

Abstract
In this study, 1,833 systemic sclerosis (SSc) cases and 3,466 controls were genotyped with the Immunochip array. Classical alleles, amino acid residues, and SNPs across the human leukocyte antigen (HLA) region were imputed and tested. These analyses resulted in a model composed of six polymorphic amino acid positions and seven SNPs that explained the observed significant associations in the region. In addition, a replication step comprising 4,017 SSc cases and 5,935 controls was carried out for several selected non-HLA variants, reaching a total of 5,850 cases and 9,401 controls of European ancestry. Following this strategy, we identified and validated three SSc risk loci, including DNASE1L3 at 3p14, the SCHIP1-IL12A locus at 3q25, and ATG5 at 6q21, as well as a suggested association of the TREH-DDX6 locus at 11q23. The associations of several previously reported SSc risk loci were validated and further refined, and the observed peak of association in PXK was related to DNASE1L3. Our study has increased the number of known genetic associations with SSc, provided further insight into the pleiotropic effects of shared autoimmune risk factors, and highlighted the power of dense mapping for detecting previously overlooked susceptibility loci.
Introduction
Systemic sclerosis (SSc, scleroderma [MIM 181750]) is an autoimmune disease characterized by three main features: (1) fibrosis of the skin and internal organs, (2) a noninflammatory vasculopathy, and (3) autoantibody production.1 It is a multiorgan system disease with considerable phenotypic heterogeneity, resulting in a broad spectrum of disease severity. From a clinical point of view, SSc is divided into limited cutaneous systemic sclerosis (lcSSc) or diffuse cutaneous systemic sclerosis (dcSSc).2 From an immunological point of view, SSc is typically classified according to mutually exclusive and disease-specific autoantibodies (anticentromere antibodies [ACAs] and antitopoisomerase antibodies [ATAs]), which are found in approximately 50% of SSc cases.3
Several studies have established that SSc is a complex genetic disease with contributions from multiple genetic loci.4,5 The evidence supporting a genetic predisposition for the disease has revealed a major contribution from the major histocompatibility complex (MHC), as well as a number of other gene regions.4–6 In fact, genome-wide association studies (GWASs) have confirmed the MHC class II region as the most significant genetic region associated with SSc.7,8 Interestingly, a recent report restricted the MHC associations to the different autoantibody subsets.9 In addition, multiple non-MHC loci, such as IRF5 (MIM 607218), STAT4 (MIM 600558), CD247 (MIM 186780), TNIP1 (MIM 607714), IRF8 (MIM 601565), IL12RB2 (MIM 601642), CSK (MIM 124095), KIAA0319L (MIM 613535), PXK (MIM 611450), JAZF1 (MIM 606246), BLK (MIM 191305), ITGAM (MIM 120980), and TNFAIP3 (MIM 191163), have been associated with this condition at the genome-wide significance level (p < 5 × 10−8).7–13 Both GWASs and candidate-gene strategies have clearly identified SSc susceptibility factors involved in different components of the immune system (innate immune response, adaptive immune response, cytokines, cytokine receptors, etc.), as well as genes involved in pathways that might play a role in vascular damage and fibrotic processes.4–6 Despite the success of these approaches, innovative strategies are needed for dealing with the remaining unmapped SSc heritability.
Hence, the next step in the genetic dissection of complex autoimmune diseases (AIDs) is to identify the causal variants for the already established susceptibility loci, as well as to discover variants with lower penetrance with the use of larger cohorts. Furthermore, these goals need to be achieved with a cost-efficient strategy.14,15 To this end, an international group of collaborators formed the Immunochip Consortium to develop and implement the Immunochip array, a custom SNP genotyping array that provides high-density mapping of AID-associated loci for large cohorts at reduced costs. The Immunochip is based on an Illumina Infinium array platform containing 196,524 variants across 186 known autoimmunity risk loci.14 The variants included in the Immunochip encompass all variants that have been previously described for white European populations (SNPdb, 1000 Genomes Project February 2010 release, and other available sequencing projects). Also, the Immunochip design includes several rare variants considered to have significant functional effects and that might have been previously overlooked.14,15
This approach has recently shown encouraging results in different AIDs, such as celiac disease (CeD [MIM 212750]), rheumatoid arthritis (RA [MIM 180300]), autoimmune thyroid disease (ATD [MIM 275000 and 140300]), psoriasis (PS [MIM 177900]), primary biliary cirrhosis (PBC [MIM 109720]), juvenile idiopathic arthritis (JIA [MIM 604302]), primary sclerosing cholangitis (PSC [MIM 613806]), narcolepsy (MIM 161400), ankylosing spondylitis (MIM 106300), atopic dermatitis (MIM 603165), and Takayasu arteritis (MIM 207600).16–27 These studies resulted in the identification of susceptibility genes and the narrowing of the associations in previously reported risk loci. Moreover, these reports increased the number of shared genetic markers between the different disorders, further supporting the common genetic component of autoimmunity.
Taking the above into consideration, the goal of this study was to explore SSc risk loci shared with other autoimmune diseases included on the Immunochip and to fine map these areas, which comprised many, but not all, previously associated SSc loci.
Subjects and Methods
Case Definition
SSc cases were defined on the basis of the 1980 preliminary classification criteria of the American Rheumatism Association (now the American College of Rheumatology)28 or the presence of at least three out of five CREST (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia) features typical of SSc. The designation of lcSSc or dcSSc was determined according to the method of Leroy.2 The SSc-specific autoantibodies, ACA and ATA, were determined by standard means as previously described.29
Sample Population
After quality-control (QC) measures were applied, the combined data set consisted of 5,850 SSc cases and 9,401 unrelated healthy controls. Table 1 shows the sample populations by sex, origin, and SSc subtype. The discovery cohort consisted of 1,833 SSc cases and 3,466 controls of white European ancestry from the US and Spain. The validation cohort was drawn from an independent group of cases and controls of similar ancestry from North America (US and Canada), Spain, Germany, the Netherlands, Italy, Sweden, and the UK (4,017 SSc cases and 5,935 controls). The populations included in this study partially overlapped with those in previously published SSc GWASs.7,9 This study was approved by the local ethics committees of all the centers that recruited the participating individuals, and all participating individuals gave written informed consent.
Table 1.
Main Clinical Features of the Studied Cohorts
| Sample Population | Total After QC |
Status and Gender |
SSc Subgroups |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Cases | % Female Cases | Controls | % Female Controls | lcSSc | dcSSc | ACA+ | ATA+ | ||
| US discovery | 3,697 | 956 | 89% | 2,741 | 65% | 594 | 327 | 281 | 155 |
| Spain discovery | 1,602 | 877 | 89% | 725 | 66% | 533 | 247 | 390 | 192 |
| US and Canada replication | 1,845 | 927 | 86% | 918 | 53% | 584 | 339 | 279 | 163 |
| Spain replication | 1,237 | 449 | 89% | 788 | 56% | 238 | 108 | 160 | 85 |
| Germany | 1,110 | 694 | 83% | 416 | 54% | 345 | 240 | 237 | 179 |
| Netherlands | 1,470 | 385 | 71% | 1,085 | 42% | 250 | 117 | 90 | 99 |
| Italy | 1,918 | 648 | 92% | 1,270 | 81% | 436 | 156 | 301 | 213 |
| Sweden | 587 | 229 | 77% | 358 | 75% | 163 | 66 | 67 | 39 |
| UK | 1,785 | 685 | 84% | 1,100 | 87% | 491 | 191 | 253 | 118 |
| Meta-analysis | 15,251 | 5,850 | 86% | 9,401 | 65% | 3,634 | 1,791 | 2,058 | 1,243 |
Note: 62.17% of cases were lcSSc, and 30.53% were dcSSc (7.3% were not designated); 35.18% of cases were ACA+, and 58.85% were ACA− (5.97% were not designated); 21.26% of cases were ATA+, and 72.15% were ATA− (6.59% were not designated); and 36.70% of cases were ACA−ATA− (6.86% were not designated). Abbreviations are as follows: ACA+, anticentromere-antibody-positive cases; ATA+, antitopoisomerase-antibody-positive cases; dcSSc, diffuse cutaneous SSc; lcSSc, limited cutaneous SSc; and SSc, systemic sclerosis cases.
The discovery population’s power to detect an association with an odds ratio (OR) = 1.5 under an additive model at the 5 × 10−6 significance level was 100% (minor allele frequency [MAF] = 0.2), 96% (MAF = 0.1), 56% (MAF = 0.05), and 1% (MAF = 0.01). The overall meta-analysis had a statistical power of 100% (MAF = 0.05) and 18% (MAF = 0.01) with an OR = 1.5 but reached a power of 99% with an OR = 2.00 (the remaining parameters were maintained as previously mentioned). Power calculations were performed as implemented in Power Calculator for Genome-wide Analyses.30
Genotyping
The genotyping for the Immunochip custom array of the discovery cohorts was performed in accordance with Illumina protocols in two centers: the Feinstein Institute for Medical Research (Manhasset) and the Translational Research Institute at the University of Queensland Diamantina Institute (Brisbane). The US and Spanish control cohorts have already been included in previous Immunochip studies.17,26
The genotyping of the replication cohorts was performed with either (1) the TaqMan SNP genotyping technology in the Applied Biosystems 7900HT Fast Real-Time PCR System according to the manufacturer’s suggestions or (2) the Immunochip platform (the controls sets overlapped with those from previous Immunochip reports).16–20,22–24,26
Genotype Calling and QC
Genotype calling on the discovery cohort was performed with the Immunochip platform according to the manufacturer’s instructions with the use of the Illumina iScan System and the Genotyping Module (v.1.8.4) of the GenomeStudio Data Analysis software. Stringent QC parameters were applied in all data sets: (1) individuals who generated genotypes at <90% loci were not considered in the analysis, (2) markers with call rates ≤ 90% were excluded, and (3) markers with allele distributions strongly deviating from Hardy-Weinberg equilibrium (HEW) in controls (p < 1 × 10−5) were discarded. After QC measures were applied, 126,270 markers passing QC (101,692 of them showed a MAF > 0.001) were included in the analysis of the discovery cohort, resulting in a genotyping rate of 99.8%. A total of 1,886 SSc cases (895 Spanish and 981 US cases) and 4,629 unaffected controls (781 Spanish and 3,848 US individuals) passed these genotyping QCs.
Individuals in the discovery phase were also excluded on the basis of inferred ethnicity by principal-component analysis (PCA) as implemented in SNP & Variation Suite v.7 (Golden Helix). The first three principal components for each individual were plotted, and those individuals deviating from the cluster centroid were discarded for further analysis. Considering the different reported ancestries in the US control set, we included HapMap Phase 2 samples as reference populations in an initial PCA. Then, we discarded those individuals who deviated more than 6 SDs from the European ancestry cluster centroid (Figure S1, available online). In addition, individuals who deviated more than 4 SDs from the centroid of their cohort were excluded from the analysis. Moreover, to identify duplicate pairs or highly related individuals among data sets, we performed pairwise comparisons by using the genome function in PLINK31 and HapMap2 populations as references (which required Pi-HAT of ≥ 0.5), and we removed one sample from each pair. By these means, 43 SSc cases (18 Spanish individuals and 25 US individuals) and 1,163 controls (56 Spanish and 1,107 US individuals) were not included in the analyzed discovery cohort.
To calculate the genomic-inflation factor (λ) while overcoming the skewed nature of the SNP selection process in the Immunochip design, we used a set of 3,120 “null” SNPs not associated with autoimmune diseases (originated by J.C. Barrett and provided by John Bowes, personal communications).19,22,23,25,26 The SNPs that did not pass QC or showed an association p value above the 90th percentile in the SSc meta-analysis were discarded. The λ for the discovery sets was estimated at 1.07 (US) and 1.085 (Spain). It has been demonstrated that λ increases with sample size.32 Therefore, it is informative to calculate a rescaled λ for an equivalent study of 1,000 cases and 1,000 controls (λ1,000).32 In this report, λ1,000 was estimated at 1.049 (US) and 1.098 (Spain). In the case of the discovery-phase meta-analysis, λ = 1.065 and λ1,000 = 1.02 (including the associations in the 90th percentile, λ = 1.209 and λ1,000 = 1.064). All loci showing one or more independent genome-wide-significant associations in the discovery phase remained significantly associated after correction for λ. However, all the identified loci (showing either genome-wide or suggestive significance, Table S1) underwent a replication step.
Imputation
We performed SNP genotype imputation of the identified SSc susceptibility loci (5 Mb regions centered in the lead SNP were considered). We included only discovery-phase genotyped SNPs that passed QC in the imputation process. Genotypes were imputed as implemented in IMPUTE2 with the use of the 1000 Genomes Phase 3 integrated reference panel.33,34 We assessed imputed SNP quality by establishing a probability threshold for merging genotypes at 0.9. Moreover, after imputation, stringent QC was applied: (1) individuals who generated genotypes at <90% loci were not considered in the analysis, (2) markers with call rates ≤ 95% were excluded, (3) markers with allele distributions deviated from HWE in controls (p < 1 × 10−3) were discarded, and (4) variants with MAF < 0.01 were not included in the analyses. A total of 390 additional variants were imputed in ATG5 (autophagy-related 5 [MIM 604261]), 541 in the SCHIP1 (schwannomin-interacting protein 1[MIM 612008])-IL12A (interleukin 12A [MIM 161560]) locus, and 628 in DNASE1L3 (deoxyribonuclease 1-like 3 [MIM 602244]).
Imputation of the HLA Region
The 5,955 SNPs obtained in the MHC after all QCs were used for the imputation process. We used as a reference panel for the imputation (1) 2,767 European-descent individuals35 with four-digit typing of human leukocyte antigen (HLA) class I and II molecules and (2) the genotypes of more than 7,500 common SNPs and indel polymorphisms across the extended MHC (xMHC).36 The imputation process was performed with Beagle software37 according to a previously described method.38 Imputed data in the xMHC for SNPs, amino acids, or the HLA four-digit code were filtered as follows: (1) variants with a success call rate below 95%, (2) variants with a MAF below 1%, and (3) all individuals with a SNP success call rate below 95% were excluded. After these filters, a total of 7,261 SNPs remained. Also, through the imputation process, a total of 449 polymorphic amino acid positions were obtained. At last, the alleles of the class I MHC (HLA-A [MIM 142800], HLA-B [MIM 142830], and HLA-C [MIM 142840]) and class II MHC (HLA-DPA1 [MIM 142880], HLA-DPB1 [MIM 142858], HLA-DQA1 [MIM 146880], HLA-DQB1 [MIM 604305], and HLA-DRB1 [MIM 142857]) were obtained. In order to assess the accuracy of the imputation, we used partial data from previously genotyped HLA alleles from 490 individuals from the US and 466 individuals from Spain for HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, and HLA-DRB1 at four digits, which were partially included in previous reports.39,40 This resulted in an accuracy of 93.51% in the US cohort and 91.70% in the Spanish cohort in the four-digit comparison (Table S2).
Statistical Analysis
The association statistics for the discovery cohorts were calculated separately in each data set via logistic regression including sex as a covariate. Then, meta-analysis combining ORs and SEs of both cohorts was performed via the inverse-variance method under the assumption of a fixed effect as implemented in PLINK v.1.07.31 Heterogeneity across the data sets was assessed with Cochran’s Q test, and those loci showing significant heterogeneity (Q < 0.05) were not considered for the validation step.
The validation SNPs were chosen for the validation phase if they (1) had a meta-analysis p < 5 × 10−6, (2) had a nominal association in both cohorts at p < 0.05, (3) had consistent OR directions in both cohorts, (4) were not located in a gene previously identified as SSc associated, (5) and mapped outside the HLA region (considered the loci located in chr6: 20,000,000–40,000,000 bp).
The possibility of different independent signals in the same locus was evaluated with a stepwise logistic regression as described in Eyre et al.17 In brief, the most associated variant in the region was considered a covariate and the association analyses were calculated for the remaining variants. Independent signals should have met the following criteria: (1) p < 5 × 10−6, (2) no high correlation with the lead SNP (r2 < 0.6), and (3) no substantial difference between the conditioned and the unconditioned p values (p < 5 × 10−4).
The combined analysis of the discovery populations with the replication cohorts was performed via the inverse-variance method under a fixed-effects model on the basis of population-specific logistic regression results. Only variants showing a genome-wide-significant association (p < 5 × 10−8), either in the whole disease or in any of the subgroup analyses, were considered confirmed associations with SSc susceptibility.
For the analyses of the HLA region, we used logistic regression to test for allelic association between the imputed variant and disease status. Individual amino acid positions were tested with a model including all the possible amino acid residues. Statistical significance was established by comparison of the deviance model to the null model.38 For the conditional analyses, we assumed that the null model comprised the previously associated variants in a stepwise manner until no genome-wide-significant associations remained in the HLA region.
Functional Prioritization of Noncoding Variants
In addition to analyzing coding variants (functional prediction was based on PolyPhen-2),41 we propose most likely functional variants that might explain the associations on the basis of noncoding element data. We used the Regulome, Genevar, and Blood eQTL browser resources to explore the evidence of transcription factor binding sites, DNase hypersensitivity sites, and expression quantitative trait loci (eQTLs) in the publicly available ENCODE database and additional eQTL databases.42–44 Molecular graphics and analyses of DNASE1L3, HLA-DPAβ1, HLA-DQα1, and HLA-DRβ1 structure were performed with the UCSF Chimera package.45
Results
The HLA region showed the most significant associations in the discovery phase, and three already reported non-HLA SSc risk factors (STAT4, PXK, and the TNPO3 [MIM 610032]-IRF5 locus)7,13,46–54 showed genome-wide-significant associations with the disease (Table S3). No independent signals as previously defined were observed in these loci. Additionally, we calculated blocks of high linkage disequilibrium (LD) for the leading signals (defined as those SNPs showing r2 > 0.9 with the lead variant according to the 1000 Genomes Project CEU [Utah residents with ancestry from northern and western Europe from the CEPH collection] population) in these loci.34 By these means, we established an association region comprising 35 kb for the leading variant in STAT4, 126 kb in the case of the TNPO3-IRF5 locus, and 91 kb in PXK. Because of their proximity, we tested for possible dependence between the signals in PXK and DNASE1L3. Interestingly, both the genome-wide association observed in our data in PXK (rs4681851) and the reported association with SSc in PXK (rs2176082)13 were dependent on the association located in DNASE1L3 (pcond rs4681851 = 0.19, pcond rs35677470 = 3.71 × 10−8; pcond rs2176082 = 0.12, pcond rs35677470 = 6.41 × 10−11). In addition, several previously described SSc susceptibility factors showed significant associations with SSc or the different subphenotypes of the disease and were confirmed in our analysis (Table S3). In this study, we performed a comprehensive analysis of the associations in the HLA region and focused on the associations previously undescribed in non-HLA loci.
Associations between the HLA Region and Systemic Sclerosis
We also conducted the imputation of the HLA region (chr6: 28,000,000–34,000,000 bp) in our data by using a large reference panel as previously described. Resulting from the imputation, we obtained 7,261 SNPs, 449 polymorphic amino acid positions, and 298 four-digit HLA alleles for both class I and class II molecules. Compared to HLA typing, the imputation had an overall accuracy of 93.08%.
Taking into account the serologically restricted HLA association in previous reports,9 we conducted different analyses to compare total SSc, ACA+, and ATA+ between cases and controls in this locus. The overall results from the analysis specific to the HLA region can be observed in Figure 1 and Table 2. After stepwise conditional multiple logistic regression analysis in the aforementioned groups, we obtained a model composed of six polymorphic amino acid positions (two and four of which were associated with ACA and ATA, respectively, Table 2) and seven SNPs (five and two of which were associated with SSc and ACA, respectively, Table 2), which successfully conditioned all observed associations in the HLA region in either SSc or any of its serological subphenotypes (Figure 1). Consequently, the identified models conditioned the known SSc-related HLA four-digit alleles (Table S4).39
Figure 1.
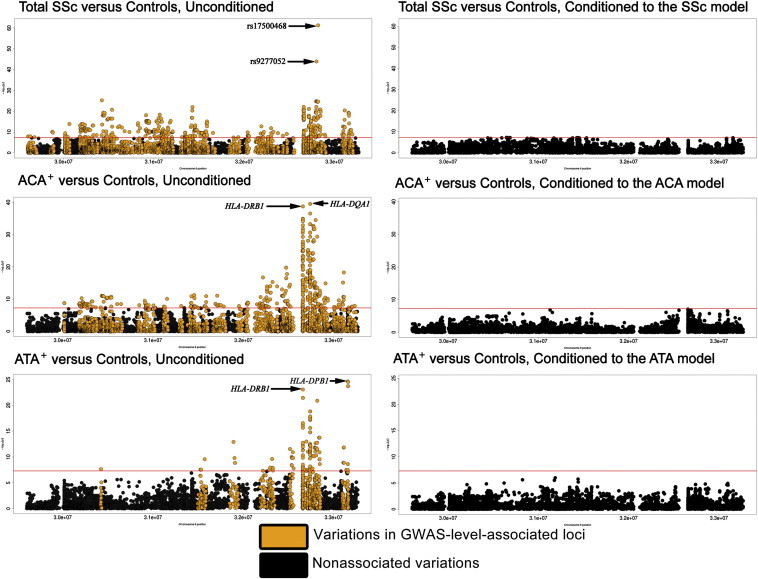
HLA Region: 28,000,000–36,000,000 bp
Manhattan plots in which each dot represents the p value of a variant (−log10(p value)) on the vertical axis and the position in chromosome 6 on the horizontal axis. The ACA model comprised HLA-DRβ1 amino acid 13, HLA-DQα1 amino acid 69, and SNPs rs12528892 and rs6933319. The ATA model comprised HLA-DRβ1 amino acids 67 and 86 and HLA-DPβ1 amino acids 76 and 96. The SSc model included the previously described ACA and ATA models and SNPs rs17500468, rs9277052, rs2442719, and rs4713605.
Table 2.
All Independent Associations Found between the HLA Region and SSc or Any of Its Subphenotypes
| SSc Subgroup | Type | Gene | Variation | AA or Nucleotide | Frequency in Cases | Frequency in Controls | p Valuea | ORb | 95% CIb |
|---|---|---|---|---|---|---|---|---|---|
| ACA+ | AA | HLA-DRB1 | AA 13c | Tyr | 0.048 | 0.139 | 1.79 × 10−39 | 0.295 | 0.22–0.38 |
| Phe | 0.232 | 0.136 | 1.817 | 1.56–2.11 | |||||
| ACA+ | AA | HLA-DQA1 | AA 69c | Leu | 0.484 | 0.581 | 4.46 × 10−23 | 0.657 | 0.58–0.75 |
| T | 0.076 | 0.020 | 3.788 | 2.85–5.02 | |||||
| ACA+ | SNP | TAP2 | rs12528892d | T | 0.064 | 0.011 | 2.74 × 10−11 | 9.137 | 6.49–12.85 |
| ACA+ | SNP | HLA-DOA | rs6933319 | T | 0.030 | 0.056 | 7.13 × 10−16 | 0.304 | 0.21–0.42 |
| SSc | SNP | TAP2 | rs17500468d | G | 0.186 | 0.082 | 5.87 × 10−62 | 2.868 | 2.52–3.25 |
| SSc | SNP | HLA-DPB1 | rs9277052d | C | 0.275 | 0.182 | 4.08 × 10−21 | 1.572 | 1.43–1.72 |
| SSc | SNP | HLA-B | rs2442719d | G | 0.491 | 0.410 | 9.55 × 10−23 | 1.512 | 1.39–1.64 |
| SSc | SNP | HLA-DPB1 | rs4713605d | A | 0.381 | 0.435 | 2.16 × 10−13 | 0.726 | 0.66–0.79 |
| SSc | SNP | HLA-DOA | rs443623 | A | 0.227 | 0.304 | 3.49 × 10−9 | 0.770 | 0.70–0.84 |
| ATA+ | AA | HLA-DRB1 | AA 67 | Leu | 0.227 | 0.388 | 3.55 × 10−22 | 0.461 | 0.38–0.55 |
| Phe | 0.311 | 0.173 | 2.158 | 1.80–2.58 | |||||
| ATA+ | AA | HLA-DPB1 | AA 96 | no Lys or Arge | 0.037 | 0.001 | 3.21 × 10−23 | 75.230 | 28.09–201.50 |
| ATA+ | AA | HLA-DRB1 | AA 86c | Val | 0.573 | 0.476 | 1.77 × 10−5 | 1.423 | 1.21–1.67 |
| ATA+ | AA | HLA-DPB1 | AA 76c | Ile | 0.074 | 0.028 | 3.05 × 10−8 | 2.577 | 1.85–3.59 |
Abbreviations are as follows: AA, amino acid; ACA+, anticentromere-antibody-positive cases; ATA+, antitopoisomerase-antibody-positive cases; CI, confidence interval; OR, odds ratio; and SSc, systemic sclerosis cases.
Logistic regression omnibus test for variations with more than two alleles.
ORs and CIs for each of the associated alleles.
Amino acid part of the binding pocket of the molecule.
Located in a putative eQTL with a p value of at least 5 × 10−8 according to Westra et al.
The absence of a lysine or an arginine in this position is the associated variant, which is very infrequent in healthy individuals of European ancestry.
Loci Revealed in the Immunochip Analyses
Eight SNPs were selected as putative SSc risk factors (Table S1). We genotyped these variants in a large multicenter replication cohort of European ancestry in order to confirm the initial associations in the discovery phase. One SNP failed to genotype in the North American replication cohort, as indicated in Table S5. As illustrated in Figure 2 and Table 3, the meta-analysis of the European and North American discovery and replication cohorts identified three non-HLA loci to be associated with overall SSc (p < 5 × 10−8, see Table S5 for results of all selected SNPs in all groups). The associated variants included a missense SNP (rs35677470) in DNASE1L3 at 3p14 (resulting in a cysteine substitution for arginine in the gene product, Figure S2), a SNP (rs77583790) in the intergenic region between SCHIP1 and IL12A at 3q25, and a SNP (rs9373839) intronic to ATG5 at 6q21. In addition, a SNP (rs7130875) in the intergenic region between TREH (trehalase [MIM 275360]) and DDX6 (DEAD-box RNA helicase 6 [MIM 600326]) at 11q23 showed suggestive association (p = 1.29 × 10−7, OR = 1.17).17
Figure 2.
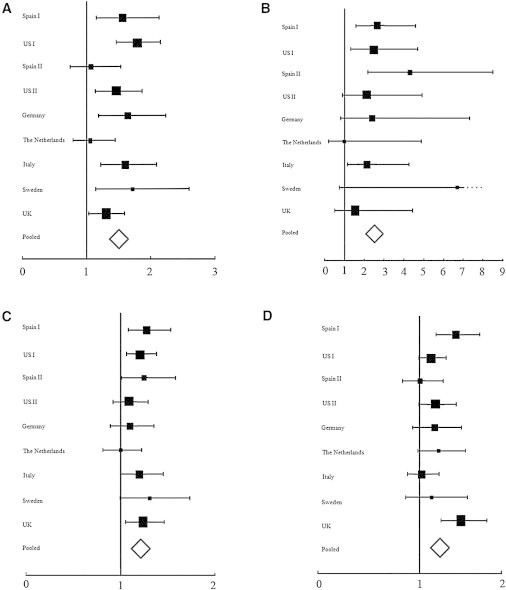
Forest Plots Showing the Population-Specific and Pooled Analyses of the Variants at Genome-wide or Suggestive Significance in the SSc versus Control Analysis
(A) rs35677470 in DNASE1L3.
(B) rs77583790 in the SCHIP1-IL12A locus.
(C) rs9373839 in ATG5.
(D) rs7130875 in the TREH-DDX6 locus.
Table 3.
Non-HLA Loci Associated with SSc and Its Subsets and Identified through Immunochip Analysis
| Locus | SNP | Chromosomal Region | Minor Allele | LD Region r2> 0.9 |
Region size |
Comments | Phenotype |
MAF |
p Value | OR |
|---|---|---|---|---|---|---|---|---|---|---|
| (kb) | Cases/Controls | |||||||||
| DNASE1L3 | rs35677470 | 3p14 | A | rs139325797, rs35677470 | 2 | missense Arg > Cys | SSc | 0.100/0.062 | 3.36 × 10−16 | 1.47 |
| lcSSc | 0.108/0.062 | 1.28 × 10−20 | 1.62 | |||||||
| ACA+ | 0.133/0.062 | 4.25 × 10−31 | 2.03 | |||||||
| SCHIP1-IL12A | rs77583790 | 3q25 | A | rs183665802, rs186702062, rs184541632, rs185514275, rs80014155, rs77583790, rs114591798, rs73154557, rs187938609, rs200875363, rs73155915, rs73155982 | 1,641 | intergenic | SSc | 0.017/0.005 | 1.22 × 10−11 | 2.57 |
| lcSSc | 0.016/0.005 | 1.53 × 10−11 | 2.81 | |||||||
| ACA+ | 0.016/0.005 | 2.40 × 10−8 | 2.76 | |||||||
| ATG5 | rs9373839 | 6q25 | G | rs34599047, rs34582442, rs1322178, rs9386514, rs62422862, rs34936565, rs9398073, rs9399978, rs9373839, rs9372120, rs2299864, rs9386516, rs62422878, rs77791277, rs3827644, rs3804329, rs34843857, rs62422881, rs9398075, rs11752888, rs10484577, rs62422886, rs3804333, rs9373842, rs9372121, rs151290510, rs9373843, rs9398078, rs66469051, rs9373846, rs7763652 | 157 | intronic | SSc | 0.241/0.185 | 3.75 × 10−8 | 1.19 |
The phenotypes with the most significant associations are shown in bold. Abbreviations are as follows: ACA+, anticentromere-antibody-positive cases; ATA+, antitopoisomerase-antibody-positive cases; dcSSc, diffuse cutaneous SSc; lcSSc, limited cutaneous SSc; LD, linkage disequilibrium; MAF, minor allele frequency; OR, odds ratio; p value, p value of the meta-analysis using the inverse variance method under a fixed effects model; and SSc, systemic sclerosis cases.
The association signals for DNASE1L3 and SCHIP1-IL12A were also significant in the lcSSc and ACA+ subgroups. Moreover, in the case of DNASE1L3, the signal was strongly related to ACA+ SSc (p = 4.25 × 10−31, OR = 2.03), and the most significant association in the SCHIP1-IL12A region was found with lcSSc (p = 1.53 × 10−11, OR = 2.81) (Table 3). Of note, there was no previously unreported genome-wide-significant association with either the dcSSc or the ATA+ subset of disease in this cohort. It is worth mentioning that the risk effects for the replicated polymorphisms were maintained in all the analyzed populations (Figure 2 and Figures S3 and S4).
In order to further dissect the associated loci, we imputed the associated regions by using the 1000 Genomes Project reference panel, as described previously. Imputation resulted in a significant increase in the number of analyzed variants, but only slight differences between the top genotyped SNPs and the top imputed SNPs were observed. Considering the dense coverage of the fine-mapped loci in the Immunochip, this low gain of information from imputation was concordant with previous Immunochip reports.22,26 As expected, the linked variants in the regions surrounding the association peaks showed concordant associations (Figure 3). However, no additional independent signals, neither SNPs nor haplotypes, showed up after conditioning on the lead variant (Figure 3).
Figure 3.
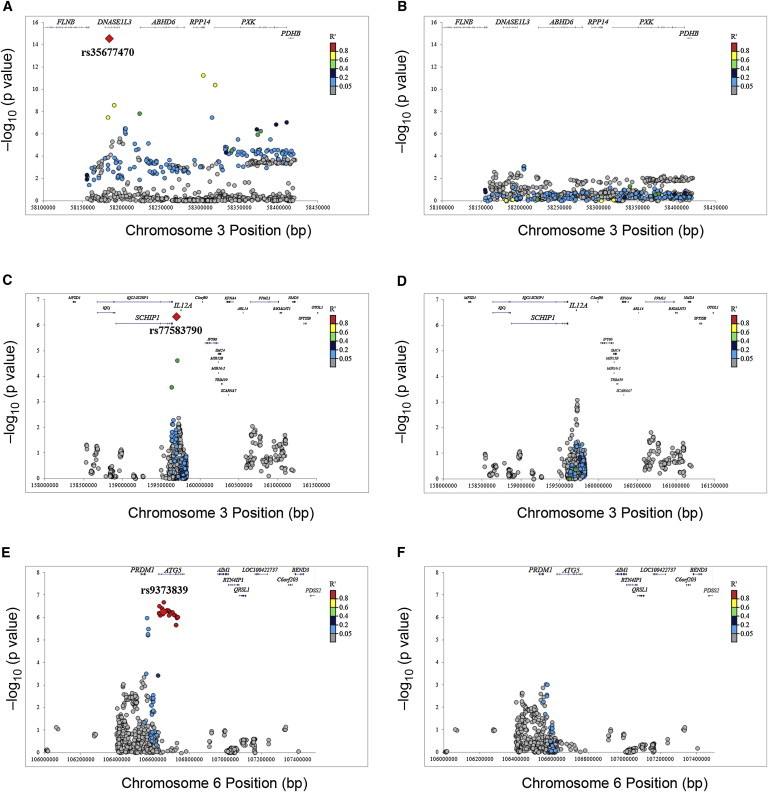
Regional Plots of the Associations Replicated at Genome-wide Significance in SSc Cases or Different Subgroups in the Overall Meta-analysis after Imputation
(A) DNASE1L3 associations in the ACA+ versus control comparison.
(B) DNASE1L3 associations in the ACA+ versus control comparison after conditioning on the lead variant (rs35677470).
(C) SCHIP1-IL12A locus associations in the lcSSc versus control comparison.
(D) SCHIP1-IL12A locus associations in the lcSSc versus control comparison after conditioning on the lead variant (rs77583790).
(E) ATG5 associations in the dcSSc versus control comparison.
(F) ATG5 associations in the dcSSc versus control comparison after conditioning on the lead variant (rs9373839).
p values correspond to the discovery phase.
In the case of DNASE1L3, the lead variant (rs35677470) encodes a probably damaging nonsynonymous change, as mentioned above. No additional variant in high LD with this SNP (r2 > 0.9) was located in a coding region or has been associated with eQTLs. Therefore, we suggest that the lead variant itself might be the most suitable causal candidate.
No variants linked with the ATG5 leading variant have protein-coding consequences or have been associated with eQTLs. Four variants located in the fifth ATG5 intron (rs62422881, rs3804333, rs1885450, and rs698029) were located in transcription factor binding sites and DNase peaks (Regulome scores = 2a–4). This region showed suggestive evidence of binding of different proteins with roles in the immune system (e.g., PRDM1, EP300, or CEBPB). In addition, rs7763652, a variant in the ATG5 promoter region, showed minimal evidence of affecting the binding of CTCF (Regulome score = 4). Further research might shed light on the functional relevance of this region.
Variants in the SCHIP1-IL12A locus showed only minimal evidence of binding. Nevertheless, this lack of functional information might be due to the low MAF.
Discussion
In the present study, we were able to further dissect the long-known association between the HLA region and SSc. We narrowed the genetic factors in this region down to a model of six amino acids and seven SNPs, all in the ACA+ and ATA+ subgroups (except for rs17500468, rs9277052, rs2442719, rs4713605, and rs443623, which presented their association in the overall disease). We further checked all associated SNPs for any functional explanation of their association. Among other methods, we checked whether any of the SNPs were in a putative eQTL according to Westra et al.44 SNPs rs17500468 and rs2442719 (associated with SSc) were in an eQTL affecting the expression of the non-HLA gene TAP2 (MIM 170261; p = 4.90 × 10−10 and 4.23 × 10−8, respectively), rs9277052 (associated with SSc) was in an eQTL affecting the expression of HLA-DPB1 (p = 2.86 × 10−69), SNP rs4713605 was in an eQTL affecting the expression of HLA-DPB1 (p = 2.26 × 10−81), and rs12528892 was also in an eQTL affecting the expression of TAP2 (p = 8.53 × 10−68). Finally, SNPs rs443623 and rs6933319 (associated with SSc and ACA production, respectively) were not predicted to be in any eQTL or a likely transcription factor binding site; the closest gene was HLA-DOA (MIM 142930), whose product forms an heterodimer with HLA-DOβ to assemble the HLA-DO molecule, found in lysosomes in B cells regulating HLA-DM-mediated peptide loading on MHC class II molecules.55 Regarding the other described genes affected by eQTL SNPs, HLA-DPB1 encodes the β chain of the HLA-DP MHC receptor complex, whereas TAP2 is involved in antigen presentation.56
Regarding the amino-acid-associated positions, we found that the associated amino acid at position 13 in HLA-DRβ1 was part of the binding pocket of this molecule with epitope,57 which was also the case with amino acid 69 in HLA-DQα1;58 both of these amino acids are associated with ACA production (Figure S5). Amino acids 86 and 76 in HLA-DRβ1 and HLA-DPβ1, respectively (both associated with ATA production), were also part of the binding pocket of their respective molecules.57,59 Conversely, amino acids 67 and 96 in HLADRβ1 and HLA-DPβ1, respectively (associated with ATA production, Table 2), were not part of the binding pocket but could affect the 3D structure of the MHC. Hence, the complexity and heterogeneity of the associations between the HLA region and SSc reflect the complex and heterogeneous nature of this disorder.
Three non-HLA variants associated with SSc susceptibility were clearly identified in this report, increasing the number of loci that have been associated with SSc and providing additional insight into SSc pathogenesis. Moreover, the nonsynonymous risk variant in DNASE1L3 showed a highly significant association with the ACA+ group considering the known non-HLA genetic associations with SSc.4–6
DNASE1L3 codes for deoxyribonuclease 1-like 3, a member of the human DNase 1 family, which is expressed primarily in the liver and spleen and secreted by macrophages.60 This protein plays a role in DNA fragmentation during apoptosis and in the generation of resected double-strand breaks in immunoglobulin-encoding genes.61–63 Moreover, it has been implicated in susceptibility to SLE and RA.17,64 The nonsynonymous rs35677470 SNP in DNASE1L3 exon 8 results in a Arg-to-Cys substitution at amino acid position 206 of the protein (p.Arg206Cys). This amino acid change results in the loss of a hydrogen bond in the protein (Figure S2), and in vitro studies have indicated that the protein constructed with this substitution lacks DNase activity; this evidence suggests a potential role in autoimmunity for this SNP.65 Moreover, the observed genome-wide associations in PXK were in modest LD with and dependent on the DNASE1L3 variant. Thus, we hypothesize that because of its functional implications, the rs35677470 polymorphism is likely to be the causal variant for the reported associations with SSc in the region. Considering that this variant is exclusive to populations with European ancestry and has a relatively low frequency and only two proxy SNPs (r2 > 0.8), it is not surprising that it remained undiscovered and that fine mapping of the region was required for identifying this highly significant association. Interestingly, vascular injury and immune deregulation, which are both SSc hallmarks, are related to cell damage and deregulated apoptosis.66 Therefore, the association between SSc (especially ACA+ SSc) and the rs35677470 loss-of-function variant in DNASE1L3 might provide a link between a defective apoptotic DNA breakdown and the production of ACA.
Utilizing the Immunochip fine-mapping platform, Eyre et al. confirmed the same SNP as the SNP most highly associated with RA in the region,17 which supports the role of this gene and this specific variant as a common autoimmunity risk factor. Direct genotyping of this polymorphism in nonfamilial SLE might shed light on the relevance of this gene beyond the currently reported association in familial SLE cases.63
As noted in the Subjects and Methods, our study reached a high statistical power to detect associations with large effects despite low frequency of the minor allele. In fact, a rare variant in the intergenic region between SCHIP1 and IL12A has been firmly associated with SSc, particularly the lcSSc subset. The same region was also identified in the Immunochip analysis in CeD.16 Trynka et al. identified three different signals (rs76830965, rs1353248, and rs2561288) in this region.16 However, these variants were not associated in our data (p > 0.05), and our lead SNP (rs77583790) was not linked to the previously mentioned polymorphisms. On the contrary, rs80014155, one of the four signals observed in PBC,20 showed r2 = 0.97 with our lead SNP. Therefore, both signals were equivalent. The location of the SSc signal in the region upstream of IL12A might point to an effect of this SNP (or a linked variant) in the regulation of IL12A expression. Moreover, different loci in the IL12 pathway have been associated with SSc (i.e., IL12RB2 and STAT4), and altered levels of IL12 have been detected in SSc-affected individuals,7,11,52,54,67 supporting a relevant role for this interleukin pathway in the disease. Nevertheless, because of the lack of functional data on this variant, its low frequency, and the high level of LD in this region, an unknown role of SCHIP1 or other surrounding loci should not be discarded.
Regarding the association located in ATG5, the ATG5-encoded protein forms a complex with ATG12 and assists in autophagosomal elongation.68 Remarkably, this signal at the genome-wide level links SSc with autophagy. Autophagy is a degradation pathway that mediates pathogen clearance and allows cells to degrade unwanted cytoplasmic material and to recycle nutrients.68 It plays a key role in both innate and adaptive immune system development and responses and has been associated with a number of AIDs.68 Along this line, several GWASs have found associations between the autophagy-related gene ATG5 and SLE susceptibility, as well as risk of childhood and adult asthma and decline in lung function.68 However, recently published fine-mapping studies in RA, CeD, PS, ATD, PBC, JIA, and narcolepsy did not find associations with this gene region.16–24 Thus, this locus appears to be restricted to some, but not all, AIDs. In any case, the intronic location and the protein binding data of the variants in the ATG5 region suggest that the functional meaning of these variants might be complex and involve distant genes (even PRDM1 [MIM 603423], which maps downstream of ATG5 and has been associated with different autoimmune diseases69–71). Moreover, there is evidence of a possible binding of CTCF in the rs7763652 region. This protein is involved in chromatin architecture and DNA-loop formation,72 which brings up the possibility of a complex DNA structure in the region. Therefore, we consider that future research will be needed for determining the functional implications of this signal.
We also identified a suggestive association in the 11q23 intergenic region between TREH and DDX6, which has been shown to be a shared susceptibility region among several AIDs. Nevertheless, the different lead SNPs in DDX6 in the Immunochip analysis of RA (rs4938573),17 CeD (rs10892258),16 and PBC (rs80065107)20 were not linked with this SSc-suggestive variant. The gene product of DDX6 (an RNA helicase that is important for efficient miRNA-induced gene silencing) has been shown to regulate vascular endothelial growth factor under hypoxic conditions, which might provide a clue to the vasculopathy and fibrosis that characterize SSc.73
The association of these loci provides genetic evidence of the possible role of defects in DNA elimination during apoptosis, introduces autophagy as a pathogenic mechanism, and reinforces the role of the IL12 pathway in the pathogenic processes that lead to SSc onset and disease progression. Therefore, we consider that the present study, together with previous knowledge about the genetic component of SSc, has contributed to the notion of this disorder as a complex condition in which several biological mechanisms, such as the innate immune response (Toll-like receptor and type I IFN pathways), the adaptive immune response (especially the IL12 pathway), tissue damage, fibrotic processes, and now DNA-clearance mechanisms during apoptosis, interact. However, neither these mechanisms nor the involved genetic loci should be considered independent compartments but rather pleiotropic players in a genetic and phenotypic continuum.
The analysis of genome-wide associations in already known loci resulted in the fine mapping of previously reported signals in STAT4 and IRF5. STAT4 is a shared susceptibility factor among RA, CeD, PBC, and JIA, but the most associated SNPs in the different conditions vary.16,17,20–22 In the case of the TNPO3-IRF5 locus, different variants have been associated with multiple autoimmune diseases and previous Immunochip analyses have reported different lead SNPs in RA and PBC.17,20,21 In our study, the leading signals in these genes were highly linked with the RA-associated variants. The dense mapping of these established SSc susceptibility factors resulted in the confirmation of previous signals and narrowed association regions in these loci (35 kb in STAT4 and 126 kb in IRF5, considering the SNPs showing r2 > 0.9 with the lead variant in the locus according to the 1000 Genomes Project CEU population).34
In the case of IL12RB2 and TNIP1, both loci showed suggestive associations but did not reach genome-wide significance. In previous reports, similar results were observed in the discovery phases in which these loci did not reach genome-wide significance and large replication cohorts were needed for confirming these associations.8,10,11 Therefore, we consider that our data reinforce the evidence of a role for these genes in SSc; however, larger cohorts should be fine mapped for narrowing down the observed signals.
As a result of design limitations, not all the known variants in CD247 and CSK were included in the Immunochip (which was planned prior to the SSc GWAS), and these regions were therefore only covered by a tag SNP strategy. Unfortunately, after QC, the tagging SNPs for the reported associations in these regions were not maintained for further analysis. Thus, the initial signals were not covered, and this study could not address the confirmation of these previous findings.
It is worth mentioning that, as intended in the Immunochip design, our data showed an expected common genetic background between SSc and other AIDs. The genetic similarities between SSc and SLE are well known,4,13 but our results reinforce the evidence of overlap with other AIDs, such as RA or PBC. The identification of common autoimmune risk factors is essential for the characterization of different pathogenic mechanisms that contribute to autoimmunity. Moreover, these common pathways might shed light on the origin of coautoimmunity and the preclinical stages of autoimmunity. In addition, relevant information can be inferred from the existence of different signals in the susceptibility regions or the lack of association between certain loci and specific diseases. Therefore, it has been suggested that platforms such as the Immunochip will help to dissect the pathogenic mechanisms underlying multiple disease states and lead to both more sensitive diagnostics and novel therapies.74
In summary, we have provided a comprehensive analysis of associations between the HLA region and SSc and its subphenotypes. Moreover, we report associations between DNASE1L3, the SCHIP1-IL12A locus, and ATG5 and SSc and a suggestive association between the TREH-DDX6 locus and SSc. The Immunochip-based interrogation of the analyzed cohorts revealed shared associations with other autoimmune diseases, which was the goal of the Immunochip Consortium, but also identified intriguing differences. Moreover, our data underline the need for direct genotyping of virtually all functional polymorphisms and rare variants in large cohorts for identifying variants that have strong effects in disease susceptibility but that might have been ignored thus far.
Consortia
The members of the Spanish Scleroderma Group are Norberto Ortego-Centeno, Raquel Ríos, José Luis Callejas, Nuria Navarrete, Rosa García Portales, María Teresa Camps, Antonio Fernández-Nebro, María F. González-Escribano, Julio Sánchez-Román, Francisco José García-Hernández, María Jesús Castillo, María Ángeles Aguirre, Inmaculada Gómez-Gracia, Benjamín Fernández-Gutiérrez, Luis Rodríguez-Rodríguez, Esther Vicente, José Luis Andreu, Mónica Fernández de Castro, Paloma García de la Peña, Francisco Javier López-Longo, Lina Martínez, Vicente Fonollosa, Gerard Espinosa, Carlos Tolosa, Anna Pros, Mónica Rodríguez Carballeira, Francisco Javier Narváez, Manel Rubio Rivas, Vera Ortiz Santamaría, Bernardino Díaz, Luis Trapiella, María del Carmen Freire, Adrián Sousa, María Victoria Egurbide, Patricia Fanlo Mateo, Luis Sáez-Comet, Federico Díaz, Vanesa Hernández, Emma Beltrán, José Andrés Román-Ivorra, Elena Grau, Juan José Alegre Sancho, Francisco J. Blanco García, Natividad Oreiro, and Luis Fernández Sueiro.
Acknowledgments
This study was supported by National Institutes of Health (NIH) National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) Centers of Research Translation (CORT) grant P50AR054144, NIH grant KL2RR024149-04, NIH NIAMS grant N01-AR02251, and NIH National Center for Research Resources grant 3UL1RR024148. This study was supported by GEN-FER from the Spanish Society of Rheumatology, SAF2009-11110 and SAF2012-34435 from the Spanish Ministry of Science, CTS-4977 from Junta de Andalucia, and RD08/0075 and RD12/0009/0004 from the Redes Temáticas de Investigación Cooperativa en Salud (Red de Investigación en Inflamación y Enfermedades Reumáticas). We are indebted to the Immunochip Consortium for provision of the control data. We are grateful to Julio Charles, Marilyn Perry, Suzanne S. Taillefer (National Study Coordinator for the Canadian Scleroderma Research Group), Sofia Vargas, Sonia García, and Gema Robledo. We also thank the Scleroderma Foundation, the Spanish Scleroderma Patient Group, the Canadian Scleroderma Patient Group, the Federation of European Scleroderma Associations, National DNA Bank Carlos III (University of Salamanca), the European League against Rheumatism Scleroderma Trials and Research group, the German Network of Systemic Sclerosis, and the affected individuals, friends, spouses, and others who generously provided the samples for these studies.
Contributor Information
Maureen D. Mayes, Email: maureen.d.mayes@uth.tmc.edu.
Lara Bossini-Castillo, Email: larabossc@gmail.com.
the Spanish Scleroderma Group:
Norberto Ortego-Centeno, Raquel Ríos, José Luis Callejas, Nuria Navarrete, Rosa García Portales, María Teresa Camps, Antonio Fernández-Nebro, María F. González-Escribano, Julio Sánchez-Román, Francisco José García-Hernández, María Jesús Castillo, María Ángeles Aguirre, Inmaculada Gómez-Gracia, Benjamín Fernández-Gutiérrez, Luis Rodríguez-Rodríguez, Esther Vicente, José Luis Andreu, Mónica Fernández de Castro, Paloma García de la Peña, Francisco Javier López-Longo, Lina Martínez, Vicente Fonollosa, Gerard Espinosa, Carlos Tolosa, Anna Pros, Mónica Rodríguez Carballeira, Francisco Javier Narváez, Manel Rubio Rivas, Vera Ortiz Santamaría, Bernardino Díaz, Luis Trapiella, María del Carmen Freire, Adrián Sousa, María Victoria Egurbide, Patricia Fanlo Mateo, Luis Sáez-Comet, Federico Díaz, Vanesa Hernández, Emma Beltrán, José Andrés Román-Ivorra, Elena Grau, Juan José Alegre Sancho, Francisco J. Blanco García, Natividad Oreiro, and Luis Fernández Sueiro
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
BEAGLE, http://faculty.washington.edu/browning/beagle/beagle.html
Blood eQTL Browser, http://genenetwork.nl/bloodeqtlbrowser/
ENCODE, http://genome.ucsc.edu/ENCODE/
Genevar (Gene Expression Variation), http://www.sanger.ac.uk/resources/software/genevar/
HLA Nomenclature, http://hla.alleles.org/
HUGO Gene Nomenclature Committee, http://www.genenames.org/
IMPUTE2, http://mathgen.stats.ox.ac.uk/impute/impute_v2.html
LocusZoom, http://csg.sph.umich.edu/locuszoom/
Online Mendelian Inheritance in Man (OMIM), http://omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
RegulomeDB, http://RegulomeDB.org/
UCSF Chimera, http://www.cgl.ucsf.edu/chimera/
Accession Numbers
The dbGaP accession number for the genotype information of the US cohort presented in this paper is phs000357.v2.p2.
References
- 1.Bolster M.B., Silver R.S. Clinical features of systemic sclerosis. In: Hochberg M.C., Silman A.J., Smolen J.S., Weinblatt M.E., Weisman M.H., editors. Fifth Edition. Mosby, Elsevier; Philadelphia: 2011. pp. 1373–1386. (Rheumatology). [Google Scholar]
- 2.LeRoy E.C., Black C., Fleischmajer R., Jablonska S., Krieg T., Medsger T.A., Jr., Rowell N., Wollheim F. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J. Rheumatol. 1988;15:202–205. [PubMed] [Google Scholar]
- 3.Steen V.D. Autoantibodies in systemic sclerosis. Semin. Arthritis Rheum. 2005;35:35–42. doi: 10.1016/j.semarthrit.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Martín J.E., Bossini-Castillo L., Martín J. Unraveling the genetic component of systemic sclerosis. Hum. Genet. 2012;131:1023–1037. doi: 10.1007/s00439-011-1137-z. [DOI] [PubMed] [Google Scholar]
- 5.Mayes M.D. The genetics of scleroderma: looking into the postgenomic era. Curr. Opin. Rheumatol. 2012;24:677–684. doi: 10.1097/BOR.0b013e328358575b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Assassi S., Radstake T.R., Mayes M.D., Martin J. Genetics of scleroderma: implications for personalized medicine? BMC Med. 2013;11:9. doi: 10.1186/1741-7015-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radstake T.R., Gorlova O., Rueda B., Martin J.E., Alizadeh B.Z., Palomino-Morales R., Coenen M.J., Vonk M.C., Voskuyl A.E., Schuerwegh A.J., Spanish Scleroderma Group Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat. Genet. 2010;42:426–429. doi: 10.1038/ng.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allanore Y., Saad M., Dieudé P., Avouac J., Distler J.H., Amouyel P., Matucci-Cerinic M., Riemekasten G., Airo P., Melchers I. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011;7:e1002091. doi: 10.1371/journal.pgen.1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorlova O., Martin J.E., Rueda B., Koeleman B.P., Ying J., Teruel M., Diaz-Gallo L.M., Broen J.C., Vonk M.C., Simeon C.P., Spanish Scleroderma Group Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genet. 2011;7:e1002178. doi: 10.1371/journal.pgen.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bossini-Castillo L., Martin J.E., Broen J., Simeon C.P., Beretta L., Gorlova O.Y., Vonk M.C., Ortego-Centeno N., Espinosa G., Carreira P., Spanish Scleroderma Group Confirmation of TNIP1 but not RHOB and PSORS1C1 as systemic sclerosis risk factors in a large independent replication study. Ann. Rheum. Dis. 2013;72:602–607. doi: 10.1136/annrheumdis-2012-201888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bossini-Castillo L., Martin J.E., Broen J., Gorlova O., Simeón C.P., Beretta L., Vonk M.C., Callejas J.L., Castellví I., Carreira P., Spanish Scleroderma Group A GWAS follow-up study reveals the association of the IL12RB2 gene with systemic sclerosis in Caucasian populations. Hum. Mol. Genet. 2012;21:926–933. doi: 10.1093/hmg/ddr522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin J.E., Broen J.C., Carmona F.D., Teruel M., Simeon C.P., Vonk M.C., van ’t Slot R., Rodriguez-Rodriguez L., Vicente E., Fonollosa V., Spanish Scleroderma Group Identification of CSK as a systemic sclerosis genetic risk factor through Genome wide Association Study follow-up. Hum. Mol. Genet. 2012;21:2825–2835. doi: 10.1093/hmg/dds099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin J.E., Assassi S., Diaz-Gallo L.M., Broen J.C., Simeon C.P., Castellvi I., Vicente-Rabaneda E., Fonollosa V., Ortego-Centeno N., González-Gay M.A., Spanish Scleroderma Group. SLEGEN consortium. U.S. Scleroderma GWAS group. BIOLUPUS A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum. Mol. Genet. 2013;22:4021–4029. doi: 10.1093/hmg/ddt248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortes A., Brown M.A. Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 2011;13:101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polychronakos C. Fine points in mapping autoimmunity. Nat. Genet. 2011;43:1173–1174. doi: 10.1038/ng.1015. [DOI] [PubMed] [Google Scholar]
- 16.Trynka G., Hunt K.A., Bockett N.A., Romanos J., Mistry V., Szperl A., Bakker S.F., Bardella M.T., Bhaw-Rosun L., Castillejo G., Spanish Consortium on the Genetics of Coeliac Disease (CEGEC) PreventCD Study Group. Wellcome Trust Case Control Consortium (WTCCC) Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011;43:1193–1201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eyre S., Bowes J., Diogo D., Lee A., Barton A., Martin P., Zhernakova A., Stahl E., Viatte S., McAllister K., Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate. Wellcome Trust Case Control Consortium High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012;44:1336–1340. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper J.D., Simmonds M.J., Walker N.M., Burren O., Brand O.J., Guo H., Wallace C., Stevens H., Coleman G., Franklyn J.A., Wellcome Trust Case Control Consortium Seven newly identified loci for autoimmune thyroid disease. Hum. Mol. Genet. 2012;21:5202–5208. doi: 10.1093/hmg/dds357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsoi L.C., Spain S.L., Knight J., Ellinghaus E., Stuart P.E., Capon F., Ding J., Li Y., Tejasvi T., Gudjonsson J.E., Collaborative Association Study of Psoriasis (CASP) Genetic Analysis of Psoriasis Consortium. Psoriasis Association Genetics Extension. Wellcome Trust Case Control Consortium 2 Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012;44:1341–1348. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J.Z., Almarri M.A., Gaffney D.J., Mells G.F., Jostins L., Cordell H.J., Ducker S.J., Day D.B., Heneghan M.A., Neuberger J.M., UK Primary Biliary Cirrhosis (PBC) Consortium. Wellcome Trust Case Control Consortium 3 Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2012;44:1137–1141. doi: 10.1038/ng.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juran B.D., Hirschfield G.M., Invernizzi P., Atkinson E.J., Li Y., Xie G., Kosoy R., Ransom M., Sun Y., Bianchi I., Italian PBC Genetics Study Group Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum. Mol. Genet. 2012;21:5209–5221. doi: 10.1093/hmg/dds359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinks A., Cobb J., Marion M.C., Prahalad S., Sudman M., Bowes J., Martin P., Comeau M.E., Sajuthi S., Andrews R., Boston Children’s JIA Registry. British Society of Paediatric and Adolescent Rheumatology (BSPAR) Study Group. Childhood Arthritis Prospective Study (CAPS) Childhood Arthritis Response to Medication Study (CHARMS) German Society for Pediatric Rheumatology (GKJR) JIA Gene Expression Study. NIAMS JIA Genetic Registry. TREAT Study. United Kingdom Juvenile Idiopathic Arthritis Genetics Consortium (UKJIAGC) Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet. 2013;45:664–669. doi: 10.1038/ng.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J.Z., Hov J.R., Folseraas T., Ellinghaus E., Rushbrook S.M., Doncheva N.T., Andreassen O.A., Weersma R.K., Weismüller T.J., Eksteen B., UK-PSCSC Consortium. International IBD Genetics Consortium. International PSC Study Group Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 2013;45:670–675. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faraco J., Lin L., Kornum B.R., Kenny E.E., Trynka G., Einen M., Rico T.J., Lichtner P., Dauvilliers Y., Arnulf I. ImmunoChip study implicates antigen presentation to T cells in narcolepsy. PLoS Genet. 2013;9:e1003270. doi: 10.1371/journal.pgen.1003270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellinghaus D., Baurecht H., Esparza-Gordillo J., Rodríguez E., Matanovic A., Marenholz I., Hübner N., Schaarschmidt H., Novak N., Michel S. High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat. Genet. 2013;45:808–812. doi: 10.1038/ng.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cortes A., Hadler J., Pointon J.P., Robinson P.C., Karaderi T., Leo P., Cremin K., Pryce K., Harris J., Lee S., International Genetics of Ankylosing Spondylitis Consortium (IGAS) Australo-Anglo-American Spondyloarthritis Consortium (TASC) Groupe Française d’Etude Génétique des Spondylarthrites (GFEGS) Nord-Trøndelag Health Study (HUNT) Spondyloarthritis Research Consortium of Canada (SPARCC) Wellcome Trust Case Control Consortium 2 (WTCCC2) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 2013;45:730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saruhan-Direskeneli G., Hughes T., Aksu K., Keser G., Coit P., Aydin S.Z., Alibaz-Oner F., Kamalı S., Inanc M., Carette S. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am. J. Hum. Genet. 2013;93:298–305. doi: 10.1016/j.ajhg.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23:581–590. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 29.Reveille J.D., Fischbach M., McNearney T., Friedman A.W., Aguilar M.B., Lisse J., Fritzler M.J., Ahn C., Arnett F.C., GENISOS Study Group Systemic sclerosis in 3 US ethnic groups: a comparison of clinical, sociodemographic, serologic, and immunogenetic determinants. Semin. Arthritis Rheum. 2001;30:332–346. doi: 10.1053/sarh.2001.20268. [DOI] [PubMed] [Google Scholar]
- 30.Skol A.D., Scott L.J., Abecasis G.R., Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 31.Purcell S.N.B., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Bakker P.I., Ferreira M.A., Jia X., Neale B.M., Raychaudhuri S., Voight B.F. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008;17(R2):R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown W.M., Pierce J., Hilner J.E., Perdue L.H., Lohman K., Li L., Venkatesh R.B., Hunt S., Mychaleckyj J.C., Deloukas P., Type 1 Diabetes Genetics Consortium Overview of the MHC fine mapping data. Diabetes Obes. Metab. 2009;11(Suppl 1):2–7. doi: 10.1111/j.1463-1326.2008.00997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Bakker P.I., McVean G., Sabeti P.C., Miretti M.M., Green T., Marchini J., Ke X., Monsuur A.J., Whittaker P., Delgado M. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia X., Han B., Onengut-Gumuscu S., Chen W.M., Concannon P.J., Rich S.S., Raychaudhuri S., de Bakker P.I. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE. 2013;8:e64683. doi: 10.1371/journal.pone.0064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnett F.C., Gourh P., Shete S., Ahn C.W., Honey R.E., Agarwal S.K., Tan F.K., McNearney T., Fischbach M., Fritzler M.J. Major histocompatibility complex (MHC) class II alleles, haplotypes and epitopes which confer susceptibility or protection in systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann. Rheum. Dis. 2010;69:822–827. doi: 10.1136/ard.2009.111906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karp D.R., Marthandan N., Marsh S.G., Ahn C., Arnett F.C., Deluca D.S., Diehl A.D., Dunivin R., Eilbeck K., Feolo M. Novel sequence feature variant type analysis of the HLA genetic association in systemic sclerosis. Hum. Mol. Genet. 2010;19:707–719. doi: 10.1093/hmg/ddp521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adzhubei I., Jordan D.M., Sunyaev S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013;Chapter 7:20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang T.P., Beazley C., Montgomery S.B., Dimas A.S., Gutierrez-Arcelus M., Stranger B.E., Deloukas P., Dermitzakis E.T. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 2010;26:2474–2476. doi: 10.1093/bioinformatics/btq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Carmona F.D., Martin J.E., Beretta L., Simeón C.P., Carreira P.E., Callejas J.L., Fernández-Castro M., Sáez-Comet L., Beltrán E., Camps M.T., Spanish Scleroderma Group The systemic lupus erythematosus IRF5 risk haplotype is associated with systemic sclerosis. PLoS ONE. 2013;8:e54419. doi: 10.1371/journal.pone.0054419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharif R., Mayes M.D., Tan F.K., Gorlova O.Y., Hummers L.K., Shah A.A., Furst D.E., Khanna D., Martin J., Bossini-Castillo L. IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann. Rheum. Dis. 2012;71:1197–1202. doi: 10.1136/annrheumdis-2011-200901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dieude P., Dawidowicz K., Guedj M., Legrain Y., Wipff J., Hachulla E., Diot E., Sibilia J., Mouthon L., Cabane J. Phenotype-haplotype correlation of IRF5 in systemic sclerosis: role of 2 haplotypes in disease severity. J. Rheumatol. 2010;37:987–992. doi: 10.3899/jrheum.091163. [DOI] [PubMed] [Google Scholar]
- 49.Ito I., Kawaguchi Y., Kawasaki A., Hasegawa M., Ohashi J., Hikami K., Kawamoto M., Fujimoto M., Takehara K., Sato S. Association of a functional polymorphism in the IRF5 region with systemic sclerosis in a Japanese population. Arthritis Rheum. 2009;60:1845–1850. doi: 10.1002/art.24600. [DOI] [PubMed] [Google Scholar]
- 50.Dieudé P., Guedj M., Wipff J., Avouac J., Fajardy I., Diot E., Granel B., Sibilia J., Cabane J., Mouthon L. Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: a new perspective for pulmonary fibrosis. Arthritis Rheum. 2009;60:225–233. doi: 10.1002/art.24183. [DOI] [PubMed] [Google Scholar]
- 51.Gourh P., Agarwal S.K., Divecha D., Assassi S., Paz G., Arora-Singh R.K., Reveille J.D., Shete S., Mayes M.D., Arnett F.C., Tan F.K. Polymorphisms in TBX21 and STAT4 increase the risk of systemic sclerosis: evidence of possible gene-gene interaction and alterations in Th1/Th2 cytokines. Arthritis Rheum. 2009;60:3794–3806. doi: 10.1002/art.24958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dieudé P., Guedj M., Wipff J., Ruiz B., Hachulla E., Diot E., Granel B., Sibilia J., Tiev K., Mouthon L. STAT4 is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum. 2009;60:2472–2479. doi: 10.1002/art.24688. [DOI] [PubMed] [Google Scholar]
- 53.Tsuchiya N., Kawasaki A., Hasegawa M., Fujimoto M., Takehara K., Kawaguchi Y., Kawamoto M., Hara M., Sato S. Association of STAT4 polymorphism with systemic sclerosis in a Japanese population. Ann. Rheum. Dis. 2009;68:1375–1376. doi: 10.1136/ard.2009.111310. [DOI] [PubMed] [Google Scholar]
- 54.Rueda B., Broen J., Simeon C., Hesselstrand R., Diaz B., Suárez H., Ortego-Centeno N., Riemekasten G., Fonollosa V., Vonk M.C. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum. Mol. Genet. 2009;18:2071–2077. doi: 10.1093/hmg/ddp119. [DOI] [PubMed] [Google Scholar]
- 55.Bellemare-Pelletier A., Tremblay J., Beaulieu S., Boulassel M.R., Routy J.P., Massie B., Lapointe R., Thibodeau J. HLA-DO transduced in human monocyte-derived dendritic cells modulates MHC class II antigen processing. J. Leukoc. Biol. 2005;78:95–105. doi: 10.1189/jlb.0105020. [DOI] [PubMed] [Google Scholar]
- 56.Bahram S., Arnold D., Bresnahan M., Strominger J.L., Spies T. Two putative subunits of a peptide pump encoded in the human major histocompatibility complex class II region. Proc. Natl. Acad. Sci. USA. 1991;88:10094–10098. doi: 10.1073/pnas.88.22.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown J.H., Jardetzky T.S., Gorga J.C., Stern L.J., Urban R.G., Strominger J.L., Wiley D.C. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 58.Tollefsen S., Hotta K., Chen X., Simonsen B., Swaminathan K., Mathews I.I., Sollid L.M., Kim C.Y. Structural and functional studies of trans-encoded HLA-DQ2.3 (DQA1∗03:01/DQB1∗02:01) protein molecule. J. Biol. Chem. 2012;287:13611–13619. doi: 10.1074/jbc.M111.320374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai S., Murphy G.A., Crawford F., Mack D.G., Falta M.T., Marrack P., Kappler J.W., Fontenot A.P. Crystal structure of HLA-DP2 and implications for chronic beryllium disease. Proc. Natl. Acad. Sci. USA. 2010;107:7425–7430. doi: 10.1073/pnas.1001772107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baron W.F., Pan C.Q., Spencer S.A., Ryan A.M., Lazarus R.A., Baker K.P. Cloning and characterization of an actin-resistant DNase I-like endonuclease secreted by macrophages. Gene. 1998;215:291–301. doi: 10.1016/s0378-1119(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 61.Shiokawa D., Tanuma S. Characterization of human DNase I family endonucleases and activation of DNase gamma during apoptosis. Biochemistry. 2001;40:143–152. doi: 10.1021/bi001041a. [DOI] [PubMed] [Google Scholar]
- 62.Errami Y., Naura A.S., Kim H., Ju J., Suzuki Y., El-Bahrawy A.H., Ghonim M.A., Hemeida R.A., Mansy M.S., Zhang J. Apoptotic DNA fragmentation may be a cooperative activity between caspase-activated deoxyribonuclease and the poly(ADP-ribose) polymerase-regulated DNAS1L3, an endoplasmic reticulum-localized endonuclease that translocates to the nucleus during apoptosis. J. Biol. Chem. 2013;288:3460–3468. doi: 10.1074/jbc.M112.423061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okamoto M., Okamoto N., Yashiro H., Shiokawa D., Sunaga S., Yoshimori A., Tanuma S., Kitamura D. Involvement of DNase gamma in the resected double-strand DNA breaks in immunoglobulin genes. Biochem. Biophys. Res. Commun. 2005;327:76–83. doi: 10.1016/j.bbrc.2004.11.142. [DOI] [PubMed] [Google Scholar]
- 64.Al-Mayouf S.M., Sunker A., Abdwani R., Abrawi S.A., Almurshedi F., Alhashmi N., Al Sonbul A., Sewairi W., Qari A., Abdallah E. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011;43:1186–1188. doi: 10.1038/ng.975. [DOI] [PubMed] [Google Scholar]
- 65.Ueki M., Takeshita H., Fujihara J., Iida R., Yuasa I., Kato H., Panduro A., Nakajima T., Kominato Y., Yasuda T. Caucasian-specific allele in non-synonymous single nucleotide polymorphisms of the gene encoding deoxyribonuclease I-like 3, potentially relevant to autoimmunity, produces an inactive enzyme. Clin. Chim. Acta. 2009;407:20–24. doi: 10.1016/j.cca.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 66.Gu Y.S., Kong J., Cheema G.S., Keen C.L., Wick G., Gershwin M.E. The immunobiology of systemic sclerosis. Semin. Arthritis Rheum. 2008;38:132–160. doi: 10.1016/j.semarthrit.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 67.Sato S., Hanakawa H., Hasegawa M., Nagaoka T., Hamaguchi Y., Nishijima C., Komatsu K., Hirata A., Takehara K. Levels of interleukin 12, a cytokine of type 1 helper T cells, are elevated in sera from patients with systemic sclerosis. J. Rheumatol. 2000;27:2838–2842. [PubMed] [Google Scholar]
- 68.Choi A.M., Ryter S.W., Levine B. Autophagy in human health and disease. N. Engl. J. Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 69.Gateva V., Sandling J.K., Hom G., Taylor K.E., Chung S.A., Sun X., Ortmann W., Kosoy R., Ferreira R.C., Nordmark G. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raychaudhuri S., Thomson B.P., Remmers E.F., Eyre S., Hinks A., Guiducci C., Catanese J.J., Xie G., Stahl E.A., Chen R., BIRAC Consortium. YEAR Consortium Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat. Genet. 2009;41:1313–1318. doi: 10.1038/ng.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ellinghaus D., Zhang H., Zeissig S., Lipinski S., Till A., Jiang T., Stade B., Bromberg Y., Ellinghaus E., Keller A. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology. 2013;145:339–347. doi: 10.1053/j.gastro.2013.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merkenschlager M., Odom D.T. CTCF and cohesin: linking gene regulatory elements with their targets. Cell. 2013;152:1285–1297. doi: 10.1016/j.cell.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 73.de Vries S., Naarmann-de Vries I.S., Urlaub H., Lue H., Bernhagen J., Ostareck D.H., Ostareck-Lederer A. Identification of DEAD-box RNA helicase 6 (DDX6) as a cellular modulator of vascular endothelial growth factor expression under hypoxia. J. Biol. Chem. 2013;288:5815–5827. doi: 10.1074/jbc.M112.420711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Voight B.F., Cotsapas C. Human genetics offers an emerging picture of common pathways and mechanisms in autoimmunity. Curr. Opin. Immunol. 2012;24:552–557. doi: 10.1016/j.coi.2012.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.