

Abstract
Introduction
A major challenge in Romania is the optimisation of antiretroviral therapy for the many HIV-infected adults with, on average, a decade of treatment experience. The RDI has developed computational models that predict virological response to therapy but these require a genotype, which is not routinely available in Romania. Moreover the models, which were trained without any Romanian data, have proved most accurate for patients from the healthcare settings that contributed the training data. Here we develop and test a novel model that does not require a genotype, with test data from Romania.
Methods
A random forest (RF) model was developed to predict the probability of the HIV viral load (VL) being reduced to <50 copies/ml following therapy change. The input variables were baseline VL, CD4 count, treatment history and time to follow-up. The model was developed with 3188 treatment changes episodes (TCEs) from North America, Western Europe and Australia. The model’s predictions for 100 independent TCEs from the RDI database were compared to those of a model trained with the same data plus genotypes and then tested using 39 TCEs from Romania in terms of the area under the ROC curve (AUC).
Results
When tested with the 100 independent RDI TCEs, the AUC values for the models with and without genotypes were 0.88 and 0.86 respectively. For the 39 Romanian TCEs the AUC was 0.60. However, when 14 cases with viral loads that may have been between 50 and 400 copies were removed, the AUC increased to 0.83.
Discussion
Despite having been trained without data from Romania, the model predicted treatment responses in treatment-experienced Romanian patients with clade F virus accurately without the need for a genotype. The results suggest that this approach might be generalisable and useful in helping design optimal salvage regimens for treatment-experienced patients in countries with limited resources where genotyping is not always available.
Keywords: HIV, treatment response prediction, computational models
Introduction
Combination antiretroviral therapy (ART) has made the long-term suppression of HIV and prevention of HIV disease progression a reality in well-resourced health care settings. Nevertheless, despite the availability of approximately 25 antiretroviral drugs from six classes, viral breakthrough, often with the emergence of drug-resistant virus, remains a significant challenge.1,2 The long-term re-suppression of drug resistant virus requires the optimal selection of the next combination of drugs. However, the complexities of the genetic basis of HIV drug resistance and the number of potential drug combinations available make optimal, individualised sequencing of therapy highly challenging.3 For physicians with limited experience or resources, treatment decisions can be even more difficult.
In well-resourced settings, a genotypic resistance test is usually performed following treatment failure.2 Interpretation of this genotype can be complex and is usually performed using rules-based software that relates point mutations to viral susceptibility to single drugs.4 However, different systems provide different interpretations and it is difficult to relate the results for individual drugs to the likely responses to potential drug combinations.5-9 Indeed, genotypic sensitivity score (derived by allocating scores for each drug in a regimen according to whether the genotype interpretation predicts the virus will be sensitive, intermediate or resistant to the drug) have been shown to be relatively weak predictors of virological response.10,11
The development of computational models to obtain, directly from the genotype and other clinical information, a quantitative prediction of virological response to any combination of drugs, rather than to individual agents, may offer a potential clinical advantage. However, this approach has an inherent challenge: the considerable quantity of data required for the modelling to incorporate a range of prognostic variables, multiple possible drug-genotype permutations and their respective drug response data.12 The HIV Resistance Response Database Initiative (RDI) was established in 2002 explicitly to address the challenge of large-scale data collection required for this approach and then to develop such models.13
Currently we have collected data from approximately 85000 patients in a standardised Oracle database. These are predominantly from Western Europe and North America but also from Africa, Australia, Japan and most recently Romania. We have trained computational models to predict virological response to treatment from genotype, viral load, CD4 cell count, and treatment history with 80% accuracy.14,15 This compares favourably with a 50-70% predictive accuracy typically achieved with genotypic sensitivity scores.15,16
This approach could be of particular clinical utility in resource-limited settings (RLS). However, the models described above require genotyping, which is not generally available in RLS. Moreover, since the great majority of the RDI’s data are from clinical practice in well-resourced countries and previous studies have shown that models are most accurate for patients from ‘familiar’ settings from which the training data were collected, a concern was that the encouraging performance of the models would not be replicated when the models were applied to cases in unfamiliar RLS.16
In this study, two computational models were developed with a training dataset excluding cases from Romania, one including and one excluding the genotype from their input variable sets. The accuracy of the models for predicting treatment responses for cases from the same settings as provided the training data was compared and the performance of the ‘no-genotype’ model tested with cases without genotypes from the ‘unfamiliar’ setting of Romania.
Methods
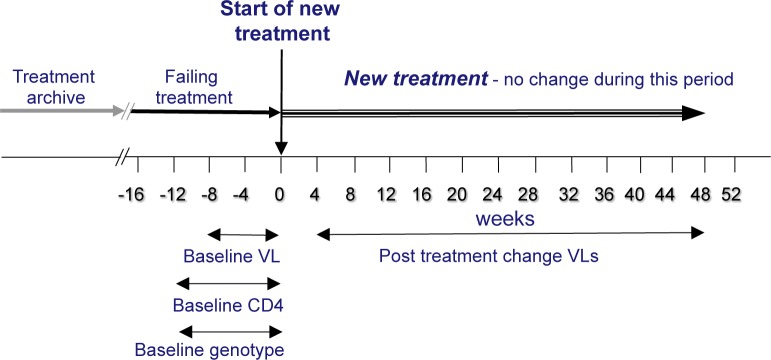
The following data were extracted from the RDI database for cases where a patient’s antiretroviral treatment was changed: baseline plasma viral load (copies/ml HIV RNA) collected up to eight weeks before the time of treatment change; baseline CD4 count (cells/ml) and genotype collected up to 12 weeks prior to the time of treatment change; antiretroviral drugs in the patient’s treatment history and in the new, changed regimen; time to follow-up viral load determination (accepted between 4-48 weeks following the treatment change) and the follow-up viral load value. The data from each case is termed a Treatment Change Episode (TCE), as illustrated in figure 1. The TCEs from Romania (39 from 19 patients) were excluded and the remaining TCEs partitioned at random into a training set of 3188 TCEs and a test set of 100. While information on viral subtype was not provided with much of the data, given the geographical location from where the data came it is reasonable to assume that most were subtype B and few, if any, were F1, the predominant subtype in Romania.
Figure 1: The Treatment Change Episode (TCE).
The training data were used to train two random forest models to estimate the probability of virological response (follow-up plasma viral load <50 copies HIV RNA/ml), using methodology described in detail elsewhere.14 The principle of RF modelling is to develop large numbers of decision trees in parallel. Then for a given sample, votes are carried out over all the trees in the forest. The individual trees are built using different sets of samples from the training dataset. In each node of a tree, the splitting feature is selected from a randomly chosen sample of the input variables. In RF modelling, the training datasets of the individual trees are built by bootstrap replication, leaving about one-third of the samples out of the bootstrap sample, which are used for validation. The injection of randomness makes RF highly resistant to over-fitting.17,18
One RF model was trained using 82 input variables, including 58 mutations from the baseline genotype, the baseline viral load and CD4 count, the drugs in the treatment history and in the new regimen and the time to follow-up. The genotype was excluded from the training of the second model, which made its predictions using the remaining 24 variables. During training, large numbers of RF models were developed and the most accurate selected for evaluation.
The accuracy of both models was evaluated using the test set of 100 RDI TCEs and the accuracy of the ‘no-genotype’ model further evaluated using three test sets derived from the 39 TCEs from Romania. Since these TCEs included viral loads with different lower limits of detection some of which were above 50 copies/ml HIV RNA, three test sets were produced as follows: Set 1: All cases with a viral load recorded as <400 were treated as being <50 (n=39); Set 2: values of <400 were omitted from the analysis but cases with a cut-off between 50 and 400 (e.g. <80, <100) were included as being <50 (n=30) and Set 3: all cases with a viral load cut-off above 50 were omitted (n=25).
The baseline data from the test TCEs were input into the model and its estimates of the probability of virological response compared to the responses actually observed in the clinic. Receiver-operator characteristic (ROC) curves were plotted and the area under the ROC curve (AUC), the principle measure of a predictive model’s performance, obtained as well as the overall accuracy, the sensitivity and the specificity. The difference between the ‘no genotype’ model’s performance (AUC) with each of the three Romanian data sets was tested for statistical significance using DeLong's test.
Results
The 3188 training TCEs had a mean (median) baseline viral load of 4.18 (4.27) and a mean (median) CD4 count of 296 (254). The patients had been exposed to a mean (median) of 2.27 (2) drug classes prior to treatment change. 31% had a virological response (viral load <50 copies/ml) following the treatment change. The 39 Romanian TCEs were from pre-treated patients with a mean (median) baseline VL of 3.62 (3.59) log and CD4 count of 453 (369) cells/ml. The patients had been exposed to a mean (median) of 2.74 (3) drug classes prior to treatment change. 33% had a virological response (viral load <50 copies/ml) following the treatment change.
The AUC for the ‘no genotype’ model when tested with the independent test set of 100 RDI TCEs was 0.86 and the overall accuracy was 78% (figure 2). Sensitivity was 71% and specificity was 89%. These figures compare with an AUC value of 0.88 and overall accuracy of 82%, sensitivity of 86% and specificity of 76% for the model that used the genotype in making its predictions.
Figure 2: ROC curves for the RF models with and without genotype.

For the Romania test set 1, the AUC was 0.60 and overall accuracy was 67%. The sensitivity was 74% and the specificity 60%. For test set 2, the AUC was 0.74 and accuracy was 70%. The sensitivity was 63% and the specificity 82%. For test set 3, the AUC was 0.83 and accuracy was 79%. The sensitivity was 75% and the specificity 88%. The differences in the AUC figures for the three datasets were not statistically significant, although there was a trend towards a statistically significant difference between set 1 and set 3 (p=0.08). The ROC curves for all three data sets are presented in figure 3 and the results summarised in table 1. Four of the nine TCEs removed in order to obtain set 3 that had follow-up viral loads recorded as <400 were predicted by the models in analysis 1 to be failures (VL ≥50 copies).
Figure 3: ROC curves for the single RF model and three Romanian test sets.

Table 1: Model performance with Romanian data.
| Romanian test data set | n | AUC | Sensitivity | Specificity | Overall accuracy |
| 1. All viral loads of <400 or <100 assumed to be <50 | 39 | 0.60 | 74% | 60% | 67% |
| 2. All viral loads of <400 omitted but VL<100 assumed to be <50 | 30 | 0.74 | 63% | 82% | 70% |
| 3. All viral loads of <400 or <100 omitted | 25 | 0.83 | 75% | 88% | 79% |
Discussion
Our study demonstrates the potential utility of the RDI computational models to help optimise therapy for treatment-experienced patients in countries with limited resources where genotyping is not always available.
The ‘no genotype’ RF model was able to predict response to cART with only marginally less accuracy than a model trained with the same TCEs, including the genotype. The difference was not statistically significant.
The model predicted treatment responses in a group of heavily pre-treated Romanian patients with clade F virus and without genotypes with a level of accuracy that was encouraging, despite having been trained without data from Romania. While the AUC for sets 1 and 2 were reduced compared to the AUC for the RDI TCEs, set 3 which excluded all those cases with unknown viral loads between 50 and 400 copies/ml, resulted in comparable accuracy. While these results suggest that the predictions of the RDI models may be generalisable beyond the clinics and settings from which the training data were obtained and apparently irrespective of HIV clade, the test sets used here were very small and it is difficult to draw firm conclusions. Certainly, previous studies by the RDI have demonstrated that the accuracy of its models is reduced when making predictions for patients from novel settings.
The differences in results between the three Romanian test sets suggests that relatively small differences in virological response near the limit of detection are important and within the scope of the models’ predictions.
In conclusion, computational models can predict virological response to ART with a high degree of accuracy. This approach has the potential to improve patient outcomes and to reduce treatment costs through the avoidance of unnecessary or premature switching to expensive, newer drugs, even in settings where genotyping is not generally available.
Optimising the accuracy and utility of the system for use in Romania, or in fact any particular setting, is likely to depend crucially on obtaining sufficient data from that setting. Such data would be used in both the training of models that are ‘familiar’ with clinical practice in that setting and for adequate testing of those models. The RDI is engaged in the further collection of data with a focus on Eastern Europe and sub-Saharan Africa, with the aim of developing models specifically for those regions.
The RDI’s HIV Treatment Response Prediction System (HIV-TRePS) is now available as a free, experimental treatment support tool at www.hivrdi.org.
Acknowledgement
RDI data and study group: The RDI wishes to thank all the following individuals and institutions for providing the data used in training and testing its models:
Cohorts: Frank De Wolf and Joep Lange (ATHENA, the Netherlands); Julio Montaner and Richard Harrigan (BC Center for Excellence in HIV & AIDS, Canada); Tobias Rinke de Wit and Raph Hamers (PASER-M cohort, The Netherlands); Brian Agan. Vincent Marconi and Scott Wegner (US Department of Defense); Wataru Sugiura (National Institute of Health, Japan); Maurizio Zazzi (MASTER, Italy).
Clinics: Jose Gatell and Elisa Lazzari (University Hospital, Barcelona, Spain); Brian Gazzard, Mark Nelson, Anton Pozniak and Sundhiya Mandalia (Chelsea and Westminster Hospital, London, UK); Lidia Ruiz and Bonaventura Clotet (Fundacion IrsiCaixa, Badelona, Spain); Schlomo Staszewski (Hospital of the Johann Wolfgang Goethe-University, Frankfurt, Germany); Carlo Torti (University of Brescia); Cliff Lane and Julie Metcalf (National Institutes of Health Clinic, Rockville, USA); Maria-Jesus Perez-Elias (Instituto Ramón y Cajal de Investigación Sanitaria, Madrid, Spain); Andrew Carr, Richard Norris and Karl Hesse (Immunology B Ambulatory Care Service, St. Vincent’s Hospital, Sydney, NSW, Australia); Dr Emanuel Vlahakis (Taylor’s Square Private Clinic, Darlinghurst, NSW, Australia); Hugo Tempelman and Roos Barth (Ndlovu Care Group, Elandsdoorn, South Africa), Carl Morrow and Robin Wood (Desmond Tutu HIV Centre, Cape Town, South Africa); Luminiţa Ene (‘Dr. Victor Babeş’ Hospital for Infectious and Tropical Diseases, Bucharest, Romania).
Clinical trials: Sean Emery and David Cooper (CREST); Carlo Torti (GenPherex); John Baxter (GART, MDR); Laura Monno and Carlo Torti (PhenGen); Jose Gatell and Bonventura Clotet (HAVANA); Gaston Picchio and Marie-Pierre deBethune (DUET 1 & 2 and POWER 3); Maria-Jesus Perez-Elias (RealVirfen).
Footnotes
ADR: designed and supervised the study and drafted the manuscript. DW developed and tested the computational models and conducted the statistical analyses. LE, DD, AP, MY and JM all contributed key data for the training and/or testing of the models and ideas for the study. LE and BL made substantial contributions to the manuscript. BL devised the concept.
Conflicts of interest: All authors - none to declare
Presented in part: 18th Conference on Retroviruses and Opportunistic Infections (CROI), Boston, USA, March 2011 (Abstract L-208)
References
- 1.Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. 2011 Jan 10;:1–166. [Google Scholar]
- 2.Thomson MA, Aberg JA, Cahn P, et al. Antiretroviral Treatment of Adult HIV Infection: 2010 Recommendations of the International AIDS Society-USA Panel. JAMA. 2010;304(3):321–33. doi: 10.1001/jama.2010.1004. [DOI] [PubMed] [Google Scholar]
- 3.Hirsch MS, Günthard HF, Schapiro JM, Brun-Vézinet F, Clotet B, Hammer SM, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society-USA panel. Clin Infect Dis. 2008;47(2):266–85. doi: 10.1086/589297. [DOI] [PubMed] [Google Scholar]
- 4.Liu TF, Shafer RW. Web Resources for HIV type 1 Genotypic-Resistance Test Interpretation. Clin Infect Dis. 2006;42(11):1608–18. doi: 10.1086/503914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schapiro JM, De Luca A, Harrigan R, Hellman N, McCreedy B, Pillay D, et al. Resistance assay interpretation systems vary widely in method and approach. Antivir Ther. 2001;6(Suppl 1):131. [Google Scholar]
- 6.Shafer RW, Gonzales MJ, Brun-Vezinet F. Online comparison of HIV-1 drug resistance algorithms identifies rates and causes of discordant interpretations. Antivir Ther. 2001;6:101. [Google Scholar]
- 7.Torti C, Quiros-Roldan E, Keulen W, Scudeller L, Caputo SL, Boucher C, et al. for the GenPherex Group of the MaSTeR Cohort Comparison between rules-based human immunodeficiency virus type 1 genotype interpretations and real or virtual phenotype: concordance analysis and correlation with clinical outcome in heavily treated patients. J Infect Dis. 2003;188(2):194–201. doi: 10.1086/376512. [DOI] [PubMed] [Google Scholar]
- 8.Stürmer M, Doerr HW, Staszewski S, Preiser W. Comparison of nine resistance interpretation systems for HIV-1 genotyping. Antivir Ther. 2003;8(3):239–44. [PubMed] [Google Scholar]
- 9.De Luca A, Cingolani A, Di Giambenedetto S, Trotta MP, Baldini F, Rizzo MG, et al. Variable prediction of antiretroviral treatment outcome by different systems for interpreting genotypic human immunodeficiency virus type 1 drug resistance. J Infect Dis. 2003;187(12):1934–43. doi: 10.1086/375355. [DOI] [PubMed] [Google Scholar]
- 10.DeGruttola V, Dix L, D’Aquila R, Holder D, Phillips A, Ait-Khaled M, et al. The relation between baseline HIV drug resistance and response to antiretroviral therapy: re-analysis of retrospective and prospective studies using a standardized data analysis plan. Antivir Ther. 2000;5(1):41–8. doi: 10.1177/135965350000500112. [DOI] [PubMed] [Google Scholar]
- 11.Frentz D, Boucher CA, Assel M, De Luca A, Fabbiani M, Incardona F, et al. Comparison of HIV-1 Genotypic Resistance Test Interpretation Systems in Predicting Virological Outcomes Over Time. PLoS One. 2010;5(7) doi: 10.1371/journal.pone.0011505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiRienzo G, DeGruttola V. Collaborative HIV resistance-response database: sample size for detection of relationships between HIV-1 genotype and HIV-1 RNA response using a non-parametric approach. Antivir Ther. 2002;7:S93. [Google Scholar]
- 13.Larder BA, DeGruttola V, Hammer S, Harrigan R, Wegner S, Winslow D, et al. The international HIV resistance response database initiative: a new global collaborative approach to relating viral genotype treatment to clinical outcome. Antivir Ther. 2002;7:S111. [Google Scholar]
- 14.Wang D, Larder BA, Revell AD, Montaner J, Harrigan R, De Wolf F, et al. A comparison of three computational modelling methods for the prediction of virological response to combination HIV therapy. Artificial Intelligence in Medicine. 2009;47(1):63–74. doi: 10.1016/j.artmed.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Revell AD, Wang D, Boyd MA, et al. The development of an expert system to predict virological response to HIV therapy as part of an online treatment support tool. AIDS. 2011;25(15):1855–63. doi: 10.1097/QAD.0b013e328349a9c2. [DOI] [PubMed] [Google Scholar]
- 16.Larder BA, Wang D, Revell A, Montaner J, Harrigan R, De Wolf F, et al. The development of artificial neural networks to predict virological response to combination HIV therapy. Antivir Ther. 2007;12(1):15–24. [PubMed] [Google Scholar]
- 17.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;((2/3)):18–22. [Google Scholar]
- 18.Breiman L. Random Forests. Machine Learning. 2001(45):5–32. [Google Scholar]