

Abstract
Neurodegeneration with brain iron accumulation (NBIA) comprises a clinically and genetically heterogeneous group of disorders with progressive extrapyramidal signs and neurological deterioration, characterized by iron accumulation in the basal ganglia. Exome sequencing revealed the presence of recessive missense mutations in COASY, encoding coenzyme A (CoA) synthase in one NBIA-affected subject. A second unrelated individual carrying mutations in COASY was identified by Sanger sequence analysis. CoA synthase is a bifunctional enzyme catalyzing the final steps of CoA biosynthesis by coupling phosphopantetheine with ATP to form dephospho-CoA and its subsequent phosphorylation to generate CoA. We demonstrate alterations in RNA and protein expression levels of CoA synthase, as well as CoA amount, in fibroblasts derived from the two clinical cases and in yeast. This is the second inborn error of coenzyme A biosynthesis to be implicated in NBIA.
Introduction
The common pathological feature of a group of genetic disorders termed “neurodegeneration with brain iron accumulation” (NBIA) is brain iron overload.1 Distinct subclasses of early-onset neurodegeneration with autosomal-recessive transmission are defined by mutations in specific genes: PANK2 (MIM 606157) causes pantothenate kinase-associated neurodegeneration (PKAN);2,3 PLA2G6 (MIM 256600) causes phospholipase A2-associated neurodegeneration (PLAN, also known as INAD);4 FA2H (MIM 611026) causes fatty acid hydroxylase-associated neurodegeneration (FAHN);5 and C19orf12 (MIM 614297) causes mitochondrial membrane protein-associated neurodegeneration (MPAN).6,7 More recently, a distinctive form of NBIA with X-linked dominant de novo mutations in WDR45 (MIM 300894), coding for a protein with a putative role in autophagy, was reported.8,9
These genes account for ∼70% of subjects with NBIA, leaving a significant fraction without an identified genetic defect. For this reason we performed exome sequence investigation in one individual with clinical presentation and neuroimaging suggestive of NBIA but without mutations in previously associated genes. By applying this approach we identified a homozygous missense mutation in COASY, coding for CoA synthase. We then performed traditional Sanger sequence analysis of a larger cohort of idiopathic NBIA cases, and we found a second individual harboring mutations in the same gene. CoA synthase is a bifunctional enzyme possessing 4′PP adenyltransferase (PPAT) and dephospho-CoA kinase (DPCK) activities, catalyzing the last two steps in the CoA biosynthetic pathway.10 The enzyme is encoded by a single gene in mammals and Drosophila,11,12 although two different genes code for PPAT and DPCK activities in yeast and bacteria.13 In human there are three splice variants: COASY alpha is ubiquitously expressed and has a molecular weight of 60 kDa; COASY beta is predominantly expressed in the brain and possesses a 29 aa extension at the N terminus;14 and COASY gamma is predicted to code for C-terminal region of CoA synthase corresponding to DPCK domain. Several studies have investigated the subcellular compartmentalization of the CoA biosynthetic pathway and have demonstrated that both PANK2, defective in the most common NBIA disorder, and CoA synthase alpha and beta are mitochondrial enzymes. PANK2 is mainly located in the intermembrane space2,15,16 whereas CoA synthase alpha and beta are anchored to the outer mitochondrial membrane by the N-terminal region17 or localized within the mitochondrial matrix.18 We here demonstrate that COASY is mainly located in the mitochondrial matrix and that the identified amino acid substitution causes instability of the protein with altered function of its enzymatic activity.
Subjects and Methods
Exome and Sanger Sequencing
Informed consent for participation in this study was obtained from all individuals involved and from their parents, in agreement with the Declaration of Helsinki, approved by the ethics committee of the Fondazione IRCCS (Istituto di Ricovero e Cura a Carattere Scientifico) Istituto Neurologico C. Besta (Milan, Italy) and by the ethics committees of the other institutes participating in the screening (Germany, UK, USA).
Exome sequencing and variant filtering was performed as described previously.8 In brief, exonic DNA fragments were enriched with the SureSelect 50 Mb kit from Agilent and sequenced as 100 bp paired-end reads on a HiSeq 2500 system from Illumina. For sequencing statistics details see Table S1 available online. We predicted that causal mutations would be very rare and would alter the protein. We therefore searched for nonsynonymous variants with a frequency <0.1% in 2,700 control exomes analyzed in Munich and public databases that, given the reported consanguinity of the parents, were anticipated to be homozygous. This analysis left a total of 12 candidate genes (Table S2). The detailed list of these 12 genes is reported in Table S3. We first excluded the following genes because of the presence of additional subjects with compound heterozygous or homozygous mutations related with different clinical phenotypes: HRNR, ADAM8, BZRAP1, C17orf47, LRP1B, EVC2, KIAA1797, and CACNB1. Moreover, variants in HRNR, CACNB1, C17orf47, and KIAA1797 were predicted to be benign by PolyPhen. Four remaining genes (GUCA2A, FBXO47, COASY, and IFNW1) were potentially good candidates carrying deleterious mutations.
By performing segregation analysis of the c.265G>T homozygous variant in GUCA2A, we found that also the healthy mother (subject I-2 of family 1) and one of the healthy sisters (subject II-4 of family 1) carried this variant.
Segregation analysis of c.490A>G in IFNW1 showed that this change was present in homozygous state in the healthy mother (subject I-2 of family 1) and in two healthy sisters (subjects II-4 and II-5 in family 1). Altogether, this observation excluded both GUCA2A and IFNW1 as potential candidate genes (see also Table S3).
FBXO47 was mainly expressed in kidney, liver, and pancreas and it was suggested to act as a tumor-suppressor gene in renal carcinoma and possibly other malignancies.19 However, because this gene carries a splice site mutation, we decided to perform sequence analysis in a subgroup of 56 NBIA-affected individuals. We did not identify any pathogenic mutation in this cohort of subjects.
Based on these data and because COASY coded for an enzyme involved in Coenzyme A biosynthesis as PANK2, we concentrated our efforts on the analysis of this gene.
RNA Extraction and Real-Time PCR
Total RNA was isolated from fibroblasts (80% confluence) with the RNeasy Mini Kit (QIAGEN). RNA quantity was measured with the Nanodrop instrument (Nanodrop Technologies). RNA was used as a template to generate complementary DNA (cDNA) by GoScript Reverse Transcriptase protocol (Promega). Reverse transcriptase products were used in real-time PCR to evaluate the expression level of COASY with the Power SYBR Green PCR Master Mix (Applied Biosystems) system. The housekeeping gene used for data normalization was GAPDH. Primer sequences are as follows: COASY, forward 5′-AGTTGCGGTTTCTCCGTTAG-3′ and reverse 5′-ATCCTGGGAGGGGGAAAT-3′; GAPDH, forward 5′-CTCTGCTCCTCCTGTTCGAC-3′ and reverse 5′-ACGACCAAATCCGTTGA-3′.
Expression and Purification of Recombinant hDPCK in Bacteria
mRNA coding for human DPCK domain (COASY amino acid sequence from 355 to 564) was expressed with the N-terminal histidine-tag from pET30-a(+) (Novagen) at 37°C in E. coli strain BL21 (DE3), after induction with 0.2 mM IPTG.
Cells were lysed by a French press in 50 mM Tris-HCl (pH 8), 0.5 M NaCl, 1% Triton X-100, 20 mM imidazole, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM β-mercaptoethanol, and Roche Complete EDTA-free protease inhibitor cocktail. After clearing, the lysate was loaded on a Ni-NTA beads (QIAGEN) column. Bound proteins were eluted with an imidazole gradient. Fractions containing His-hDPCK were pooled, desalted, and loaded onto an anion-exchange (AE) Resource-S column (GE Healthcare) equilibrated in 50 mM Tris-HCl (pH 7.4), 2.5% glycerol, 20 mM β-mercaptoethanol. The protein was eluted with a NaCl gradient, concentrated by ultrafiltration, and further separated by size exclusion chromatography (SEC) on a Superdex-200 column (GE Healthcare) equilibrated in 10 mM Tris-HCl (pH 7.4), 0.15 M NaCl, 2.5% glycerol, 0.1 mM EDTA, and 1 mM DTT. The entire purification scheme was carried out at 4°C.
Mitochondria and Mitoplast Isolation from Cultured Cells
Isolated mitochondria from cultured cells were obtained according to the protocol described by Fernández-Vizarra.20
For mitoplast purification, mitochondria were dissolved in 1 ml Buffer A (MOPS 20 mM, sucrose 0.25 M [pH 7.4]). A total of 1 ml of 200 μg/ml digitonin in Buffer A was added to each sample. Samples were mixed and incubated on ice 5 min, then centrifuged 3 min at 8,000 rpm at 4°C. Supernatant was discarded and pellet dissolved in 1 ml Buffer B (MOPS 20 mM, sucrose 0.25 M, EDTA Na4 1 mM [pH 7.4]). Samples were incubated on ice for 5 min, then centrifuged at 12,000 rpm at 4°C for 3 min. Separate fractions of mitochondria and mitoplasts were also treated with 0.04 μg of proteinase K (PK) for 15 min at 4°C or 37°C; PK digestion was blocked with PMSF. In some samples of mitochondria and mitoplasts, 0.1% Triton X-100 was added followed by incubation for 15 min at 37°C.
Immunoblot Analysis
Approximately 1 × 106 fibroblasts, grown in DMEM (EuroClone) were trypsinized, centrifuged at 1,200 rpm for 3 min, and solubilized in 200 μl of RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP40, 0.5% NaDOC, 5 mM EDTA) with 1× Complete Mini Protease Inhibitor Cocktail Tablets (Roche) for 40 min at 4°C. 30 μg of proteins were used for each sample in denaturing sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Immunoblot analysis was performed as described21 with the ECL-chemiluminescence kit (Amersham).
Antibodies
A rabbit monoclonal anti-COASY antibody was used at 1:1,000 dilution (EPR8246-Abcam). A mouse monoclonal anti-β-TUBULIN antibody was used at a final concentration of 1 μg/ml (Sigma-Aldrich). Secondary anti-rabbit and anti-mouse antibodies were used at 1:2,000 and 1:7,000 dilution, respectively.
HPLC Analysis of Dephospho-CoA, CoA, and AcetylCoA
HPLC analysis was performed on recombinant wild-type and mutant DPCK proteins, on fibroblast lysates derived from control and subjects carrying COASY variants, and on isolated yeast mitochondria.
The method employed a column (Kinetex 5u C18 100A New Column 250 × 4.6 mm from Phenomenex) eluted with 100 mmol/l NaH2PO4 and 75 mmol/l CH3COONa (pH was adjusted to 4.6 by the addition of concentrated H3PO4)-acetonitrile (94:6, v/v) at a flow rate of 1.0 ml/min. The ultraviolet (UV) detector was set at 259 nm. To obtain standard solutions of 5 μM, dephospho-CoA and CoA were dissolved in 50 mmol/l KH2PO4-K2HPO4 buffer (pH 7.0). CoA and dephospho-CoA standards were eluted at approximately 4.5 and 8 min, respectively, and CoA compounds were quantified by comparison of peak areas with those of authentic standards.
In Vitro DPCK Activity
1 μg of purified wild-type or mutant protein was incubated for 2 hr at 37°C in 50 μl of reaction mixture containing 50 mM Tris-HCl (pH 8), 5 mM MgCl2, 1 mM ATP, and 0.1 mM dephospho-CoA. After incubation, sample was treated with perchloric acid (PCA) 3%, vortexed, and centrifuged at 13,000 rpm at 4°C. Trietanolamine was added to the supernatant to a final concentration of 100 mM, and then the sample was neutralized with 5 M K2CO3.
Fibroblast Analysis
Fibroblasts, grown on 10 cm plates (approx. 80%–90% confluent), were washed with PBS and collected by trypsinization. 40 μl of ice-cold PCA (5%) was added to cells and samples were vortexed and centrifuged at 18,000 × g for 5 min at 4°C. The supernatant was collected and triethanolamine was added to a final concentration of 100 mM. The pH was adjusted to 6.5 with 5 M K2CO3 before centrifuging again at 18,000 × g for 3 min at 4°C to remove potassium perchlorate. Neutralized PCA extract was made up to 100 μl with Na2H2PO4 (150 mM), Tris-(2-carboxyethyl) phosphine hydrochloride (TCEP) (10 mM), EDTA (5 mM), and methanol (9%) and filtered through a 0.2 μm PVDF filter, and 50 μl was injected for HPLC analysis of CoA compounds.
In Vitro PPAT/DPCK Assay of Cell Homogenates
Fibroblasts grown on 10 cm plates (approx. 80%–90% confluent) were washed with PBS and collected by trypsinization. Cells were homogenized in 150 μl buffer containing 50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 10 mM 2-glycerophosphate, 1 mM EDTA, 0.5 mM TCEP, and protease inhibitor cocktail (Roche). Total protein concentration in the homogenate was measured by Bradford assay. 65 μg of homogenate protein was incubated with 2 mM ATP, 5 mM MgCl2, and 5 mM 4-phosphopantetheine in a total volume of 50 μl at 30°C for 1 hr. 4-phosphopantetheine was prepared by phosphorylating pantetheine with bacterially expressed pantothenate kinase 1b. For control incubation, ATP and MgCl2 were added to homogenate, but 4-phosphopantetheine was omitted. After the incubation, PCA (3.5% final) was added to the reaction mixtures before centrifugation at 18,000 × g for 5 min at 4°C. The pH of the PCA-soluble fraction was adjusted to 6.5 with TEA/K2CO3 and CoA compounds formed were analyzed by HPLC as described above.
Yeast Mitochondria Analysis
Mitochondrial suspensions were diluted to obtain about 0.5 μg/μl in a final volume of 150 μl of 5% 5-sulfosalicylic acid containing 50 μmole/l DTT and vortexed. The homogenates were centrifuged at 12,000 × g for 10 min at 4°C. The supernatant was passed through a 0.45 μm filter (Millipore) and the filtrate (40 μl) was injected directly into the HPLC system. We loaded equal amount of yeast mitochondrial proteins (40 μg) and we performed CoA quantification by evaluating peak’s areas as compared to known concentration of internal standard.
Yeast Strains and Media
Yeast strains used in this study were W303-1B (MATαade2-1 leu2-3,112 ura3-1 his3-11,15 trp1-1 can1-100) ade2-1 leu2-3,112 ura3-1 trp1-1 his3-11,15, its isogenic strain cab5::KanMx4 that harbors plasmid pFl38-CAB5 or pFl39-CAB5, and the strain cab5::KanMx4 that harbors plasmid pYEX-BX-COASY (see below). Cells were cultured in minimal medium 40 supplemented with appropriate amino acids and bases for auxotrophy as previously described.22 To obtain medium lacking pantothenate (40-Pan), a mixture of vitamins without pantothenate was prepared. Various carbon sources (Carlo Erba Reagents) were added at the indicated concentration. YP medium contained 1% Bacto-yeast extract and 2% Bacto-peptone (ForMedium). Media were solidified with 20 g/l agar (ForMedium) and strains were incubated at 23°C, 30°C, or 37°C.
Cloning Procedures and Plasmid Vectors
pFL38-CAB5 was obtained by PCR amplification of CAB5, including the upstream and the downstream regions, from genomic DNA of strain W303-1B with primers as follows.
For CAB5 (forward 5′-GGGGGGATCCCCATTGCTTAGAATGGGCGG-3′ and reverse 5′-CCGCGGTACCGAGAACCCATAGAATTCGAC-3′), the oligos were modified at 5′ end in order to insert restriction sites for cloning in the centromeric plasmid pFL38 carrying the URA3 marker.23 pFL39-CAB5 was obtained by subcloning CAB5 into pFL39 vector carrying the TRP1 marker.23 Human COASY and human COASYArg499Cys were amplified by PCR from pcDNA3.1 constructs, containing wild-type and mutant cDNA, respectively, with primers described below.
For COASY (forward 5′-GGGGGGATCCATGGCCGTATTCCGGTCG-3′ and reverse 5′-CCGCGTCGACTCAGTCGAGGGCCTGATGAGTC-3′), the oligonucleotides contained appropriate restriction sites to allow cloning in the BamHI-SalI-digested pYEX plasmid under the control of CUP1 promoter. All cloned fragments were sequenced to check the absence of mutations. Restriction-enzyme digestions, Escherichia coli transformation, and plasmid extractions were performed with standard methods.24
Site-Directed Mutagenesis and Generation of Yeast cab5 Strains
The conserved human arginine 499 residue (RefSeq accession number NM_025233.6), which is replaced by a cysteine in human COASY, corresponds to arginine 146 (RefSeq NM_001180504.3) in the yeast protein. The CAB5 mutant allele was obtained by site-directed mutagenesis (QuikChange II Site-Directed Mutagenesis Kit Stratagene) by introducing an AGA>TGT codon substitution, resulting in p.Arg146Cys amino acid change. The corresponding modified primers used to generate mutated allele are as follows.
For COASYArg146Cys (forward 5′-CGCAAGAATTGCAACTAGAATGTTTAATGACAAGAAATCCTG-3′ and reverse 5′-CAGGATTTCTTGTCATTAAACATTCTAGTTGCAATTCTTGCG-3′), mutagenized insert was verified by sequencing of both strands.
The pFL38 plasmid-borne CAB5 was transformed in the W303-1B by the lithium-acetate method25 to allow cell viability and the resident CAB5 was deleted with the KanMX4 cassette amplified from plasmid pCXJKan by primers described below.
For CAB5-Kan (forward 5′-CAGATAGCCACAATTAAAATATGCTGGTAGTGGGATTGACAGGTCGTACGCTGCAGGTCGAC-3′ and reverse 5′-GTAATTATAAGATATCAACCTTATACCCGCTGAAGACTTTTTATTTTGAAGATCGATGAATTGAGCTCG-3′), both of them contained a 5′ complementary stretch for an internal sequence of CAB5 ORF and a 3′ complementary stretch (underlined in the sequences) for the extremities of the KanMX4 cassette. Strains with engineered CAB5 were selected on YP supplemented with 200 μg/ml geneticin and gene rearrangement was confirmed by PCR. Transformation of cab5::KanR/pFLl38CAB5 strain with pfl39-CAB5 and pFL39-CAB5Arg146Cys constructs and plasmid shuffling on media supplemented with 5-FOA in order to select spontaneous events of Ura+ constructs loss were finally performed. Similarly, cab5::KanR/pFL39CAB5 strain was transformed with pYEX-BX-COASY and pYEX-BX-COASYArg499Cys constructs; loss of Trp+ plasmids was induced by growing the transformants with tryptophan in the medium.
Yeast Mitochondria Isolation
Mitochondria were purified as previously reported.26 In brief, cells cultured at 28°C in the 40 medium supplemented with 0.6% glucose were collected and washed. Spheroplasts were obtained after Zymoliase20T digestion (Nacalai Tesque) and disrupted with a glass-teflon potter homogenizer. Mitochondria were purified by differential centrifugation. Total protein concentrations were quantified according to Bradford (Bio-rad).
Results
Molecular and Biochemical Investigations
Exome-NGS analysis of one individual (subject II-3, family 1, Figure 1) affected by idiopathic NBIA resulted in the identification of 12 genes (Table S3) that carried variants potentially relevant for the disease. However, as described in detail in the Subjects and Methods section, several of these genes were not investigated further because (1) the identified variants were present in additional individuals and associated with other clinical phenotypes; (2) genetic segregation analysis was not compatible with clinical presentation of the subjects of family 1; or (3) gene function and tissue-specific expression could hardly explain the neurological presentation. The homozygous mutation in COASY, coding for a bifunctional enzyme converting 4′-phosphopantetheine into dephospho-CoA and then to Coenzyme A, was considered as potentially relevant for the disease and further investigated. By Sanger sequencing we confirmed the presence of the homozygous missense COASY mutation in the affected individual (Figure 1: subject II-3, family 1). The family had no history of neurological disorders and this subject was the youngest and the only affected of five siblings. She was born to consanguineous parents after an uneventful pregnancy and normal delivery. Birth weight was 3,850 g. There was no history of perinatal complications and she attained normal early developmental milestones. From 24 months of age, parents reported gait difficulties and persistent toe walking. At age 6, when she started primary school, she showed poor academic ability. At age 15, general physical examination was normal. Neurological evaluation showed mild oro-mandibular dystonia with dysarthria and also spastic dystonic paraparesis, but she was still able to walk unaided. Neuropsychological evaluation demonstrated cognitive impairment (total IQ = 49). The disease continued to progress slowly and at the age of 20 she became unable to ambulate independently. During the most recent examination at age 25, the clinical picture was dominated by a severe spastic bradykinetic-rigid syndrome associated with mild dystonia and with distal areflexia in the lower limbs. There were no clinical or psychometric data suggesting mental deterioration but behavioral disturbances with obsessive-compulsive symptoms and depression was evident. Funduscopic examination and visual evoked potential studies were normal and on electroretinogram there were no signs of retinopathy. Electromyographic and nerve conduction studies were consistent with a mild motor axonal neuropathy. Serial brain MRI showed bilateral hypointensity in the globi pallidi associated with a central region of hyperintensity in the antero-medial portion (Figure 1).
Figure 1.
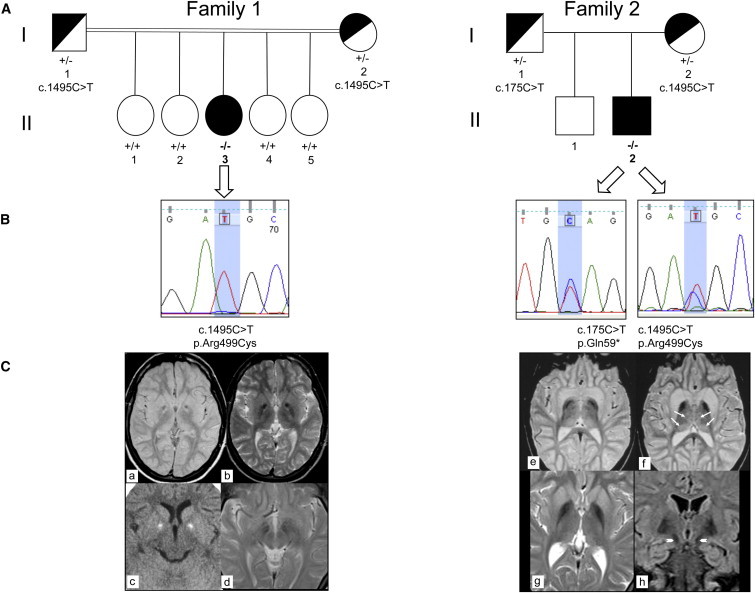
Genetics and MRI of Subjects Carrying COASY Mutations
(A) Pedigrees of family 1 (left) and family 2 (right). II-3, affected individual in family 1; II-2, affected individual in family 2. The presence of homozygous or compound heterozygous mutation is indicated by −/−; wild-type sequence by +/+; heterozygous mutation by +/−.
(B) Electropherograms show sequence variations in individual II-3 of family 1 (left) and in individual II-2 of family 2 (right).
(C) Left: MRI of individual II-3 of family 1 at 11 years of age (a–c). Axial MR (1.5 T) proton density and T2-weighted images (a, b) show bilateral low signal intensity in the globi pallidi (clearly visible in b) with a central region of high signal intensity located in the antero-medial portion of the nuclei (“eye-of-the-tiger” sign) and with a large central spot of low signal intensity. Axial CT (c) shows bilateral hyperdensities consistent with calcifications and corresponding to the central spot visible on MRI. Six years later (d), no changes were found. The hypointensity in the medial portion of the substantia nigra was also unchanged. Right: MRI of individual II-2 of family 2 at 9 years of age (e, f) and at age 19 (g, h). Axial T2-weighted 1.5 T MR images (e, f) reveal hypointensity in the pallida. Both caudate nuclei and putamina are swollen and hyperintense. Slight hyperintensity is also present in both medial and posterior thalami (arrows). Axial T2-weighted MR image (g) confirms bilateral symmetric low signal intensity and atrophy in the pallida. Both putamina and caudate nuclei are still slightly hyperintense with minimal swelling. Coronal FLAIR image (h) demonstrates low signal in both pallida and in the medial portion of the substantia nigra (arrowheads).
Identification of one Italian subject carrying COASY mutation prompted us to analyze the nine exons of this gene in a cohort of 280 NBIA-affected individuals of different ethnicity by using polymerase chain reaction and direct Sanger sequencing. Primer sequences and PCR conditions are described in Table S4. By this analysis we identified a second Italian case carrying COASY mutations (Figure 1: subject II-2, family 2). He is 20 years old and he was born at term of uneventful pregnancy from healthy nonconsanguineous parents. Psychomotor development was normal in the first year of life, but he was delayed in walking as a result of instability and toe walking. At age 3 the neurological picture was characterized by spastic tetraparesis with moderate mental and language impairment. The disease was progressive, with worsening of the motor signs in the lower limbs and progressive involvement of the upper limbs and oro-mandibular region. He lost independent ambulation at age 15. At age 17, the neurological examination showed mild oro-mandibular dystonia with dysarthria, spastic-dystonic tetraparesis with prevalent involvement of lower limbs, and parkinsonian features (rigidity and abnormal postural reflexes). Distal amyotrophia and areflexia with pes cavus were also evident. Cognitive impairment was severe (total IQ < 40) with obsessive-compulsive behavior and complex motor tics. On follow-up, 2 years later, the neurological picture was unchanged. Nerve conduction study and electromyography detected a motor axonal neuropathy more prominent in the lower limbs. There was no retinal or optic nerve involvement, as demonstrated by normal funduscopic and evoked potential studies.
The first brain MRI performed at age 5 demonstrated hyperintensity and swelling of both caudate nuclei and putamina and mild hyperintensity in both thalami. Globi pallidi were normal. At ages 9 and 19, hypointensity in the globi pallidi was evident and no significant changes were found in the caudate nuclei, putamina, and thalami (Figure 1).
Subject II-3 of family 1 (Figure 1) carried a homozygous missense mutation, a c.1495C>T transition causing an amino acid change p.Arg499Cys (referral sequence NM_025233.6; numbering starts from the first methionine). Segregation analysis performed in family 1 indicated heterozygous state in the parents (Figure 1), and the four healthy sisters showed wild-type sequence (Figure 1).
Subject II-2 of family 2 (Figure 1) turned out to be a compound heterozygote for the same mutation, c.1495C>T (p.Arg499Cys), identified in subject II-3, and for a c.175C>T transition, resulting in a premature p.Gln59∗ stop codon in the N-terminal regulatory region of the protein. Segregation analysis in the parents demonstrated that the two mutations were on different alleles: one inherited from the mother and one from the father (Figure 1). The healthy brother was not available for genetic testing.
The missense substitution affected an amino acid residue Arg499, which is highly conserved in all available animal, plant, and yeast species, including S. cerevisiae, and is localized in the nucleotide-binding site of the DPCK domain (Figure 2). Furthermore, mutational analysis of Arg140, equivalent to Arg499, in the mycobacterial dephospho-CoA kinase (CoaE) revealed the importance of this residue in ATP binding and phosphotransfer reaction.27,28
Figure 2.
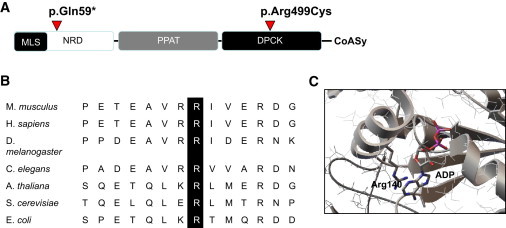
COASY: Conserved Domains, Phylogenetic Conservation, and Crystal Structure
(A) Schematic domain organization of human CoA synthase and location of point mutations. Abbreviations are as follows: MLS, mitochondrial localization signal; NRD, N terminus regulatory domain; PPAT, 4′PP adenylyltransferase domain; DPCK, dephospho-CoA kinase domain.
(B) Amino acid sequence alignment showing conservation of Arg499 across species.
(C) Crystal structure of E. coli DPCK (CoaE) (PDB ID 1VHL) showing the position of Arg140 (equivalent to Arg499 in human DPCK) in the nucleotide-binding site.
The substitution was predicted to be pathogenic by in silico analysis according to Polyphen2 (p = 1) and MutPred (p = 0.909). Frequency of the mutation derived from the Exome Variant Server and calculated on European, American, and African population was 1 out of 13,005 analyzed cases.
To evaluate the impact of the two mutations on the stability of the transcript, we extracted mRNA from fibroblasts of subjects II-3 (family 1) and II-2 (family 2) and reverse transcribed it into cDNA. Quantitative real-time PCR showed that although in individual II-3 the amount of mutant COASY transcript was similar to that of the control sample (Figure 3A), it was reduced to 50% in individual II-2, suggesting RNA decay.
Figure 3.
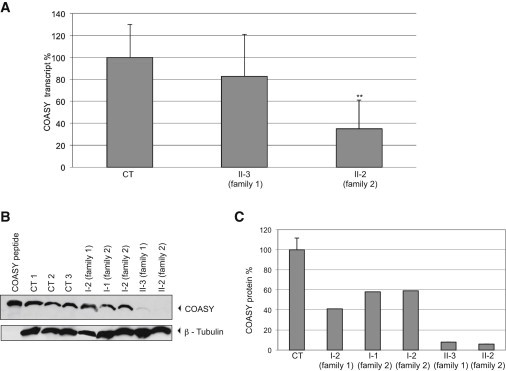
COASY mRNA Expression and Protein Accumulation in Skin Fibroblasts
(A) Quantification of COASY mRNA levels by real-time PCR in fibroblasts of subject II-3 and II-2 relative to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The amount of COASY transcript is reduced in subject II-2 versus control samples (CT), indicating mRNA decay. Data are represented as mean ± SD. Statistically significant differences with CT were determined by the Student’s t test; ∗∗p < 0.02.
(B) Immunoblot analysis of COASY in fibroblasts derived from three healthy subjects (CT 1, CT 2, CT 3), individual I-2 (family 1), individuals I-1 and I-2 (family 2), and affected subjects (II-3 and II-2). The same amount of protein (30 μg) was loaded. β-tubulin was used as a loading control. As a control, COASY in vitro translation product (COASY peptide) was loaded.
(C) Relative quantification of the protein amount: mean ± SD of three controls (CT); of individual I-2 (family 1); of individuals I-1 and I-2 (family 2); and of affected subjects II-3 and II-2. Histogram shows COASY amount quantified by densitometry and normalized on β-tubulin level.
Next, we analyzed COASY level in total cell lysates obtained from both mutants and control fibroblasts by using a monoclonal anti-COASY antibody. We first tested the antibody specificity by verifying its cross-reactivity with the 62 kDa COASY alpha in vitro translation product (Figure 3B).
Immunoblot analysis revealed the presence of a normal protein content in three different control fibroblasts whereas a significant reduction of the protein amount was detected in fibroblasts of subject II-2 (family 2) carrying the premature stop codon and the missense p.Arg499Cys (Figure 3B). Interestingly, we also observed a minimally detectable immunoreactive band corresponding to COASY in subject II-3 (family 1) carrying the homozygous p.Arg499Cys substitution (Figure 3B). This suggests that the p.Arg499Cys change is associated with instability or accelerated degradation of the protein. Immunoblot analysis of fibroblasts derived from subject I-2 of family 1 and from both parents of family 2 (Figure 3B) showed a partial reduction of the protein level. As reported in Figure 3C, protein amount quantified by densitometry analysis with three different controls as standard resulted to be around 50% in subject I-2 of family 1 and in the parents of family 2 and less than 5% in both affected individuals.
Submitochondrial Localization of COASY
To better determine submitochondrial localization of COASY, we carried out immunoblot analysis of mitochondria and submitochondrial fractions derived from HeLa cells, using a commercially available antibody (see Subjects and Methods).
Immunoblotting of different cellular fractions revealed the presence of a band of the expected molecular weight in total lysate and intact mitochondria (Figure 4A). To determine whether the protein was present on the outer mitochondrial membrane, we treated mitochondria with proteinase K (PK) and demonstrated COASY resistance to degradation (Figure 4A). Efficiency of PK activity was demonstrated by treating mitochondria with Triton X-100, which dissolves the membranes and makes the protein accessible to PK digestion (Figure 4A). This result was further supported by hybridizing the same filter with control antibodies against proteins such as CORE1 or ETHE1, which are located in the inner mitochondrial membrane and in the mitochondrial matrix, respectively, or against VDAC1, which is located in the outer mitochondrial membrane (Figure 4A) facing the intermembrane space. We observed that COASY was also present in total lysates and not enriched in the mitochondrial fraction, suggesting that its localization might not be exclusively in mitochondria.
Figure 4.
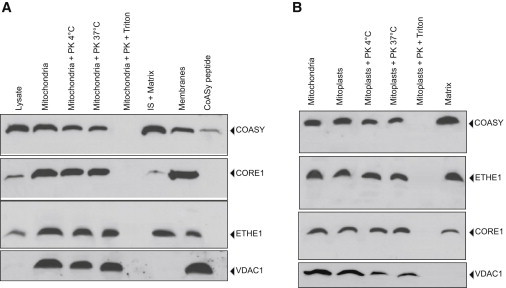
Mitochondrial Localization of COASY
(A) Immunoblot analysis on mitochondria and different submitochondrial fractions derived from HeLa cells. Mitochondria were treated for 15 min at 4°C or 37°C with proteinase K (PK) in presence or absence of triton. The filter was incubated with anti-COASY, anti-CORE1, anti-ETHE1, and anti-VDAC1 antibodies. As a control, COASY in vitro translation product (COASY peptide) was loaded.
(B) Immunoblot analysis on mitoplasts, matrix, and inner membrane isolated from HeLa cells. Mitoplasts were treated for 15 min at 4°C or 37°C with PK in presence or absence of triton. The filter was sequentially incubated with anti-COASY, anti-ETHE1, anti-CORE1, and anti-VDAC antibodies.
The protein was found in mitoplasts and was resistant to PK digestion. Further fractionation of mitoplasts demonstrated that the protein was mainly present in the matrix, probably anchored to the inner mitochondrial membrane (Figure 4B). The presence of trans-membrane domains was predicted by TMpred and PSIPRED software. We also observed the presence of VDAC in mitoplasts, suggesting that the outer mitochondrial membrane was not completely removed. However, VDAC was partially digested with PK and, most importantly, it was completely absent in the mitochondrial matrix.
HPLC Assays on Recombinant Protein and Fibroblasts Derived from Affected Subjects
To assess the effect of the p.Arg499Cys substitution on the DPCK activity, we expressed mRNA corresponding to the wild-type and mutant DPCK domain in bacteria as His-tag fusion proteins. Recombinant proteins were purified by NTA chromatography and 1 μg of each protein was loaded on an SDS-PAGE and stained with Coomassie blue (Figure 5A, top). To demonstrate that the recombinant proteins were recognized by the anti-COASY antibody, the gel was blotted and incubated with the specific antibody (Figure 5A, bottom). Activities of wild-type DPCK and of the mutant DPCK-Arg499Cys were measured in vitro by HPLC analysis via 1 μg of recombinant proteins.
Figure 5.
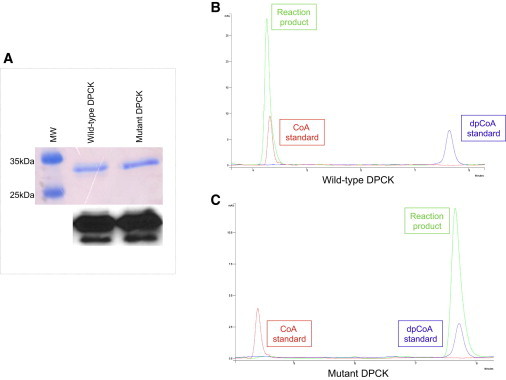
HPLC Analysis of CoA Production by Wild-Type and Mutant DPCK Recombinant Proteins
(A) Top: equal amount of purified wild-type and mutant DPCK proteins were loaded on a 12% SDS page and stained with Coomassie blue. Bottom: immunoblot analysis on the same gel showing that anti-COASY antibody is able to recognize both the wild-type and the mutant protein.
(B) Chromatogram showing the peak corresponding to the reaction product (green) obtained from incubation of wild-type DPCK recombinant protein with ATP and dephospho-CoA.
(C) Chromatogram showing the peak corresponding to the reaction product (green) obtained from incubation of mutant DPCK-Arg499Cys recombinant protein with ATP and dephospho-CoA. Red peak, CoA standard; blue peak, dephospho-CoA standard.
This analysis evaluates dephospho-CoA conversion into CoA after incubation of wild-type and mutant DPCK proteins with ATP and dephospho-CoA. As indicated by the chromatogram in Figure 5B, the wild-type enzyme was able to completely convert dephospho-CoA into CoA, as demonstrated by the coincidence of the reaction mixture peak with that of CoA standard. On the contrary, the DPCK-Arg499Cys mutant did not have this enzymatic activity and the peak corresponding to CoA was not observed (Figure 5C). This finding suggests that CoA biosynthesis might be abolished in the presence of the p.Arg499Cys change.
We then analyzed CoA levels in fibroblasts derived from healthy and affected subjects by HPLC, but we did not observe a significant difference. However, a general reduction of acetyl-CoA and total CoA was observed in both affected individuals as compared to control, and this difference was statistically significant for acetyl-CoA in subject II-3 of family 1 (Figure 6A).
Figure 6.
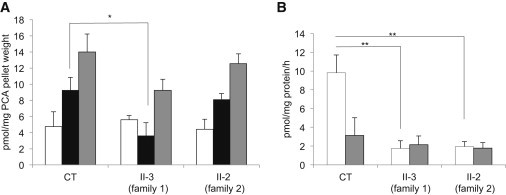
HPLC Analysis of CoA and CoA Derivatives in Fibroblasts
(A) CoA (white bar), acetyl-CoA (black bar), and total CoA (gray bar) levels in primary skin fibroblasts derived from a healthy control (CT) and from the two affected individuals (II-3, family 1; II-2, family 2). Results shown are mean ± SEM of four independent experiments. Statistically significant differences in acetyl-CoA amount between CT and subject II-3 (family 1) were determined by the Student’s t test; ∗p < 0.05. This subject also shows a reduction in acetyl-CoA, which is not statistically significant. A reduction of total CoA was observed in both affected individuals, although not statistically significant.
(B) De novo synthesis of CoA and dephosphoCoA (dpCoA) in primary skin fibroblasts derived from a healthy control (CT) and from the two affected individuals (II-3, family 1; II-2, family 2). CoA (white bar) and dpCoA (gray bar) produced from 4′PP as substrate were quantified by HPLC after deproteinization of reaction mixture with PCA (3% final). Results shown are mean ± SEM of values from three independent experiments. Statistically significant differences with CT were determined by the Student’s t test; ∗∗p < 0.02.
To examine whether skin fibroblasts from affected individuals were able to synthesize CoA, we performed an in vitro assay to evaluate CoA biosynthesis in cell homogenates with 4′PP (4′-phosphopantetheine) as substrate. HPLC analysis of reaction mixtures showed that dephospho-CoA and CoA were efficiently produced de novo from 4′PP in control fibroblasts (Figure 6B). We also observed residual de novo production of dephospho-CoA and CoA in skin fibroblasts from affected subjects, although the level of CoA was approximately 20% of that produced by control fibroblasts (Figure 6B). These findings suggest the existence of an alternative as yet uncharacterized pathway for CoA biosynthesis. However, we cannot exclude the possibility that the remaining COASY aberrant protein present in fibroblasts may still retain some catalytic activity.
Studies in Yeast Saccharomyces cerevisiae
To further test the pathogenic role of the COASY missense mutation, we used the yeast Saccharomyces cerevisiae. Biosynthesis of CoA in S. cerevisiae follows the same pathway described for mammalian cells: pantothenate, formed de novo from several amino acids or taken up from outside the cell, is converted in CoA in five reactions catalyzed by enzymes encoded by CAB1 through CAB5.29 With the exception of CAB1, the other genes of the pathway have been identified because of sequence similarity and their function in CoA biosynthesis assessed by heterologous complementation with bacterial genes. The only difference with human is that in yeast, as in E. coli, the PPAT and DPCK activities reside on different proteins encoded by CAB4 and CAB5 genes, respectively. Deletion of each CAB gene results in a lethal phenotype, indicating an essential role for this pathway in yeast.
Sequence analysis indicated that Arg499 is highly conserved from yeast to human and corresponds to Arg146 in the yeast Cab5p (see also Figure 2). By using the plasmid shuffling method, deletion strains expressing either the mutant alleles cab5Arg146Cys and COASYArg499Cys or the CAB5 and COASY wild-type genes were generated. The Δcab5 lethal phenotype was rescued by the re-expression of either human COASY wild-type or human COASYArg499Cys and yeast cab5Arg146Cys. No major defects of growth on different substrates or at different temperatures were observed (data not shown).
However, we noticed that the mutant cab5Arg146Cys as well as the strain expressing COASYArg499Cys became auxotrophic for pantothenate and showed growth reduction. In fact, wild-type yeast can form colonies regardless of the presence of pantothenate at all tested temperatures (Figure 7A); by contrast, in the absence of pantothenate both mutants cab5Arg146Cys and COASYArg499Cys failed to form colonies at 37°C and a significant impairment of growth was observed at both 23°C and 28°C when compared with that of the strain expressing the wild-type alleles (Figure 7B). This result supports the pathogenicity of the substitution p.Arg499Cys and suggests that the mutant enzyme requires a higher concentration of pantothenate to produce enough CoA to sustain yeast growth.
Figure 7.
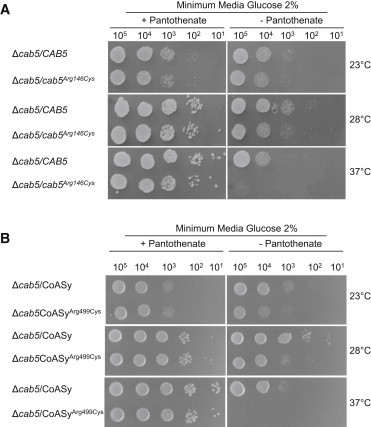
Growth of Yeast Strains in Presence or Absence of Pantothenate
The strain Δcab5 was transformed with pFL39 plasmid carrying the wild-type CAB5 and the mutant allele cab5Arg146Cys (A) or with pYEX-BX plasmid carrying COASY and COASYArg499Cys (B). Equal amounts of serial dilutions of cells from exponentially grown cultures (105, 104, 103, 102, 101 cells) were spotted onto minimum medium 40 plus 2% glucose, with or without pantothenate 1 mg l−1. The growth was scored after 3 days of incubation at 23°C, 28°C, or 37°C. Each experiment of serial dilution grow test was done in triplicate starting from independent yeast cultures.
Because Cab5p as COASY is located into the mitochondria,30 we measured the level of CoA in mitochondria isolated from wild-type, COASYArg499Cys, and cab5Arg146Cys transformed yeasts grown in complete medium at 28°C with 0.6% glucose. We first verified, by immunoblot analysis, that COASYArg499Cys was expressed in yeast at a comparable level as in the wild-type enzyme (not shown). We could not verify cab5Arg146Cys expression because the available antibody did not cross-react with the yeast protein. We observed that the level of CoA was reduced to 40% in yeast transformed with both the human COASYArg499Cys and yeast cab5Arg146Cys mutant versions as compared to wild-type (Figure 8).
Figure 8.
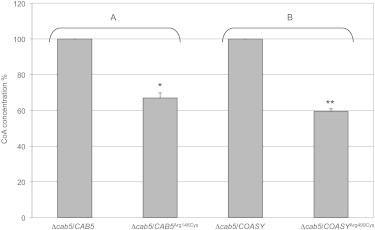
HPLC Analysis of CoA in Yeast Mitochondria
CoA level in mitochondria isolated from Δcab5 yeast transformed with wild-type (WT) or mutant (p.Arg146Cys) yeast CAB5 (A), and with wild-type or mutant (p.Arg499Cys) human COASY (B). Equal amount of mitochondrial proteins (40 μg) were used in each assay. Results shown are mean ± SD of values from three independent experiments. Values of mutant samples are expressed as percentage of values obtained in wild-type samples taken as 100%. Statistically significant differences were determined by the Student’s t test; ∗p < 0.05; ∗∗p < 0.02.
Discussion
We here report the second inborn error of CoA synthesis leading to a neurodegenerative disorder. The first defect discovered was due to PANK2 mutations, causing the most prevalent NBIA subtype, PKAN.2
Coenzyme A (CoA) is a crucial cofactor in all living organisms and is involved in several enzymatic reactions. It is a key molecule for the metabolism of fatty acids, carbohydrates, amino acids, and ketone bodies. Its biosynthesis proceeds through a pathway conserved from prokaryotes to eukaryotes, involving five enzymatic steps, which utilize pantothenate (vitamin B5), ATP, and cysteine.
In the first step, catalyzed by pantothenate kinase, the product of PANK2, pantothenic acid is phosphorylated to generate 4′-phosphopantothenic acid. Then, this intermediate is converted into 4′-phosphopantothenoylcysteine, which is subsequently decarboxylated to 4′-phosphopantetheine. The last two steps are carried out by the bifunctional enzyme CoA synthase, which converts 4′-phosphopantetheine into dephospho-CoA and then CoA.31
We have identified mutations in the COASY in two subjects with clinical and MRI features typical of NBIA. They displayed a strikingly similar phenotype, more severe in subject II-2 of family 2, presenting with early-onset spastic-dystonic paraparesis with a later appearance of parkinsonian features, cognitive impairment, and pronounced obsessive-compulsive disorder. The disease was slowly progressive with loss of ambulation during adolescence and adulthood. This phenotype overlaps with other NBIA disorders, including the presence of an axonal neuropathy, which is commonly reported in phospholipase A2-associated neurodegeneration (PLAN) and also in mitochondrial membrane protein-associated neurodegeneration (MPAN) cases.32
In subject II-3 of family 1, MR images are reminiscent of the “eye-of-the-tiger” sign even if with subtle features, which differentiate it from the typical appearance present in PKAN.33,34 In subject II-2 of family 2, an isolated involvement of neostriatum, which usually hallmarks a metabolic rather than degenerative disorder, preceded the evidence of the typical increase of pallida iron content. Such features have not been previously reported, expanding the MR spectrum of NBIA disorders.
Both individuals presented with a severe neurological disorder but they have survived up to the third decade of life, suggesting the presence of residual amount of CoA as observed in cultured fibroblasts. The complete absence of CoA would be probably incompatible with life, and organisms have developed alternative strategies to counteract deleterious effects of mutations in CoA enzymatic pathway. For instance, mammals possess four closely related PANK isoforms,2 1α, 1β, 2, and 3, which exhibit a tissue-specific pattern of expression. This redundancy could explain why PKAN patients can survive into the first or second decade of life. Probably, the different isoforms can compensate each other to maintain adequate CoA level. This was clearly demonstrated in mice by the simultaneous knockout of two different Pank genes.35
COASY has been reported to code for three transcript variants resulting in tissue-specific isoforms.14 The existence and functional significance of these variants are presently unknown but both mutations found in this study affect the protein sequence common to isoforms alpha and beta, predicting overall impairment of COASY function. Considering the ubiquitous presence of the enzymatic COASY activity, it remains unexplained why only the brain is affected and other organs are preserved. It is possible that a more severe impairment of CoA levels occurs in this organ, thus explaining the prevalence of neurological symptoms. At the cellular level CoA concentration is regulated by numerous factors, including hormones, glucocorticoids, nutrients, and cellular metabolites,36,37 and a link between the complex signaling mTOR pathway, which is implicated in numerous metabolic and signaling processes, and CoA biosynthesis has been proposed.38 Moreover, it is relevant to notice that the mutations targeted genes coding for pantothenate kinase39 and PPAT activity of CoA synthase36 are the two rate-limiting steps in CoA biosynthesis. All together these factors could contribute to modulate the clinical presentation of individuals carrying COASY mutations.
It is still unknown how mutations in genes involved in Coenzyme A enzymatic pathway cause neurodegeneration with iron accumulation in specific areas of the brain but whereas for PANK2 it was hypothesized that cysteine accumulation may chelate iron and catalyze free radical formation,40 a different mechanism could be involved in case of COASY mutations.
In Drosophila it has been demonstrated that abolishing the different genes of CoA biosynthetic pathway including the fumble/PANK2 and PPAT-DPCK activities causes a neurological phenotype characterized by brain vacuolization without iron accumulation.12
Identification of mutations in CoA synthase strongly reinforces the essential role of CoA biosynthetic pathway for the development and functioning of the nervous system. This also underlines the importance of further investigations on different subcellular pools of CoA available, because a specific mitochondrial pathway could exist considering that both PANK2 and CoA synthase are mitochondrial enzymes.16–18 At present it is not understood whether CoA can pass from cytosol to mitochondria, even if a CoA-specific carrier has been identified in the inner mitochondrial membrane.41 Moreover, it is not clear whether the regulation of the different pools is coordinated and whether the utilization could be modulated in response to different physiological or pathological conditions.
In conclusion, we have demonstrated that COASY mutations cause a distinctive NBIA subtype. This finding will require further investigation to understand the connection linking CoA metabolism to neurodegeneration, iron accumulation, and mitochondrial bioenergetics. We propose CoPAN, standing for COASY protein-associated neurodegeneration, as the acronym for NBIA caused by CoA synthase mutations to conform with the current nomenclature in use to classify these disorders.
Acknowledgments
We would like to thank Mario Savoiardo and Federica Zibordi for helpful neuroradiological and clinical support and Fabrizio Villa for experimental advice. The financial support of Telethon GGP11088 to V.T. is gratefully acknowledged. This work was supported by TIRCON project of the European Commission’s Seventh Framework Programme (FP7/2007-2013, HEALTH-F2-2011, grant agreement no. 277984). We thank the Cell line and DNA bank of paediatric movement disorders of the Telethon Genetic Biobank Network (project no. GTB07001) and the Bank for the Diagnosis and Research of Movement Disorders (MDB) of the EuroBiobank. The financial support of Mariani Foundation of Milan is gratefully acknowledged. T.B.H. and S.H. were supported by the NBIA Disorders Association. M.A.K. is a Wellcome Trust Intermediate Clinical Fellow. H.H. and C.B. are grateful to the MRC UK (grant number G0802870) and Backman-Strauss Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
MutPred, http://mutpred.mutdb.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
PSIPRED, http://bioinf.cs.ucl.ac.uk/psipred/
References
- 1.Gregory A., Hayflick S.J. Genetics of neurodegeneration with brain iron accumulation. Curr. Neurol. Neurosci. Rep. 2011;11:254–261. doi: 10.1007/s11910-011-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou B., Westaway S.K., Levinson B., Johnson M.A., Gitschier J., Hayflick S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 3.Hörtnagel K., Prokisch H., Meitinger T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum. Mol. Genet. 2003;12:321–327. doi: 10.1093/hmg/ddg026. [DOI] [PubMed] [Google Scholar]
- 4.Morgan N.V., Westaway S.K., Morton J.E., Gregory A., Gissen P., Sonek S., Cangul H., Coryell J., Canham N., Nardocci N. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 2006;38:752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruer M.C., Paisán-Ruiz C., Boddaert N., Yoon M.Y., Hama H., Gregory A., Malandrini A., Woltjer R.L., Munnich A., Gobin S. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA) Ann. Neurol. 2010;68:611–618. doi: 10.1002/ana.22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartig M.B., Iuso A., Haack T., Kmiec T., Jurkiewicz E., Heim K., Roeber S., Tarabin V., Dusi S., Krajewska-Walasek M. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2011;89:543–550. doi: 10.1016/j.ajhg.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panteghini C., Zorzi G., Venco P., Dusi S., Reale C., Brunetti D., Chiapparini L., Zibordi F., Siegel B., Garavaglia B. C19orf12 and FA2H mutations are rare in Italian patients with neurodegeneration with brain iron accumulation. Semin. Pediatr. Neurol. 2012;19:75–81. doi: 10.1016/j.spen.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Haack T.B., Hogarth P., Kruer M.C., Gregory A., Wieland T., Schwarzmayr T., Graf E., Sanford L., Meyer E., Kara E. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012;91:1144–1149. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saitsu H., Nishimura T., Muramatsu K., Kodera H., Kumada S., Sugai K., Kasai-Yoshida E., Sawaura N., Nishida H., Hoshino A. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013;45:445–449. doi: 10.1038/ng.2562. e1. [DOI] [PubMed] [Google Scholar]
- 10.Aghajanian S., Worrall D.M. Identification and characterization of the gene encoding the human phosphopantetheine adenylyltransferase and dephospho-CoA kinase bifunctional enzyme (CoA synthase) Biochem. J. 2002;365:13–18. doi: 10.1042/BJ20020569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosveld F., Rana A., Lemstra W., Kampinga H.H., Sibon O.C. Drosophila phosphopantothenoylcysteine synthetase is required for tissue morphogenesis during oogenesis. BMC Res. Notes. 2008;1:75. doi: 10.1186/1756-0500-1-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosveld F., Rana A., van der Wouden P.E., Lemstra W., Ritsema M., Kampinga H.H., Sibon O.C. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum. Mol. Genet. 2008;17:2058–2069. doi: 10.1093/hmg/ddn105. [DOI] [PubMed] [Google Scholar]
- 13.Daugherty M., Polanuyer B., Farrell M., Scholle M., Lykidis A., de Crécy-Lagard V., Osterman A. Complete reconstitution of the human coenzyme A biosynthetic pathway via comparative genomics. J. Biol. Chem. 2002;277:21431–21439. doi: 10.1074/jbc.M201708200. [DOI] [PubMed] [Google Scholar]
- 14.Nemazanyy I., Panasyuk G., Breus O., Zhyvoloup A., Filonenko V., Gout I.T. Identification of a novel CoA synthase isoform, which is primarily expressed in the brain. Biochem. Biophys. Res. Commun. 2006;341:995–1000. doi: 10.1016/j.bbrc.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 15.Johnson M.A., Kuo Y.M., Westaway S.K., Parker S.M., Ching K.H., Gitschier J., Hayflick S.J. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann. N Y Acad. Sci. 2004;1012:282–298. doi: 10.1196/annals.1306.023. [DOI] [PubMed] [Google Scholar]
- 16.Alfonso-Pecchio A., Garcia M., Leonardi R., Jackowski S. Compartmentalization of mammalian pantothenate kinases. PLoS ONE. 2012;7:e49509. doi: 10.1371/journal.pone.0049509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhyvoloup A., Nemazanyy I., Panasyuk G., Valovka T., Fenton T., Rebholz H., Wang M.L., Foxon R., Lyzogubov V., Usenko V. Subcellular localization and regulation of coenzyme A synthase. J. Biol. Chem. 2003;278:50316–50321. doi: 10.1074/jbc.M307763200. [DOI] [PubMed] [Google Scholar]
- 18.Rhee H.W., Zou P., Udeshi N.D., Martell J.D., Mootha V.K., Carr S.A., Ting A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon-Kayser B., Scoul C., Renaudin K., Jezequel P., Bouchot O., Rigaud J., Bezieau S. Molecular cloning and characterization of FBXO47, a novel gene containing an F-box domain, located in the 17q12 band deleted in papillary renal cell carcinoma. Genes Chromosomes Cancer. 2005;43:83–94. doi: 10.1002/gcc.20170. [DOI] [PubMed] [Google Scholar]
- 20.Fernández-Vizarra E., Ferrín G., Pérez-Martos A., Fernández-Silva P., Zeviani M., Enríquez J.A. Isolation of mitochondria for biogenetical studies: An update. Mitochondrion. 2010;10:253–262. doi: 10.1016/j.mito.2009.12.148. [DOI] [PubMed] [Google Scholar]
- 21.Tiranti V., Galimberti C., Nijtmans L., Bovolenta S., Perini M.P., Zeviani M. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum. Mol. Genet. 1999;8:2533–2540. doi: 10.1093/hmg/8.13.2533. [DOI] [PubMed] [Google Scholar]
- 22.Magni G.E., Von Borstel R.C. Different rates of spontaneous mutation during mitosis and meiosis in yeast. Genetics. 1962;47:1097–1108. doi: 10.1093/genetics/47.8.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonneaud N., Ozier-Kalogeropoulos O., Li G.Y., Labouesse M., Minvielle-Sebastia L., Lacroute F. A family of low and high copy replicative, integrative and single-stranded S. cerevisiae/E. coli shuttle vectors. Yeast. 1991;7:609–615. doi: 10.1002/yea.320070609. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J., Russel D.W. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 25.Gietz R.D., Schiestl R.H. Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007;2:35–37. doi: 10.1038/nprot.2007.14. [DOI] [PubMed] [Google Scholar]
- 26.Glick B.S., Pon L.A. Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol. 1995;260:213–223. doi: 10.1016/0076-6879(95)60139-2. [DOI] [PubMed] [Google Scholar]
- 27.Walia G., Gajendar K., Surolia A. Identification of critical residues of the mycobacterial dephosphocoenzyme a kinase by site-directed mutagenesis. PLoS ONE. 2011;6:e15228. doi: 10.1371/journal.pone.0015228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walia G., Surolia A. Insights into the regulatory characteristics of the mycobacterial dephosphocoenzyme A kinase: implications for the universal CoA biosynthesis pathway. PLoS ONE. 2011;6:e21390. doi: 10.1371/journal.pone.0021390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olzhausen J., Schübbe S., Schüller H.J. Genetic analysis of coenzyme A biosynthesis in the yeast Saccharomyces cerevisiae: identification of a conditional mutation in the pantothenate kinase gene CAB1. Curr. Genet. 2009;55:163–173. doi: 10.1007/s00294-009-0234-1. [DOI] [PubMed] [Google Scholar]
- 30.Reinders J., Zahedi R.P., Pfanner N., Meisinger C., Sickmann A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 2006;5:1543–1554. doi: 10.1021/pr050477f. [DOI] [PubMed] [Google Scholar]
- 31.Leonardi R., Zhang Y.M., Rock C.O., Jackowski S. Coenzyme A: back in action. Prog. Lipid Res. 2005;44:125–153. doi: 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Hogarth P., Gregory A., Kruer M.C., Sanford L., Wagoner W., Natowicz M.R., Egel R.T., Subramony S.H., Goldman J.G., Berry-Kravis E. New NBIA subtype: genetic, clinical, pathologic, and radiographic features of MPAN. Neurology. 2013;80:268–275. doi: 10.1212/WNL.0b013e31827e07be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayflick S.J., Westaway S.K., Levinson B., Zhou B., Johnson M.A., Ching K.H., Gitschier J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N. Engl. J. Med. 2003;348:33–40. doi: 10.1056/NEJMoa020817. [DOI] [PubMed] [Google Scholar]
- 34.Kruer M.C., Boddaert N., Schneider S.A., Houlden H., Bhatia K.P., Gregory A., Anderson J.C., Rooney W.D., Hogarth P., Hayflick S.J. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am. J. Neuroradiol. 2012;33:407–414. doi: 10.3174/ajnr.A2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia M., Leonardi R., Zhang Y.M., Rehg J.E., Jackowski S. Germline deletion of pantothenate kinases 1 and 2 reveals the key roles for CoA in postnatal metabolism. PLoS ONE. 2012;7:e40871. doi: 10.1371/journal.pone.0040871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tahiliani A.G., Beinlich C.J. Pantothenic acid in health and disease. Vitam. Horm. 1991;46:165–228. doi: 10.1016/s0083-6729(08)60684-6. [DOI] [PubMed] [Google Scholar]
- 37.Smith C.M., Savage C.R., Jr. Regulation of coenzyme A biosynthesis by glucagon and glucocorticoid in adult rat liver parenchymal cells. Biochem. J. 1980;188:175–184. doi: 10.1042/bj1880175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemazanyy I., Panasyuk G., Zhyvoloup A., Panayotou G., Gout I.T., Filonenko V. Specific interaction between S6K1 and CoA synthase: a potential link between the mTOR/S6K pathway, CoA biosynthesis and energy metabolism. FEBS Lett. 2004;578:357–362. doi: 10.1016/j.febslet.2004.10.091. [DOI] [PubMed] [Google Scholar]
- 39.Rock C.O., Calder R.B., Karim M.A., Jackowski S. Pantothenate kinase regulation of the intracellular concentration of coenzyme A. J. Biol. Chem. 2000;275:1377–1383. doi: 10.1074/jbc.275.2.1377. [DOI] [PubMed] [Google Scholar]
- 40.Gregory A., Hayflick S.J. Neurodegeneration with brain iron accumulation. Folia Neuropathol. 2005;43:286–296. [PMC free article] [PubMed] [Google Scholar]
- 41.Fiermonte G., Paradies E., Todisco S., Marobbio C.M., Palmieri F. A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme A and adenosine 3′,5′-diphosphate in human mitochondria. J. Biol. Chem. 2009;284:18152–18159. doi: 10.1074/jbc.M109.014118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.