

Abstract
Richieri-Costa-Pereira syndrome is an autosomal-recessive acrofacial dysostosis characterized by mandibular median cleft associated with other craniofacial anomalies and severe limb defects. Learning and language disabilities are also prevalent. We mapped the mutated gene to a 122 kb region at 17q25.3 through identity-by-descent analysis in 17 genealogies. Sequencing strategies identified an expansion of a region with several repeats of 18- or 20-nucleotide motifs in the 5′ untranslated region (5′ UTR) of EIF4A3, which contained from 14 to 16 repeats in the affected individuals and from 3 to 12 repeats in 520 healthy individuals. A missense substitution of a highly conserved residue likely to affect the interaction of eIF4AIII with the UPF3B subunit of the exon junction complex in trans with an expanded allele was found in an unrelated individual with an atypical presentation, thus expanding mutational mechanisms and phenotypic diversity of RCPS. EIF4A3 transcript abundance was reduced in both white blood cells and mesenchymal cells of RCPS-affected individuals as compared to controls. Notably, targeting the orthologous eif4a3 in zebrafish led to underdevelopment of several craniofacial cartilage and bone structures, in agreement with the craniofacial alterations seen in RCPS. Our data thus suggest that RCPS is caused by mutations in EIF4A3 and show that EIF4A3, a gene involved in RNA metabolism, plays a role in mandible, laryngeal, and limb morphogenesis.
Main Text
Richieri-Costa-Pereira syndrome (RCPS [MIM 268305]) is an autosomal-recessive syndrome characterized by a midline cleft mandible in addition to Robin sequence, laryngeal abnormalities, and radial and tibial deficiencies associated with clubfeet1 (Figure 1). Learning and language disabilities have been reported in more than 50% of the affected individuals.1 All but one of the RCPS-affected families described to date are from Brazil, suggesting a founder effect.1 Previous attempts to identify the genetic cause of RCPS were unsuccessful,2 possibly because the causative mutation is ancient and the shared flanking region has been reduced by multiple recombinations. We performed homozygosity mapping analysis with the Affymetrix 50K Xba1 SNP array in seven individuals belonging to four consanguineous families (Table S1 available online, individuals 1, 5–7, 9, 12, and 19 and Figure S1) and one unaffected sibling for each of the probands 6, 9, and 19.1 Although these families came from the same geographic region, they have no knowledge of shared ancestry. Approval for research on human subjects was obtained from the ethics committee of the HRAC-USP-Bauru. All samples were collected after individuals or their relatives had provided informed consent. The region of homozygosity was selected with the Homozygosity Mapper online program assuming a rare-recessive model of inheritance. The only extended region of homozygosity unique to affected individuals was observed at 17q25.3 (Figure S1). This region was further genotyped with 9 microsatellite markers and 10 SNPs (Table S2) in 20 affected individuals from 17 apparently unrelated families and 46 unaffected relatives (31 parents and 15 unaffected siblings). LOD scores were estimated by the Merlin3 software under a parametric model including data from 8 families (families of the probands 2–5, 7, 9, 10, and 13). Segregation analysis with the above-mentioned markers confirmed linkage to 17q25.3 (Figure S2; maximum LOD score: 9.533 at θ = 0.0 for marker rs2289534). Additional recombinants at rs2289534 (patients 8 and 17) and rs3829612 (patient 11) (Figure S1B) refined the disease locus to a 122 kb region (UCSC Genome Browser human reference genome build hg19, chr17: 78,039,369–78,161,152) containing CCDC40 (MIM 613799), GAA (MIM 606800), EIF4A3 (MIM 608546), and CARD14 (MIM 607211). Mutations in GAA cause glycogen storage disease type II (GSD2 [MIM 232300]), and therefore GAA was excluded as candidate for RCPS. Most recently, heterozygous mutations in CARD14 have been associated with psoriasis (PSOR2 [MIM 602723])4 and biallelic CCDC40 loss-of-function mutations were associated with ciliary dyskinesia, primary 15 (CIDL5 [MIM 613808]).5 Because there is no clinical overlap between the phenotypes in RCPS and those above syndromes, EIF4A3 is the most likely candidate for RCPS.
Figure 1.
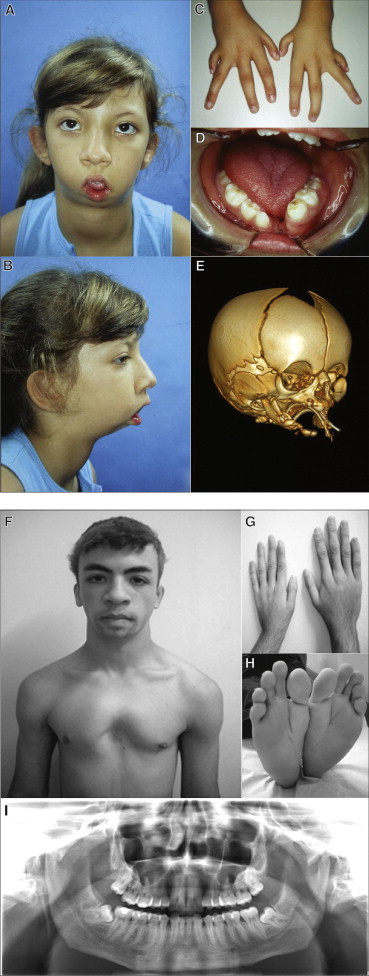
Clinical and Radiographic Aspects of Typical and Milder Forms of RCPS
(A–D) Affected individual 2 at 9 years of age, illustrating (A, B) typical facial features of the syndrome including micrognathia and microstomia, (C) hypoplasia of fingers and clinodactyly, and (D) absence of lower central incisors and median mandibular cleft.
(E) CT scan of the skull of affected individual 23 at 12 days of age. Note the very rudimentary mandibular formation with large medial cleft, micrognathia, and incomplete zygomatic arches.
(F–I) Affected individual 25, illustrating (F) absence of microstomia and presence of pectus excavatum, (G) short left hand with hypoplastic thumbs, (H) feet with characteristically abnormal shape, and (I) normal fusion of mandible.
Except for exon 1 of EIF4A3, which proved difficult to sequence, Sanger sequencing of the remaining exons of EIF4A3, of all exons of CCDC40 and CARD14, and of their exon-intron boundaries did not reveal any nonconservative coding substitutions or obvious splice site disruptions (Table S3). Whole-exome sequencing (WES) was performed in affected individual 17 according to previously standardized protocols.6 WES revealed only seven homozygous nonsynonymous changes not described in the available SNP database at that time (NCBI dbSNP v.132) but none of them mapped to 17q25.3.
By re-examining the WES data for the candidate region, we found that exon 1 of EIF4A3 had low coverage (mean coverage = 3.6) compared to the other exons (mean coverage of the remaining exons = 17.2); in addition, the EIF4A3 5′ UTR is GC rich, which could explain our technical difficulties in sequencing this region. PCR amplification of exon 1 showed a larger homozygous allele only among affected individuals (Figure S3). This larger 5′ UTR allele was not observed in any of the 520 Brazilian control individuals, further confirming its association with the disease. To better characterize the 5′ UTR of EIF4A3, we sequenced 140 control alleles from 70 unrelated Brazilian individuals and discovered multiple allelic patterns, which varied in size and organization of motifs containing 18 or 20 nucleotides (nt). These motifs could be divided in three types: (1) a 20-nt motif, TCGGCAGCGGCACAGCGAGG, termed CACA-20-nt; (2) a 18-nt motif, TCGGCAGCGGCAGCGAGG, termed CA-18-nt; and (3) another 20-nt motif that possessed a G instead of an A, TCGGCAGCGGCGCAGCGAGG, termed CGCA-20-nt. The most prevalent (97%) allelic pattern among controls was characterized by an initial CACA-20-nt repeated between 2 and 9 times, followed by one CA-18-nt, another CACA-20-nt, and one final CA-18-nt (total repeats = 5 to 12), ending 43 bases upstream of the first ATG (Figure 2). In turn, affected individuals exhibited the following pattern: an initial CACA-20-nt, followed by 12 to 13 repeats of CGCA-20-nt, one CACA-20-nt, and one final CA-18-nt (total number of repeats = 15 or 16). To maintain clarity, the identified alleles are hereafter referred by the absolute number of repeats, which varied from 3 to 12 in controls and were either 15 or 16 in these RCPS-affected individuals. We observed that 17 affected probands were homozygous for the 16-repeat allele, and 3 apparently unrelated affected individuals (6, 14, and 18; Figure S2) were compound heterozygotes (15 or 16 repeats). All tested parents were heterozygous for the 16-repeat allele, and unaffected siblings either lacked the expanded allele or were heterozygotes (Figure S2). Accordingly, the expanded alleles segregated perfectly with the disease, following an autosomal-recessive model.
Figure 2.
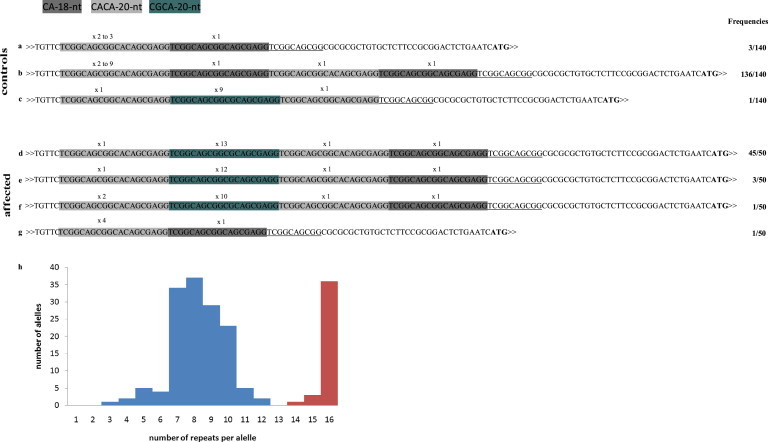
Representation of the 5′ UTR of EIF4A3 Alleles According to the Pattern of the Repeats and Their Distribution Frequency
(A–C) Three patterns of repeats in control alleles: note that the number of CA-18-nt motifs varied among the different allele patterns and that the CGCA-20-nt motif was present in only 1 out of 140 alleles.
(D–F) Three patters of repeats in affected alleles: the 15- and 16-repeat alleles have the same underlying structure (D, E), whereas the 14-repeat allele contains an additional copy of the CACA-20-nt motif (F).
(G) The nonexpanded 5′ UTR in cis with the nucleotide substitution c.809A>G (p.Asp270Gly) allele in individual 25 corresponds to one of the patterns of the control individuals (A).
The first thymidine (T) at the 5′ end corresponds to position +38 of the transcription initiation site and the ATG corresponds to the translation initiation codon. Underlined sequence downstream of the last 18-nt motif represents a partially conserved motif in all alleles.
(H) Distribution of the alleles per repeat size. Control sample alleles are shown in blue and affected alleles shown in red. The counting of the alleles in affected individuals took into consideration relatedness.
Comparative analysis of the 5′ UTR of EIF4A3 mammalian orthologs showed that only anthropoid primates, which present mandible fusion as one of their morphological autapomorphies,7 share a repetitive sequence highly similar to the human CA-18-nt motif (Figure S4). It is thus possible that the most ancient allele in humans might have had one or few CA-18-nt motifs and that the 20-nt motif has arisen more recently by a CA or, more rarely, by a CG insertion. We speculate that an increased instability of the region is possibly being driven by the CGCA-20-nt motif; however, current data are not sufficient to predict the mechanisms responsible for the appearance of the 16-repeat allele linked to the disorder. Even though the 16-repeat allele seems to be relatively stable through meiosis and to have a unique pattern of organization, we classified this mutation as an expansion and added RCPS to the growing group of disorders caused by noncoding repeat expansions, which includes Friedreich ataxia8 (FRDA [MIM 229300]), myotonic dystrophies9 (DM1 [MIM 60900]; DM2 [MIM 602668]), fragile X-associated tremor/ataxia syndromes10 (Fragile X syndrome [MIM 300624], FXTAS [MIM 300623]), several spinocerebellar ataxias,11–14 and frontotemporal dementia and amyotrophic lateral sclerosis15 (FTDALS [MIM 105550]) (reviewed in Table S4).
EIF4A3 encodes a DEAD box helicase (eIF4AIII), the core protein of the exon junction complex (EJC). It coordinates the control of downstream processes of mRNA splicing and nonsense-mediated mRNA decay (NMD) and is also involved in rRNA biogenesis.16–20 eIF4AIII interacts directly with mRNA and forms the minimally stable core of the EJC by interacting with Y14, MAGOH, UPF3, MLN51, and RNPS1.21,22
Sequencing of EIF4A3 in five additional Brazilian affected individuals ascertained elsewhere23,24 revealed that four of them (individuals 21–24, including 2 siblings) were homozygous for the 16-repeat allele. In contrast, the fifth (individual 25) was a compound heterozygote, possessing a 14-repeat allele in trans with a nucleotide change, c.809A>G (EIF4A3 transcript, RefSeq accession number NM_014740.3), which leads to an amino acid substitution at Asp270 (p.Asp270Gly) (Figure 3). The 14-repeat allele showed a pattern of motifs distinct from that of the typical 16-repeat allele (Figure 2). These two mutations were not identified in any database nor in our control sample (n = 520/expansion allele; n = 285/substitution mutation). Strong functional constraint of Asp270 was suggested by its location in the C-terminal helicase RecA2 domain and by its complete conservation not only in eIF4AIII orthologs from seven divergent vertebrate species, plant, and yeast but also in eIF4A paralogs (Figure 3). SIFT and PolyPhen analyses predicted that Gly at position p.270 perturbs the structure and function of eIF4AIII (Figure 3). The side chain of aspartate 270 forms a hydrogen bond with Tyr429 in UPF3B (Figure 3), an EJC protein involved in NMD,26–28 and mutation of this residue also affects eIF4AIII recruitment by CWC22 to the spliceosome.29 Furthermore, glycine strongly destabilizes helices compared to aspartate,25 so the helical structure downstream of the mutated residue may also be affected (Figure 3C). Together, these observations suggest that p.Asp270Gly is probably pathogenic and that RCPS is caused by different mutational mechanisms: expansion of the 5′ UTR (14 to 16 repeats) or missense mutation.
Figure 3.
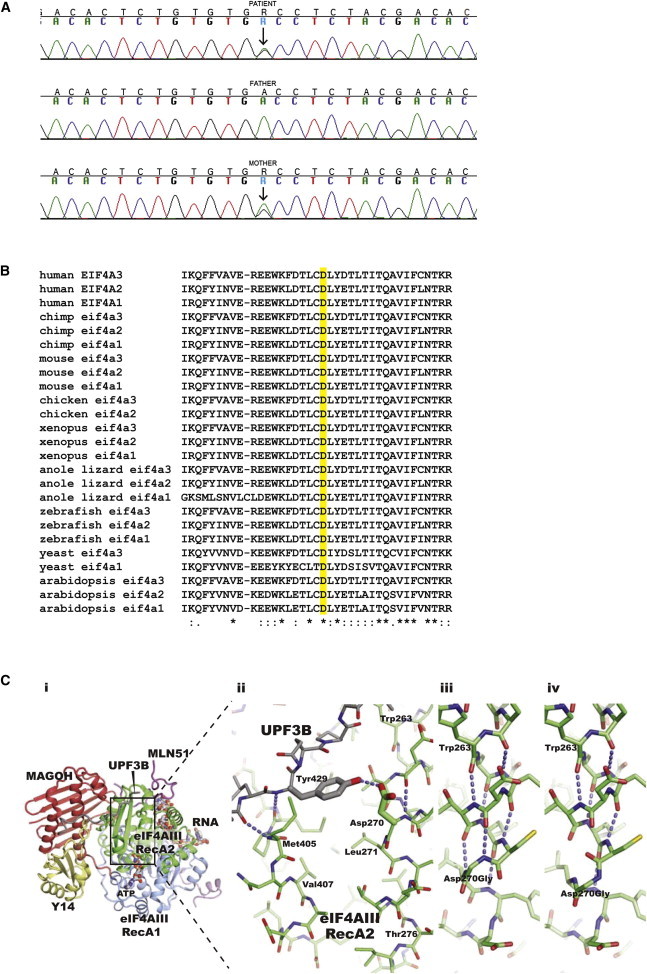
Description of the Missense Mutation in EIF4A3, Evolutionary Comparative Analysis, and Structural Analysis
(A) Sequence analysis of exon 8 showing the A to G substitution at position c.809 (indicated by an arrow), leading to the amino acid substitution p.Asp270Gly. The online tool PolyPhen-2 predicts this mutation as possibly damaging with a score of 0.860 (sensitivity: 0.72 and specificity: 0.89).
(B) Comparative sequence analysis of EIF4A3 encoded orthologous proteins and its paralogs, showing that the Asp270 (highlighted D) is highly conserved throughout evolution.
(C) Structure of the core human exon junction complex bound to a C-terminal fragment of UPF3B (PDB entry 2XB2 for RCSB). (i) Overall view: the RNA and ATP binding eIF4A3 is shown with its RecA1 and RecA2 domains colored blue and green, respectively. A fragment of MLN51 (magenta) encloses both RecA domains and also forms part of the binding site for the 5′ end of the RNA. MAGOH (red) contacts both domains of eIF4AIII, the MLN51 fragments, and positions Y14 (yellow) in the complex. The C-terminal UPF3B fragment (gray) interacts with both Y14 and the eIF4AIII RecA2 domain. (ii) Close-up on the UPF3B Tyr429 interaction with the Asp270 in the eIF4A3 RecA2 domain. Selected putative hydrogen bonds of relevance for the Asp-Gly mutation are indicated with blue dashed lines. (iii) Selected model of the glycine mutant with a secondary structure similar to that of the experimental structures of wild-type eIF4AIII. (iv) Selected model of the glycine mutant with a disrupted secondary structure compared to the experimental structures of wild-type eIF4AIII. Models were prepared with Modeler.25
The 15- and 16-repeat allele haplotypes spanning EIF4A3 are consistent with a common origin (Figures S2 and S5) and corroborate our previous founder effect hypothesis for most Brazilian RCPS-affected individuals. The 15-repeat alleles might represent a retraction of the 16-repeat allele or alternatively might have arisen by unequal crossing over (Figure S2), as suggested in polyalanine expansion disorders.30 The 14-repeat allele is embedded in the same 42 kb haplotype observed in the recombinant 16-repeat alleles (Figure S5); however, its origin remains unclear given its distinct motif organization and structure (Figure 2). In contrast, the c.809A>G mutation is embedded in a distinct haplotype (Figures S2 and S5), suggesting multiple pathogenic mutational origins in EIF4A3.
The phenotype of RCPS-affected individuals varies in expressivity even within families. The main clinical craniofacial characteristic fully penetrant in all 16-repeat allele homozygous or 15/16-repeat allele compound heterozygous individuals is midline mandibular involvement, ranging from abnormal fusion observed radiologically to complete lack of fusion leading to a wide gap at the mandibular symphysis (Figure 1 and Table S1).1 The three affected compound heterozygous individuals for the 15/16-repeat alleles presented a very similar phenotype to the homozygous 16/16 affected individuals (Table S1). In contrast, individual 25, who is a compound heterozygote for a 14-repeat allele and c.809A>G (p.Asp270Gly), was the only individual with mandible fusion (Table S1; Figure 1). The milder phenotype in individual 25 might be due to a weaker effect of the amino acid substitution mutation or of the 14-repeat allele in comparison to the 16-repeat allele. Atypical or milder phenotypes have been associated with substitution mutations in Friedreich ataxia,31 an expansion disorder with an autosomal-recessive inheritance pattern (Table S4).
To further understand the functional effect of the EIF4A3 expanded alleles, we first investigated EIF4A3 transcripts. Through cDNA sequencing analysis, we showed that the 5′ UTR repeats are included in the EIF4A3 transcripts from white blood cells from both controls and affected individuals (data not shown). We did not observe any evidence of EIF4A3 alternative splicing either in controls or affected individuals (Figure S6). Next, we performed real-time quantitative PCR (RT-qPCR) for EIF4A3 with mRNA obtained from white blood cells (four affected and nine control individuals) and from mesenchymal cells (2 affected and 11 control individuals) by standardized methods.32,33 EIF4A3 transcript abundance was about 30%–40% lower in affected individuals than in controls in both cell types tested (white blood cells, p = 0.0193; mesenchymal cells, p = 0.001; Figure S6). It is likely that the expanded allele does not alter the splicing of EIF4A3 mRNA but it seems to reduce its abundance in the RCPS cell types investigated. Further studies, including quantification of expressed protein in different cell types, will be necessary to confirm whether this expansion represents a partial loss-of-function mutation. We also cannot exclude the possibility that this expanded allele leads to RNA toxicity affecting other proteins, as has been shown for full expansions at DMPK (MIM 605377) and at CNBP1 (MIM 116955).34 The substitution p.Asp270Gly is likely to weaken the interaction between eIF4AIII and UPF3B, thus resulting in a less-efficient NMD and transcriptional or translation regulation. It is thus possible that the mechanism behind the RCPS phenotype might be a partial loss-of-function of EIF4A3 (hypomorphic mutation), which would be the expected model considering the autosomal-recessive inheritance pattern of the disease.
To further investigate the role of EIF4A3 in craniofacial development as well as its deficiency as a putative mechanism for RCPS, we modeled eif4a3 deficiency in zebrafish embryos by using specific morpholinos (MO). Three different MOs along with their corresponding mispaired controls were purchased from Gene Tools (MO sequences in Figures 4, S7, and S8). MOs were designed to block either eif4a3 mRNA translation (TRA1-MO and TRA2-MO) or eif4a3-pre-mRNA splicing (Spl-MO). Zebrafish embryos at 1–4 cell stage were injected with 5 nl of each MO at the indicated concentration (ranging from 0.01 to 0.1 mM) depending on the MO, and development was allowed to proceed.35,36 Because a slight delay in morphant development was observed, developing fish were staged taking into account the presence of typical developmental structures. Morphants with consistent and reproducible craniofacial phenotypes were scored as a unique category (Table S5 and Figures 4, S7, and S8). Similar craniofacial phenotypes were observed regardless of the nature of the injected MO. Morphants staged at 24 hr postfertilization (hpf) showed eyes reduced in size and dark and opaque zones in all brain structures. In addition, the otic vesicle and the midbrain/hindbrain border regions were barely detectable. Anomalies in the trunk and tail were observed in some embryos, but these anomalies were not further characterized because they were variable and inconsistent among different MOs (Figures 4, S7, and S8). Acridine orange staining37 performed on 24-hpf staged morphant and control embryos suggested extensive apoptosis in morphants in comparison to controls, which was even more intense in the anterior-most regions of morphants (Figures 4, S7, and S8). Cartilage and bone staining revealed underdevelopment of craniofacial cartilage, bone alterations, and clefting of the lower jaw (Figures 5, S7, and S8). Furthermore, the third through sixth pharyngeal arches were underdeveloped in morphants. Therefore, morphant fish displayed multiple defects in craniofacial structures analogous to those in RCPS-affected individuals. Importantly, most of the abnormalities were rescued when an in vitro synthesized mRNA encoding for the zebrafish eIF4AIII translationally fused to EGFP was coinjected with each of the three MOs tested (Table S5 and Figures 4, 5, and S7–S9). The TRA1-MO and TRA2-MO anneal on two different regions of the 5′ UTR and, thus, do not anneal to the injected mRNA, ruling out MO off-targeting effects. Because craniofacial cartilage and bones mainly derive from the cranial neural crest (CNC), we assessed the expression of typical CNC marker genes in treated and control 24-hpf staged embryos. RT-qPCR revealed that eif4a3 knockdown adversely affects the transcription of typical neural crest gene markers, such as sox9b, foxd3, sox10, and tbx2 (Figure S10). Therefore, eif4a3 depletion might result in failure of the EJC assembly, which ultimately would lead to CNC cell death and underdevelopment of the pharyngeal arches.
Figure 4.
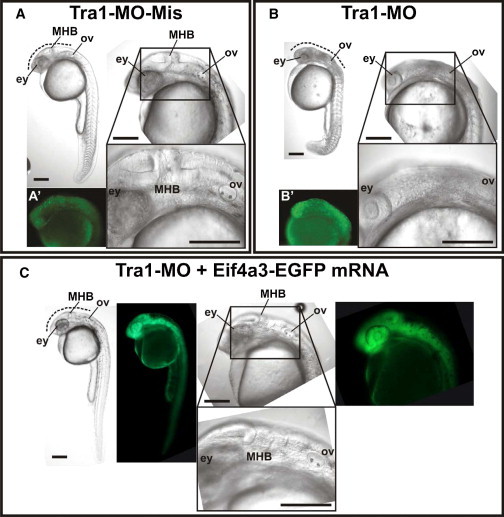
Zebrafish eif4a3 Knockdown via Morpholinos
Microinjected zebrafish embryos staged at 24 hr postfertilization (hpf) presented an observable phenotype.
(A) Control embryo microinjected with mispaired MO (TRA1-MO-Mis).
(B) Morphant embryo microinjected with MO designed to block eif4a3-mRNA translation (TRA1-MO).
(C) Rescued embryo microinjected with TRA1-MO and mRNA coding for eif4a3 fused to EGFP (eif4a3-EGFP mRNA).
Lateral views of embryos were registered under stereoscopic microscope (whole body at the upper left of each panel) or with two different magnifications under differential interference contrast microscope (anterior-most region at the upper and lower right of each panel). Alterations in the morphants’ trunk were observed but not reproducible and only phenotyping at the craniofacial level was done. Dotted lines mark regions between eyes (ey) and otic vesicle (ov), which include the midbrain-hindbrain border (MHB) and showed darkened tissue in morphants, suggesting the presence of apoptotic cells. Lateral views of 24 hpf embryos after acridine orange staining, by standardized protocols,35 are shown in (A′) and (B′). Green fluorescence panels in (C) show expression of eif4a3-EGFP in rescued embryo. Scale bars represent 200 μm. TRA1-MO sequence: TGTGACGGATTTCGGTGTAAATTAC. TRA1-MO-Mis sequence: TGTCACCGATTTCCGTCTAAAATAC.
Figure 5.
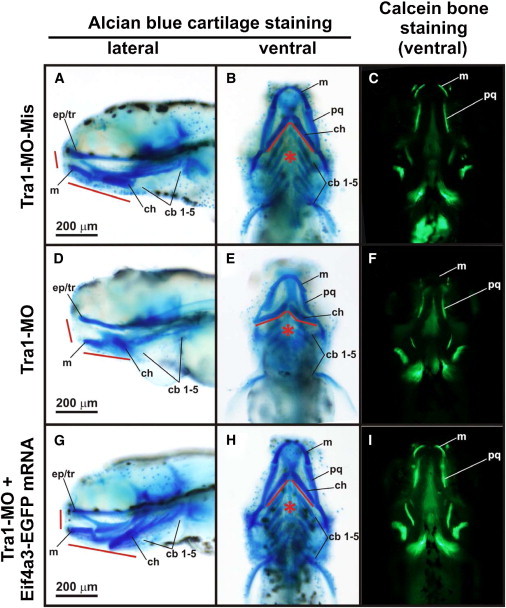
Craniofacial Phenotype in Zebrafish eif4a3 Morphant Larvae
Five days postfertilization (dpf) larvae microinjected with TRA1-MO-Mis (A–C), TRA1-MO (D–F), and TRA1-MO + eif4a3-EGFP mRNA (G–I) were stained with alcian blue36 to observe cartilage structures (A, D, G, lateral views; B, E, H, ventral views) or with calcein to observe bone structures (C, F, I, ventral views). Eyes were removed to register alcian blue-stained larvae. Craniofacial precursors in morphant larvae were severely affected showing hypoplasia of numerous craniofacial cartilages (red lines), which is evident in the rostral and jaw elements. Impairment of pharyngeal arches development was also observed (asterisks). Abbreviations are as follows: cb1–5, ceratobranchial arches 1–5; ch, ceratohyal; ep/tr, ethmoid plate/trabecula; m, Meckel’s cartilage; pq, palatoquadrate. Scale bars represent 200 μm.
In summary, our findings suggest that EIF4A3 deficiency leads to abnormal development of most pharyngeal arches, resulting in altered mandible and laryngeal morphogenesis. Although not explored in this manuscript, deficiency of EIF4A3 also interferes in limb development and is associated with learning and language disabilities observed in a high proportion of RCPS patients. The altered neurodevelopmental phenotype in RCPS, which should be further studied, is unsurprising given the role of eiF4AIII in regulating transcription abundance of neuronal effector genes that underlie learning and memory processes.38,39 In addition, heterozygous deletions of EIF4A3 associated with intellectual disability and autism have recently been reported.40 RCPS belongs to the growing list of craniofacial syndromes caused by loss-of-function mutations of genes encoding proteins involved in RNA metabolism and ribosome biosynthesis, such as TCOF1 ([MIM 606847]; Treacher Collins syndrome, TCS1 [MIM 154500]),41 SF3B4 ([MIM 605593]; Nager syndrome, AFD1 [MIM 154400]),42 and EFTUD2 ([MIM 603892]; mandibular dysostosis and microcephaly, MFDM [MIM 610536]).43 Apoptosis of CNC cells might be a common mechanism underlying all these syndromes22,44. However, considering the complexity of RNA metabolism, it is possible that dysregulation of distinct pathways might explain their specific phenotypes. Of these other syndromes, only EFTUD2-mutated individuals present cognitive impairment; notably, the U5-116-kD spliceosomal GTPase protein encoded by EFTUD2 directly interacts with eIF4AIII.22 Further studies will be necessary to investigate the functional relationship between EIF4A3 and the above-mentioned genes and to understand how mutations in EIF4A3 lead to the pleiotropic phenotype of RCPS.
Acknowledgments
We are thankful to Alessandra Splendore and Gerson S. Kobayashi for critical discussion on the manuscript, to Regina Bueno for art support, and to the Human Genome Research Center sequencing service for technical support. This work was funded by CEPID/FAPESP, CNPq and INCT/CNPq-FAPESP, and by the Wellcome Trust (093329 to A.O.M.W. and S.R.F.T.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Favaro F.P., Zechi-Ceide R.M., Alvarez C.W., Maximino L.P., Antunes L.F.B.B., Richieri-Costa A., Guion-Almeida M.L. Richieri-Costa-Pereira syndrome: a unique acrofacial dysostosis type. An overview of the Brazilian cases. Am. J. Med. Genet. A. 2011;155A:322–331. doi: 10.1002/ajmg.a.33806. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira de Lima R.L., Moretti-Ferreira D., Richieri-Costa A., Murray J.C. Identity by descent and candidate gene mapping of Richieri-Costa and Pereira syndrome. Am. J. Med. Genet. A. 2003;122A:56–58. doi: 10.1002/ajmg.a.20270. [DOI] [PubMed] [Google Scholar]
- 3.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 4.Jordan C.T., Cao L., Roberson E.D., Duan S., Helms C.A., Nair R.P., Duffin K.C., Stuart P.E., Goldgar D., Hayashi G. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am. J. Hum. Genet. 2012;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Twigg S.R., Lloyd D., Jenkins D., Elçioglu N.E., Cooper C.D., Al-Sannaa N., Annagür A., Gillessen-Kaesbach G., Hüning I., Knight S.J. Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet. 2012;91:897–905. doi: 10.1016/j.ajhg.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott J.E., Lack J.B., Ravosa M.J. On the reversibility of mandibular symphyseal fusion. Evolution. 2012;66:2940–2952. doi: 10.1111/j.1558-5646.2012.01639.x. [DOI] [PubMed] [Google Scholar]
- 8.Campuzano V., Montermini L., Moltò M.D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 9.Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 10.Galloway J.N., Nelson D.L. Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol. 2009;4:785. doi: 10.2217/fnl.09.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daughters R.S., Tuttle D.L., Gao W., Ikeda Y., Moseley M.L., Ebner T.J., Swanson M.S., Ranum L.P. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi H., Abe K., Matsuura T., Ikeda Y., Hitomi T., Akechi Y., Habu T., Liu W., Okuda H., Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moseley M.L., Zu T., Ikeda Y., Gao W., Mosemiller A.K., Daughters R.S., Chen G., Weatherspoon M.R., Clark H.B., Ebner T.J. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 14.Sato N., Amino T., Kobayashi K., Asakawa S., Ishiguro T., Tsunemi T., Takahashi M., Matsuura T., Flanigan K.M., Iwasaki S. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan C.C., Dostie J., Diem M.D., Feng W., Mann M., Rappsilber J., Dreyfuss G. eIF4A3 is a novel component of the exon junction complex. RNA. 2004;10:200–209. doi: 10.1261/rna.5230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibuya T., Tange T.O., Stroupe M.E., Moore M.J. Mutational analysis of human eIF4AIII identifies regions necessary for exon junction complex formation and nonsense-mediated mRNA decay. RNA. 2006;12:360–374. doi: 10.1261/rna.2190706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandrov A., Colognori D., Steitz J.A. Human eIF4AIII interacts with an eIF4G-like partner, NOM1, revealing an evolutionarily conserved function outside the exon junction complex. Genes Dev. 2011;25:1078–1090. doi: 10.1101/gad.2045411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haremaki T., Sridharan J., Dvora S., Weinstein D.C. Regulation of vertebrate embryogenesis by the exon junction complex core component Eif4a3. Dev. Dyn. 2010;239:1977–1987. doi: 10.1002/dvdy.22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunz J.B., Neu-Yilik G., Hentze M.W., Kulozik A.E., Gehring N.H. Functions of hUpf3a and hUpf3b in nonsense-mediated mRNA decay and translation. RNA. 2006;12:1015–1022. doi: 10.1261/rna.12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z., Krainer A.R. Splicing remodels messenger ribonucleoprotein architecture via eIF4A3-dependent and -independent recruitment of exon junction complex components. Proc. Natl. Acad. Sci. USA. 2007;104:11574–11579. doi: 10.1073/pnas.0704946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh G., Kucukural A., Cenik C., Leszyk J.D., Shaffer S.A., Weng Z., Moore M.J. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–764. doi: 10.1016/j.cell.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Souza J., dal Vesco K., Tonocchi R., Closs-Ono M.C., Passos-Bueno M.R., da Silva-Freitas R. The Richieri-Costa and Pereira syndrome: report of two Brazilian siblings and review of literature. Am. J. Med. Genet. A. 2011;155A:1173–1177. doi: 10.1002/ajmg.a.33975. [DOI] [PubMed] [Google Scholar]
- 24.Raskin S., Souza M.B., Medeiros M.C., Manfron M., Chong-Silva D.C. Richieri-Costa and Pereira syndrome: case report of a severe phenotype. Am. J. Med. Genet. A. 2013;161A:1999–2003. doi: 10.1002/ajmg.a.35989. [DOI] [PubMed] [Google Scholar]
- 25.Fiser A., Do R.K., Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen C.B., Ballut L., Johansen J.S., Chamieh H., Nielsen K.H., Oliveira C.L., Pedersen J.S., Séraphin B., Le Hir H., Andersen G.R. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 27.Bono F., Ebert J., Lorentzen E., Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–725. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Buchwald G., Ebert J., Basquin C., Sauliere J., Jayachandran U., Bono F., Le Hir H., Conti E. Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc. Natl. Acad. Sci. USA. 2010;107:10050–10055. doi: 10.1073/pnas.1000993107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steckelberg A.L., Boehm V., Gromadzka A.M., Gehring N.H. CWC22 connects pre-mRNA splicing and exon junction complex assembly. Cell Rep. 2012;2:454–461. doi: 10.1016/j.celrep.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 30.Warren S.T. Polyalanine expansion in synpolydactyly might result from unequal crossing-over of HOXD13. Science. 1997;275:408–409. doi: 10.1126/science.275.5298.408. [DOI] [PubMed] [Google Scholar]
- 31.Martelli A., Napierala M., Puccio H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis. Model. Mech. 2012;5:165–176. doi: 10.1242/dmm.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi G.S., Alvizi L., Sunaga D.Y., Francis-West P., Kuta A., Almada B.V., Ferreira S.G., de Andrade-Lima L.C., Bueno D.F., Raposo-Amaral C.E. Susceptibility to DNA damage as a molecular mechanism for non-syndromic cleft lip and palate. PLoS ONE. 2013;8:e65677. doi: 10.1371/journal.pone.0065677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masotti C., Ornelas C.C., Splendore-Gordonos A., Moura R., Félix T.M., Alonso N., Camargo A.A., Passos-Bueno M.R. Reduced transcription of TCOF1 in adult cells of Treacher Collins syndrome patients. BMC Med. Genet. 2009;10:136. doi: 10.1186/1471-2350-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Udd B., Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- 35.Eimon P.M., Ashkenazi A. The zebrafish as a model organism for the study of apoptosis. Apoptosis. 2010;15:331–349. doi: 10.1007/s10495-009-0432-9. [DOI] [PubMed] [Google Scholar]
- 36.Nasevicius A., Ekker S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 37.Solomon K.S., Kudoh T., Dawid I.B., Fritz A. Zebrafish foxi1 mediates otic placode formation and jaw development. Development. 2003;130:929–940. doi: 10.1242/dev.00308. [DOI] [PubMed] [Google Scholar]
- 38.Giorgi C., Yeo G.W., Stone M.E., Katz D.B., Burge C., Turrigiano G., Moore M.J. The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell. 2007;130:179–191. doi: 10.1016/j.cell.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 39.Barker-Haliski M.L., Pastuzyn E.D., Keefe K.A. Expression of the core exon-junction complex factor eukaryotic initiation factor 4A3 is increased during spatial exploration and striatally-mediated learning. Neuroscience. 2012;226:51–61. doi: 10.1016/j.neuroscience.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen L.S., Kim H.G., Rosenfeld J.A., Shen Y., Gusella J.F., Lacassie Y., Layman L.C., Shaffer L.G., Gécz J. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet. 2013;22:1816–1825. doi: 10.1093/hmg/ddt035. [DOI] [PubMed] [Google Scholar]
- 41.Dauwerse J.G., Dixon J., Seland S., Ruivenkamp C.A., van Haeringen A., Hoefsloot L.H., Peters D.J., Boers A.C., Daumer-Haas C., Maiwald R. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 2011;43:20–22. doi: 10.1038/ng.724. [DOI] [PubMed] [Google Scholar]
- 42.Bernier F.P., Caluseriu O., Ng S., Schwartzentruber J., Buckingham K.J., Innes A.M., Jabs E.W., Innis J.W., Schuette J.L., Gorski J.L., FORGE Canada Consortium Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 2012;90:925–933. doi: 10.1016/j.ajhg.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lines M.A., Huang L., Schwartzentruber J., Douglas S.L., Lynch D.C., Beaulieu C., Guion-Almeida M.L., Zechi-Ceide R.M., Gener B., Gillessen-Kaesbach G., FORGE Canada Consortium Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am. J. Hum. Genet. 2012;90:369–377. doi: 10.1016/j.ajhg.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dixon J., Brakebusch C., Fässler R., Dixon M.J. Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum. Mol. Genet. 2000;9:1473–1480. doi: 10.1093/hmg/9.10.1473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.