

Abstract
The use of acyl hydrazides as peptoid sub-monomers is investigated. We demonstrate here that azapeptoids derived entirely from acyl hydrazides can be made conveniently and efficiently using standard peptoid sub-monomer chemistry. Structural studies reveal that the main chain amide bond in these molecules predominantly adopts a trans conformation. A high-quality one bead one compound library of tetramers was made by split and pool synthesis and we found that the identity of the molecule on a single bead could be determined by tandem MALDI mass spectrometry.
Our laboratory is interested in the synthesis of large combinatorial libraries of synthetic oligomers from which interesting protein ligands might be isolated. Towards this end we have focused considerable attention on peptoids (oligomers of N-alkyl or N-aryl substituted glycine).1 Using one bead one compound (OBOC)2 libraries of this class of compounds as a convenient vehicle, we have developed powerful screening assays for the isolation of protein ligands of potential interest as “tool compounds”, therapeutic leads and diagnostic reagents. Peptoids have many advantages in this regard. Most importantly, their synthesis by the “sub-monomer” route is highly efficient and employs primary amines as the diversity element in a split and pool synthesis,2c of which thousands are available. In this sense, they are nearly ideal molecules for combinatorial chemistry. Moreover, they have superior serum stability3 and cell permeability4 when compared to their peptide cousins. On the other hand, standard peptoids lack hydrogen-bonding moieties unless these are specifically incorporated into the side chain. Moreover, they are “floppy” molecules, which probably limit their affinity for protein targets. Unlike peptides, the tertiary amide bond in peptoids does not exhibit a strong preference for the trans geometry, nor are there significant constraints to rotation about the carbonyl-Cα or the Cα-N bonds in the backbone.
Hence, strategies to introduce structural constraints into peptoid oligomers have attracted significant attention in recent years5–8. Recently, our group has also turned its attention to this issue, with a companion goal of introducing greater chemodiversity into the molecules. Here, we report preliminary results of one such effort, the creation of N-azapeptoid libraries. It is to be noted that the azapeptoids we report here are structurally distinct from hydrazino azapeptoids9 and retro hydrazino azapeptoids10 reported previously (Fig. 1). We anticipated that these molecules could be derived from the use of acyl hydrazides in the sub-monomer synthesis protocol (Fig. 2), thus placing hydrogen-bonding groups close to the backbone, which is not the case in typical peptoids. Given that many acyl hydrazides are commercially available and hundreds more can be made easily by condensing carboxylic acids with hydrazine, the building blocks for these compounds are as rich and varied as the primary amines employed to make typical peptoids.
Fig. 1.
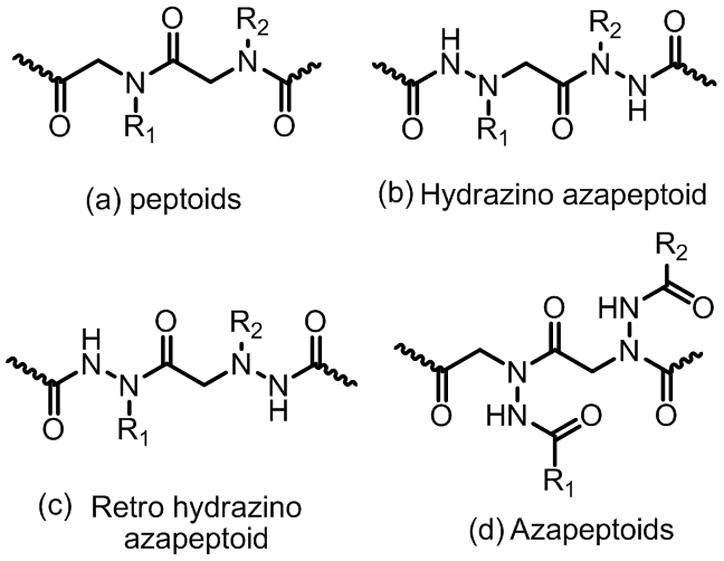
Structural features of (a) peptoids, (b) hydrazino azapeptoids, (c) retro hydrazino azapeptoids and (d) azapeptoids.
Fig. 2.
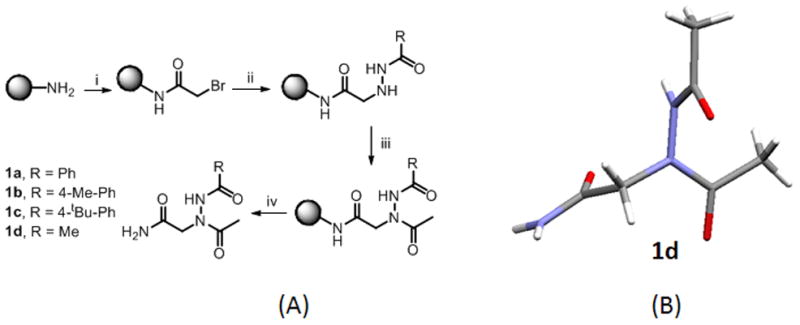
(A) Proposed sub-monomer synthesis of N-azapeptoids 1a–1d. For 100 mg resin (i) Bromoacetic acid (2M, 1mL) and DIC (3.4 M, 1mL), 37 ºC, 10 mins; (ii) Acyl hydrazide (2M, 2mL), 37 ºC, 1h (iii) acetic acid (2M, 1mL), DIC (3.4M, 1mL), 30 mins (iv) TFA:TIS:water (96:2:2) (2mL). (B) Single crystal X-ray structure of 1d shows the trans-amide geometry of both the main chain and side chain amide bonds.
Although the possibility of using acyl hydrazides as an alternative to primary amines in peptoid synthesis was pointed out by Zuckermann and co-workers in a patent application,11 there has been, to our knowledge, no experimental work in the area. Since our goal is to create large, high-quality combinatorial libraries, which requires that all reactions proceed in very high yield, the concern is that the reduced nucleophilicity of acyl hydrazides relative to amines might be problematic. To begin to probe this issue, we synthesized compounds 1a–1d (Fig. 2) on Knorr Amide MBHA resin by first coupling activated 2-bromoacetic acid to the amine group on the beads followed by displacement of the bromide with acyl hydrazides. Finally, the N-terminus was capped using acetic acid and diisopropylcarbodiimide (DIC). After cleavage from the beads, HPLC and NMR analysis of the crude reaction mixtures showed highly efficient conversion to a single product when the displacement reaction was carried out at 37 °C for one hour (see Supplementary Information). The crystal structure of 1d (Fig. 2B) shows that both the main chain and side chain amide bonds adopt a trans configuration. It is also interesting to note that the acyl hydrazide-derived side chain is oriented almost perpendicular to the peptoid main chain. There is no indication of a possible hydrogen bond between the hydrazide N-H and either of the main chain carbonyl groups.
The 1H NMR spectra of 1a–1d are dominated by a single amide bond isomer in solution (>90%; see Table S1 in the Supplementary Information) so, based on the crystal structure of 1d, we assume that these all contain trans amides as well. As mentioned before, most common peptoids exhibit a mixture of cis and trans amide bonds, so this marks a clear difference between traditional peptoids and those derived from acyl hydrazides.
We also synthesized dimers 2a–2b (Fig. 3 and Supplementary Information, Fig. S2). After constructing a first monomer from benzoic hydrazide on the resin, we optimized the subsequent acylation and displacement reactions, which were monitored by reverse phase HPLC. The resin bound benzoic hydrazide was acylated by using either chloro or bromo acetic acid (1M) in the presence of DIC (1.4M) at 37 ºC. The acylation step was complete within 10 minutes. The chloride or bromide was then displaced with a second molecule of benzoic hydrazide (2M) under different conditions. We found that benzoic hydrazide is not sufficiently reactive to completely displace the resin bound chloride at 37 ºC when the reaction was carried out for one hour. Microwave irradiation (three times for 15 sec (10% of 1.5kV)) also failed to provide complete conversion. In contrast, displacement of resin bound bromide was very efficient and 98% conversion was achieved at 37 ºC in one hour. Using these conditions (bromoacetic acid (1M) and DIC (1.4M) for 10 minutes at 37 ºC for the acylation reaction and 2M acyl hydrazide for one hour at 37 ºC for the displacement reaction) compounds 2b and 3 were also synthesized in high purity (>95%).
Fig. 3.
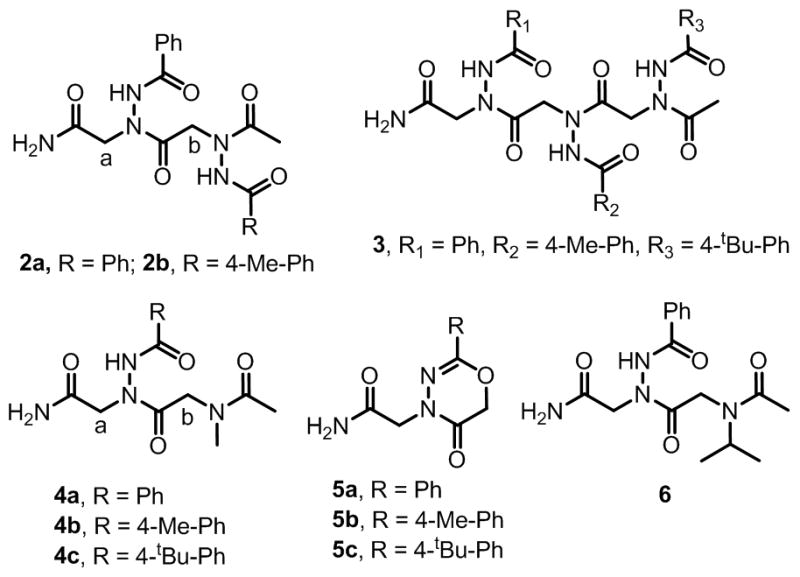
Chemical Structures of azapeptoids 2a–2b, 3; peptoid-azapeptoid hybrids 4a–4c, 6; and 4-substituted 2-aryl-4H-1,3,4-oxadiazin-5(6H)-ones 5a–5c.
Finally, we synthesized four tetramer azapeptoids using 10 different acyl hydrazides, which were obtained in more than 95% purity (see Supplementary Information, Fig. S51–Fig. S54). With these data in hand, we proceeded to use solid-phase split and pool synthesis to create an OBOC library of tetramers on TentaGel resin with a diversity of 10,000 compounds using the aforementioned 10 different acyl hydrazides (see Supplementary Information). Several individual beads were picked at random, separated into wells of a microtiter plate, and the compounds were released into solution. MALDI TOF/TOF MS/MS analysis12 showed strong molecular ions that, when fragmented, provided unequivocal sequence data (see Supplementary Information). We conclude that this is a high quality library suitable for screening experiments.
In addition to the synthesis of libraries of peptoids derived completely from acyl hydrazides, we were interested in embedding these residues into standard peptoids. Therefore, we attempted to synthesize compounds 4a–4c by using methylamine in place of the second acyl hydrazide moiety in a standard sub-monomer protocol (Fig. 3). In each case we obtained ~75% of the expected compounds 4a–4c along with ~25% of 5a–5c (Fig. 3). Although the amount of 5a–5c could be minimized to ~15% by using chloroacetic acid instead of bromoacetic acid in the second monomer, formation of the heterocycle could not be suppressed completely (see Supporting Information). The identities of the compounds 5a–5c were confirmed by independent synthesis of compound 5a by reacting the bromo acylated resin with diisopropylethyl amine (DIEA) for one hour followed by cleavage with TFA.
The formation of compounds 5a–5c can be rationalized by the base-mediated intramolecular cyclization of 1-acyl-2-(α-chloroacyl)hydrazides, which is well precendented in the literature.13 As expected from a competition between this reaction and displacement of the halogen by the amine, the ratios of the displacement and cyclization products were dependent on the amine employed. When isopropyl amine was used instead of methylamine (compound 6), compound 5a was observed as the major by HPLC (~70%). Therefore, the use of amines and acyl hydrazides to make hybrid peptoid oligomers and their libraries should be avoided.
This intramolecular cyclization reaction could be used to advantage to create libraries of peptoids or azapeptoids capped with diverse 4-substituted 2-aryl-4H-1,3,4-oxadiazin-5(6H)-ones, which have garnered attention as inhibitors of monoamine oxidase (MAO) B.14 To demonstrate this point, we added an additional acyl hydrazide moiety (10 different acyl hydrazides) to the tetramer library described above and then acylated that by using bromoacetic acid. Then the library was treated with DIEA to obtain a library where the 5th position contains ten different 4-substituted 2-aryl-4H-1,3,4-oxadiazin-5(6H)-ones. The structures of compounds displayed on single beads could again be determined easily from the tandem MALDA MS/MS spectra (see Supplementary Information).
It should be noted that the formation of 4-substituted 2-aryl-4H-1,3,4-oxadiazin-5(6H)-ones was not observed when acyl hydrazides were employed as the nucleophile in the halide displacement step. This suggests that while acyl hydrazides are sufficiently nucleophilic to participate in the desired bromide displacement reaction, they are not sufficiently basic to deprotonate the hydrazide nitrogen on the previous residue.
We have not yet attempted to study in detail the three-dimensional conformations of acyl hydrazide-derived peptoids, other than to probe the amide bond geometry and to learn from the crystal structure of 1d that, at least in the solid state, the side chain prefers to sit almost orthogonal to the peptoid main chain, as one might have expected from inspection from models. However, it should be mentioned that some unusual characteristics of the NMR spectra of these compounds hint that they likely exhibit additional interesting structural features. The 1H NMR spectra of compounds 1a–1d were comprised of sharp, well-defined peaks with a striking exception. At room temperature, the methylene (CH2) proton resonances were very broad in CDCl3 and were not observed at all in DMSO-d6 (see Supplementary Information). This cannot be ascribed to exchange with solvent. When the compounds were incubated in D2O and then analyzed by high-resolution mass spectrometry, no detectable exchange of these methylene protons was observed (see Supplementary Information).
The 1H NMR spectrum of compound 2a and 2b showed that one set of methylene hydrogens (set b, Fig. 3) appear as a broad singlet at 25 ºC in CDCl3 whereas the other set of methylene protons (set a) were completely missing. However, low temperature 1H NMR experiments clearly showed both the sets (a and b) methylene protons. Interestingly, both these sets of protons were found to be diastereotopic and geminal couplings were clearly evident from the 1H spectra.
The 1H NMR spectra of the compounds 4a–4c shows a similar behaviour as that of compounds 1 and 2. The methylene protons (set a) were not observed for 4a–4c in CDCl3. As the temperature was lowered, the methylene protons became evident. Similar to 2a, the methylene hydrogens of 4c, for example, were observed as two doublets (16.8 Hz at 0 ºC), which clearly suggest diastereotopicity of these protons. The lack of exchange with solvent would appear to rule out this behaviour being due to chemical exchange phenomena. Therefore, while we do not completely understand the molecular basis of this effect, it indicates the presence of two or more conformational states for N-azapeptoids that are in intermediate exchange on the NMR time scale at room temperature.
In conclusion, we have shown that acyl hydrazides can be used efficiently as sub-monomers to generate peptoid oligomers with trans-amide bond geometries. The presence of hydrogen bond donor and acceptor units in the side chain may prove useful in engaging protein targets. While N-azaacyl units cannot be imbedded into a chain of standard peptoids efficiently due competing ring closure, one can take advantage of this reaction to create N-azaacyl peptoids capped with diverse 2-aryl-4H-1,3,4-oxadiazin-5(6H)-ones. We have demonstrated that all of this chemistry is compatible with the synthesis of high quality OBOC libraries and that the structure of a compound displayed on a single bead can be determined by tandem mass spectrometry. Thus, these new libraries can be employed in the various powerful screening platforms developed over the last few years for TentaGel-displayed OBOC libraries.15
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: [Experimental details, HPLC traces, NMR and mass spectra of different compounds and X-ray crystallographic data for 1d (CCDC 827861) are provided.]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, Spellmeyer DC, Tan R, Frankel AD, Santi DV, Cohen FE, Bartlett PA. Proc Natl Acad Sci U S A. 1992;89:9367. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]; (b) Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Nature. 1991;354:84. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]; (c) Furka A, Sebestyen F, Asgedom M, Dibo G. Int J Pept Protein Res. 1991;37:487. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]; (d) Lam KS, Krchnak V, Lebl M. Chem Rev. 1997;97:411. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 3.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Bioorg Med Chem Lett. 1994;4:2657. [Google Scholar]
- 4.(a) Yu P, Liu B, Kodadek T. Nat Biotechnol. 2005;23:746. doi: 10.1038/nbt1099. [DOI] [PubMed] [Google Scholar]; (b) Kwon YU, Kodadek T. J Am Chem Soc. 2007;129:1508. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KTV, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. Proc Natl Acad Sci U S A. 1998;95:4309. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, Truong KTV, Dill KA, Cohen FE, Zuckermann RN. Proc Natl Acad Sci U S A. 1998;95:4303. doi: 10.1073/pnas.95.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, Dill KA, Zuckermann RN, Barron AE. J Am Chem Soc. 2003;125:13525. doi: 10.1021/ja037540r. [DOI] [PubMed] [Google Scholar]; (d) Shah NH, Butterfoss GL, Nguyen K, Yoo B, Bonneau R, Rabenstein DL, Kirshenbaum K. J Am Chem Soc. 2008;130:16622. doi: 10.1021/ja804580n. [DOI] [PubMed] [Google Scholar]; (e) Lee JH, Zhang Q, Jo S, Chai SC, Oh M, Im W, Lu H, Lim HS. J Am Chem Soc. 2011;133:676. doi: 10.1021/ja108230s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang K, Wu CW, Sanborn TJ, Patch JA, Kirshenbaum K, Zuckermann RN, Barron AE, Radhakrishnan I. J Am Chem Soc. 2006;128:1733. doi: 10.1021/ja0574318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pokorski JK, Miller Jenkins LM, Feng H, Durell SR, Bai Y, Appella DH. Org Lett. 2007;9:2381. doi: 10.1021/ol070817y. [DOI] [PubMed] [Google Scholar]; (b) Shin SBY, Kirshenbaum K. Org Lett. 2007;9:5003. doi: 10.1021/ol702207n. [DOI] [PubMed] [Google Scholar]
- 8.Stringer JR, Crapster JA, Guzei IA, Blackwell HE. J Org Chem. 2010;75:6068. doi: 10.1021/jo101075a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheguillaume A, Lehardy F, Bouget K, Baudy-Floc’h M, Grel PL. J Org Chem. 1999;64:2924. doi: 10.1021/jo981487l. [DOI] [PubMed] [Google Scholar]
- 10.Aubin S, Martin B, Delcros J, Arlot-Bonnemains Y, Baudy-Floc’h M. J Med Chem. 2005;48:330. doi: 10.1021/jm049455f. [DOI] [PubMed] [Google Scholar]
- 11.Zuckerman RN, Goff DN, Simon NG, Spear K, Scott B, Sigmund AG, Goldsmith RA, Marlowe CK, Pei Y, Richter L, Simon R. WO1996040202A1 PCT Int Appl.
- 12.(a) Heerma W, Versluis C, de Koster CG, Kruijtzer JAW, Zigrovic I, Liskamp RMJ. Rapid Commun Mass Spectrom. 1996;10:459. doi: 10.1002/(SICI)1097-0231(19960315)10:4<459::AID-RCM501>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; (b) Heerma W, Boon JPJL, Versluis C, Kruijtzer JAW, Hofmeyer LJF, Liskamp RMJ. J Mass Spectrom. 1997;32:697. [Google Scholar]; (c) Ruijtenbeek R, Versluis C, Heck AJR, Redegeld FAM, Nijkamp FP, Liskamp RMJ. J Mass Spectrom. 2002;37:47. doi: 10.1002/jms.245. [DOI] [PubMed] [Google Scholar]
- 13.(a) Van Alphen J. Rec Trav Chim. 1928;47:673. [Google Scholar]; (b) Van Alphen J. Rec Trav Chim. 1929;48:417. [Google Scholar]
- 14.Mazouz F, Lebreton L, Milcent R, Burstein C. Eur J Med Chem. 1988;23:441. doi: 10.1021/jm00024a006. [DOI] [PubMed] [Google Scholar]
- 15.(a) Alluri PG, Reddy MM, Bacchawat-Sikder K, Olivos HJ, Kodadek T. J Am Chem Soc. 2003;125:13995. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]; (b) Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Am Chem Soc. 2008;130:5744. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]; (c) Reddy MM, Wilson R, Wilson J, Connell S, Gocke A, Hynan L, German D, Kodadek T. Cell. 2011;144:132. doi: 10.1016/j.cell.2010.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.