activates ERK via an Src-dependent pathway that is inhibited by PP1, whereas the Iso-dependent pathway for ERK activation persists in PP1-treated cells. These results indicate that Iso and carvedilol stabilize β1ARs in slightly different active conformations that couple to distinct signal transduction pathways and signaling responses.
Fig. 3.

Iso and Carv recruit the ERK phosphorylation response via distinct pathways. HEK293 cells stably expressing similar amounts of S49G389-, G49G389-, or S49R389-β1ARs were pretreated for 30 min with vehicle AG1478 (AG, 10 μM), PP1 (10 μM), or H89 (10 μM) prior to a 5 min challenge with 1 μM Iso, 1 μM Carv, or EGF (100 ng/ml). The results of immunoblot analyses from a single representative experiment are depicted on top, with the data from 3 separate experiments (means ± SE) quantified at bottom. *P < 0.05 vs. cognate agonist response in G49G389 or S49R389 cells. #P < 0.05 vs. agonist alone.
We previously demonstrated that β1ARs partition to lipid raft membranes (19). Cell lysates were solubilized in detergent-free alkaline sodium carbonate buffers and subjected to isopycnic centrifugation on a discontinuous sucrose gradient to determine whether S49G or G389R polymorphisms alter signaling phenotypes by influencing β1AR partitioning to buoyant lipid raft membranes. Figure 4 shows that β1AR variants are recovered almost exclusively in the light sucrose gradient fraction that contains the bulk of the cellular flotillin immunoreactivity (a lipid raft recovery marker) and excludes >98% of total cell protein (including Golgi and other intracellular organelle markers). We did not identify any gross allele-specific differences in β1AR partitioning to lipid raft microdomains.
Fig. 4.

S49G389-, G49G389-, S49R389-, and G49R389-β1AR haplotypes display similar basal localization to lipid raft membranes. Cav, caveolin; F, fraction.
DISCUSSION
There is recent evidence that polymorphisms at positions 49 and 389 in the coding region of the β1AR act as genetic modifiers of clinical outcome in various cardiovascular disorders. Most studies have focused on the role of the R389G polymorphism as a predictor of pharmacologic responsiveness to β-AR inhibitor therapy, showing that R389-β1ARs (the more common allele in individuals of European descent) adopt a more active conformation, display enhanced coupling to the Gs-cAMP pathway, and confer enhanced sensitivity to β-blocker therapy in HF, relative to G389-β1ARs. The position 389 polymorphism could in theory also impact on clinical outcome by influencing signaling via cardioprotective pathways involving ERK, but a role for the R389G polymorphism as a modifier of signaling via G protein-independent signaling pathways has not previously been considered. The S49G polymorphism has received considerably less attention, with the analysis largely confined to epidemiologic studies that implicate the S49G polymorphism as a genetic determinant of clinical outcome in HF. Two laboratories have identified allele-specific differences in agonist-dependent downregulation kinetics in studies performed in reductionist models (6, 18). A single study from Levin et al. (6) also showed that a position 49 glycine residue confers enhanced basal and agonist-dependent coupling to the AC/cAMP pathway (i.e., the β1AR harboring a position 49 glycine residue is a constitutively active receptor), but this finding was not replicated by others (18). Studies to date also have not rigorously considered a possible interplay between the polymorphisms at positions 49 and 389. Therefore, we examined the signaling profiles of β1AR polymorphic variants harboring either serine or glycine at position 49 and either glycine or arginine at position 389.
Studies reported herein show that 1) agonist-dependent cAMP/PKA and ERK responses are more robust in R389-β1AR cells, than in G389-β1AR cells and that 2) the effect of the S459G polymorphism is most obvious on a G389-β1AR background; G389-β1ARs with a glycine at position 49 display enhanced agonist-dependent cAMP/PKA and ERK responses, compared with S49G389-β1ARs. Of note, the two previous studies that linked the S49G polymorphism to changes in β1AR trafficking and desensitization kinetics also were performed on a G389 background (6, 18); effects of a position 49 SNP on signaling by R389-β1ARs have never been considered. This oversight may be important, since clinical studies to date do not provide an entirely coherent picture of the importance of β1AR polymorphisms as genetic modifiers of cardiac phenotypes. It is tempting to speculate that discrepancies in the literature could be due to the stratification of patients according to a single polymorphic locus (either position 49 or position 389), rather than the haplotypes that exist in the general population. This is important for two reasons. First, glycine is the minor allele at both loci, but there are important racial differences in allele frequencies for both S49G and R389G polymorphisms that could confound an analysis. Second, there is strong linkage disequilibrium between S49G and R389G loci that functions to limit the haplotypes (combinations of specific polymorphisms) in the general population. Most studies identify S49/R389-, S49/G389-, and G49/R389-β1ARs as the most prevalent haplotypes in Caucasians; the G49/G389 haplotype appears to be quite rare (14, 22). Hence, while the results of this and previous studies in reductionist models that characterize the signaling properties of the S49G polymorphism on a G389 background are of interest from a theoretical standpoint, the physiological relevance of these findings will require further study.
Since the S49G polymorphism has been linked to changes in β1AR trafficking, and the spatial relationship between the βAR and its downstream effectors can function to either facilitate or restrict signaling responses, we examined whether polymorphic variants of the β1AR compartmentalize differently between lipid raft and nonraft membranes. Lipid rafts are a buoyant membrane fraction that are highly enriched in βARs, Gs, several AC isoforms, EGFRs, and components of the ERK-MAPK pathway; lipid rafts have been implicated as platforms that “launch” signaling to the ERK cascade. Our fractionation studies show that the various polymorphic variants of the β1AR are recovered exclusively in the buoyant lipid raft membrane fraction; we did not identify an obvious difference in the partitioning of β1AR variants between lipid raft and nonraft membranes that might explain their distinct signaling phenotypes. However, these studies were performed on resting cells that had not been exposed to agonist ligands; they do not rule out possible allele-specific differences in β1AR trafficking patterns following agonist exposure that would influence signaling responses.
HF is associated with elevated catecholamine levels and chronic/persistent β1AR stimulation; this heightened sympathetic tone contributes to the pathogenesis of various HF syndromes. The cardiotoxic actions of β1AR have been attributed to Gs-dependent activation of AC and enhanced accumulation of cAMP. Drugs that inhibit cardiotoxic β1AR-mediated responses (such as metoprolol, bisoprolol, and carvedilol) have been approved for the treatment of HF syndromes. However, their clinical efficacy is not equivalent, with evidence that carvedilol affords survival advantage over other approved pharmacologic inhibitors (15). While this has been ascribed to carvedilol's ancillary antioxidant, anti-inflammatory, antiproliferative, or anti-α1-AR actions (2, 13, 29), a mechanism related to carvedilol's unique pharmacologic profile at β1ARs also is possible. Carvedilol acts like other βAR inhibitors to block the βAR-Gs-cAMP pathway, but carvedilol also exhibits an additional action as a partial agonist for the G protein-independent cardioprotective pathway that activates ERK (28). Previous literature examined carvedilol's pharmacologic actions in cells that overexpress only certain molecular forms of the β1AR. This study is the first to extend the analysis and show that carvedilol exerts a similar action as a biased partial agonist for the ERK pathway at all β1AR variants. However, we identified allele-specific differences in the magnitude of the carvedilol-dependent ERK phosphorylation response that mirror the allele-specific differences in ERK phosphorylation by Iso. We also identified an agonist-dependent difference in the mechanism for β1AR-dependent activation of ERK. Notably, while previous studies showed that Iso and carvedilol recruit similar EGFR-/Src-dependent pathways to activate ERK in HEK293 cells that heterologously overexpress both β1ARs and EGFRs (12), we show that β1ARs activate ERK via an EGFR-independent mechanism at endogenous levels of EGFR expression. However, an agonist-dependent difference in the mechanism for β1AR activation of ERK activation was identified under these conditions. We show that carvedilol activates ERK via a Src-dependent mechanism, whereas the Iso-dependent ERK pathway is Src independent; the pharmacologic profiles for Iso- and carvedilol-dependent ERK activation are similar across the various β1AR polymorphic variants. The EGFR-independent mechanisms for ERK activation by β1ARs, which may or may not involve Src and presumably are obscured at high levels of EGFR overexpression, may contribute to β1AR-dependent cardioprotection in the heart and are the focus of further study.
In summary, this study identifies allele-specific differences in signaling phenotypes for different β1AR polymorphic variants. These differences in the cellular actions of individual β1AR variants could contribute to the pathogenesis of clinical HF phenotypes and may have bearing on the success of β-blocker therapy.
GRANTS
This work was supported by U.S. Public Health Service National Heart, Lung, and Blood Institute Grants HL-93343 and HL-95605.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.Z. performed experiments; F.Z. and S.F.S. analyzed data; F.Z. and S.F.S. interpreted results of experiments; F.Z. and S.F.S. prepared figures; F.Z. and S.F.S. drafted manuscript; F.Z. and S.F.S. approved final version of manuscript; S.F.S. conception and design of research; S.F.S. edited and revised manuscript.
REFERENCES
- 1.Borjesson M, Magnusson Y, Hjalmarson A, Andersson B. A novel polymorphism in the gene coding for the β1-adrenergic receptor associated with survival in patients with heart failure. Eur Heart J 21: 1853–1858, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Bristow MR, Larrabee P, Minobe W, Roden R, Skerl L, Klein J, Handwerger D, Port JD, Muller-Beckmann B. Receptor pharmacology of carvedilol in the human heart. J Cardiovasc Pharmacol 19, Suppl 1: S68–S80, 1992 [DOI] [PubMed] [Google Scholar]
- 3.Dorn GW, 2nd, Liggett SB. Mechanisms of pharmacogenomic effects of genetic variation within the cardiac adrenergic network in heart failure. Mol Pharmacol 76: 466–480, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev 12: 181–188, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, Rockman HA. β-Blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA 105: 14555–14560, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levin MC, Marullo S, Muntaner O, Andersson B, Magnusson Y. The myocardium-protective Gly-49 variant of the β1-adrenergic receptor exhibits constitutive activity and increased desensitization and down-regulation. J Biol Chem 277: 30429–30435, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, Weber SA, Greene SM, Hodne D, Nelson B, Morrison J, Domanski MJ, Wagoner LE, Abraham WT, Anderson JL, Carlquist JF, Krause-Steinrauf HJ, Lazzeroni LC, Port JD, Lavori PW, Bristow MR. A polymorphism within a conSved β1-adrenergic receptor motif alters cardiac function and β-blocker response in human heart failure. Proc Natl Acad Sci USA 103: 11288–11293, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of β-adrenergic signaling in heart failure? Circ Res 93: 896–906, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Magnusson Y, Levin MC, Eggertsen R, Nystrom E, Mobini R, Schaufelberger M, Andersson B. Ser49Gly of β1-adrenergic receptor is associated with effective β-blocker dose in dilated cardiomyopathy. Clin Pharmacol Therapeut 78: 221–231, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Mason DA, Moore JD, Green SA, Liggett SB. A gain-of-function polymorphism in a G-protein coupling domain of the human β1-adrenergic receptor. J Biol Chem 274: 12670–12674, 1999 [DOI] [PubMed] [Google Scholar]
- 11.Mialet Perez J, Rathz DA, Petrashevskaya NN, Hahn HS, Wagoner LE, Schwartz A, Dorn GW, Liggett SB. β1-Adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med 9: 1300–1305, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest 117: 2445–2458, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohlstein EH, Douglas SA, Sung CP, Yue TL, Louden C, Arleth A, Poste G, Ruffolo RR, Jr, Feuerstein GZ. Carvedilol, a cardiovascular drug, prevents vascular smooth muscle cell proliferation, migration, and neointimal formation following vascular injury. Proc Natl Acad Sci USA 90: 6189–6193, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pacanowski MA, Gong Y, Cooper-Dehoff RM, Schork NJ, Shriver MD, Langaee TY, Pepine CJ, Johnson JAInvestigators I β-Adrenergic receptor gene polymorphisms and β-blocker treatment outcomes in hypertension. Clin Pharmacol Therapeut 84: 715–721, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poole-Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, Lubsen J, Lutiger B, Metra M, Remme WJ, Torp-Pedersen C, Scherhag A, Skene A, Carvedilol Or Metoprolol European Trial Investigators Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 362: 7–13, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Port JD, Bristow MR. Altered β-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol 33: 887–905, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Ranade K, Jorgenson E, Sheu WH, Pei D, Hsiung CA, Chiang FT, Chen YD, Pratt R, Olshen RA, Curb D, Cox DR, Botstein D, Risch N. A polymorphism in the β1-adrenergic receptor is associated with resting heart rate. Am J Hum Genet 70: 935–942, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rathz DA, Brown KM, Kramer LA, Liggett SB. Amino acid 49 polymorphisms of the human β1-adrenergic receptor affect agonist-promoted trafficking. J Cardiovasc Pharmacol 39: 155–160, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J Biol Chem 275: 41447–41457, 2000 [DOI] [PubMed] [Google Scholar]
- 20.Rybin VO, Xu X, Steinberg SF. Activated protein kinase C isoforms target to cardiomyocyte caveolae: stimulation of local protein phosphorylation. Circ Res 84: 980–988, 1999 [DOI] [PubMed] [Google Scholar]
- 21.Sandilands A, Yeo G, Brown MJ, O'Shaughnessy KM. Functional responses of human beta1-adrenoceptors with defined haplotypes for the common 389R>G and 49S>G polymorphisms. Pharmacogenetics 14: 343–349, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Small KM, Mialet-Perez J, Liggett SB. Genetic variation within the β1-adrenergic receptor gene results in haplotype-specific expression phenotypes. J Cardiovasc Pharmacol 51: 106–110, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Small KM, Wagoner LE, Levin AM, Kardia SL, Liggett SB. Synergistic polymorphisms of β1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med 347: 1135–1142, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Steinberg SF, Zhang H, Pak E, Pagnotta G, Boyden PA. Characteristics of the β-adrenergic receptor complex in the epicardial border zone of the 5-day infarcted canine heart. Circulation 91: 2824–2833, 1995 [DOI] [PubMed] [Google Scholar]
- 25.Terra SG, Hamilton KK, Pauly DF, Lee CR, Patterson JH, Adams KF, Schofield RS, Belgado BS, Hill JA, Aranda JM, Yarandi HN, Johnson JA. β1-adrenergic receptor polymorphisms and left ventricular remodeling changes in response to β-blocker therapy. Pharmacogenet Genomics 15: 227–234, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Terra SG, Pauly DF, Lee CR, Patterson JH, Adams KF, Schofield RS, Belgado BS, Hamilton KK, Aranda JM, Hill JA, Yarandi HN, Walker JR, Phillips MS, Gelfand CA, Johnson JA. β-adrenergic receptor polymorphisms and responses during titration of metoprolol controlled release/extended release in heart failure. Clin Pharmacol Therapeut 77: 127–137, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Wenzel K, Felix SB, Bauer D, Heere P, Flachmeier C, Podlowski S, Kopke K, Hoehe MR. Novel variants in 3 kb of 5′UTR of the β1-adrenergic receptor gene (−93C>T, −210C>T, and −2146T>C): −2146C homozygotes present in patients with idiopathic dilated cardiomyopathy and coronary heart disease. Hum Mutat 16: 534, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of β-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA 104: 16657–16662, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yue TL, Cheng HY, Lysko PG, McKenna PJ, Feuerstein R, Gu JL, Lysko KA, Davis LL, Feuerstein G. Carvedilol, a new vasodilator and β-adrenoceptor antagonist, is an antioxidant and free radical scavenger. J Pharmacol Exp Ther 263: 92–98, 1992 [PubMed] [Google Scholar]