

Abstract
Inherited deafness is clinically and genetically heterogeneous. We recently mapped DFNB86, a locus associated with nonsyndromic deafness, to chromosome 16p. In this study, whole-exome sequencing was performed with genomic DNA from affected individuals from three large consanguineous families in which markers linked to DFNB86 segregate with profound deafness. Analyses of these data revealed homozygous mutation c.208G>T (p.Asp70Tyr) or c.878G>C (p.Arg293Pro) in TBC1D24 as the underlying cause of deafness in the three families. Sanger sequence analysis of TBC1D24 in an additional large family in which deafness segregates with DFNB86 identified the c.208G>T (p.Asp70Tyr) substitution. These mutations affect TBC1D24 amino acid residues that are conserved in orthologs ranging from fruit fly to human. Neither variant was observed in databases of single-nucleotide variants or in 634 chromosomes from ethnically matched control subjects. TBC1D24 in the mouse inner ear was immunolocalized predominantly to spiral ganglion neurons, indicating that DFNB86 deafness might be an auditory neuropathy spectrum disorder. Previously, six recessive mutations in TBC1D24 were reported to cause seizures (hearing loss was not reported) ranging in severity from epilepsy with otherwise normal development to epileptic encephalopathy resulting in childhood death. Two of our four families in which deafness segregates with mutant alleles of TBC1D24 were available for neurological examination. Cosegregation of epilepsy and deafness was not observed in these two families. Although the causal relationship between genotype and phenotype is not presently understood, our findings, combined with published data, indicate that recessive alleles of TBC1D24 can cause either epilepsy or nonsyndromic deafness.
Main Text
Hearing loss occurs in approximately 0.2% of newborns, and two-thirds of these cases appear to have a genetic cause.1 The prevalence of hearing loss increases with age, and there are hundreds of syndromes that include deafness as one feature of a complex phenotype (OMIM, see Web Resources). Additionally, 114 loci have been genetically mapped for nonsyndromic deafness segregating as either a dominant (DFNA) or a recessive (DFNB) trait (Hereditary Hearing Loss Homepage, see Web Resources).1,2 For approximately half of these loci, the genes with mutations that result in nonsyndromic deafness have been identified. The wild-type alleles of these genes subserve a myriad of cellular functions that span the gamut from transcription factors to extracellular matrix proteins,2,3 and one-third of them encode proteins that interact with actin and are crucial for mechanotransduction of sound in the inner ear.4,5 Despite unique anatomical structures and physiological functions described in the auditory system,6,7 there are only a few examples of genes that have a pattern of expression limited to the inner ear.8,9 In fact, many genes with mutations associated with nonsyndromic deafness do not encode inner-ear-cell-specific molecules but rather are expressed in a variety of organ systems.10–12 For example, ACTG1 (MIM 102560), encoding cytoplasmic γ-actin,13 and HGF (MIM 142409), encoding hepatocyte growth factor (HGF), are associated with nonsyndromic deafness DFNA20 (MIM 604717) and DFNB39 (MIM 608265), respectively, and show widespread expression.14 An inference from these observations is that a mutation in any gene, no matter how widely expressed in the body, could be a cause of a phenotypically restricted human disorder.15,16
Genome-wide homozygosity mapping of DFNB in a large consanguineous Pakistani family (PKDF799) identified the locus DFNB86 on chromosome 16p with a maximum LOD score of 8.5.17 In this study, three additional consanguineous pedigrees (DEM4221, DEM4587, and DEM4476) from Pakistan were ascertained (inbreeding coefficients, Table S1, available online), and their recessive deafness was linked to DFNB86 with maximum LOD scores of 6.90, 3.26, and 5.97, respectively. The overlapping region of homozygosity of these four pedigrees spans 2.05 Mb and contains 121 annotated genes (Figure 1). Here, we report that recessive mutations in TBC1D24 (TBC1 domain family, member 24 [MIM 613577]) are the cause of the nonsyndromic deafness (DFNB86) segregating in these four families.
Figure 1.
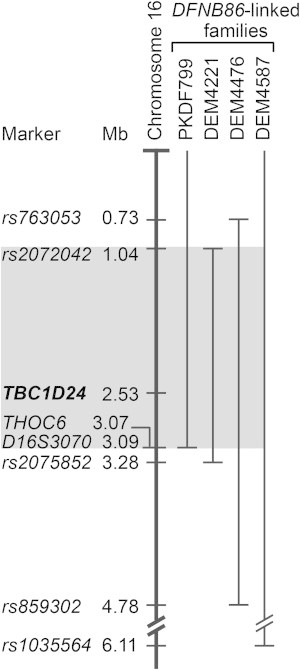
Refinement of the DFNB86 Linkage Interval
The thick vertical bar represents human chromosome 16p. The linkage interval for the deafness segregating in each of the four families is indicated by a thin vertical bar. The gray shaded region highlights the DFNB86 linkage interval shared by the four families.
Approval for this study was obtained from the institutional review boards of the Baylor College of Medicine and Affiliated Hospitals (Houston), the National Centre of Excellence in Molecular Biology (University of the Punjab, Lahore), and Quaid-I-Azam University (Islamabad) and from the Combined Neuroscience Institutional Review Board (protocol OH93-DC-0016) at the National Institutes of Health (Bethesda). Written informed consent was obtained from all family members participating in this study.
Pure-tone audiometric evaluations of 4 of the 11 affected individuals from family PKDF799 revealed profound deafness (hearing threshold ≥ 90 dB) at all test frequencies, whereas obligate carriers had normal hearing thresholds.17 Genomic DNA samples from three affected individuals (IV-23 from family PKDF799, IV-11 from family DEM4221, and IV-6 from family DEM4476) were processed for whole-exome sequencing (WES; Figure 2). For family PKDF799, a TargetSeq Exome Enrichment Kit (Applied Biosystems) was used for capturing the whole exome (45.1 Mb), which was sequenced on an Applied Biosystems SOLiD5500 platform. For families DEM4221 and DEM4476, an EZ Exome v.3.0 kit (NimbleGen) was used for capturing ∼64 Mb of protein-coding plus untranslated expressed sequences, and massively parallel sequencing was performed on an Illumina HiSeq. Sequence reads generated from these libraries were filtered for quality and were mapped to the hg19 human reference genome (UCSC Genome Browser). For family PKDF799, mapping and variant calling were performed with LifeScope (Applied Biosystems). ANNOVAR was used for variant analysis.18 For families DEM4221 and DEM4476, sequence alignment and variant calling were performed with the Burrows-Wheeler Aligner and Genome Analysis Toolkit, respectively. Depth of coverage and the number of DNA variants in the three WES data sets are summarized in Table S2. Assuming locus homogeneity, and because there is significant evidence of linkage between markers for DFNB86 and the deafness segregating in these families, we focused our evaluation of the WES data sets only on DNA variants in the smallest DFNB86 linkage interval defined by meiotic recombinations (Figure 1). Additional criteria for filtering data for identifying the pathogenic variants were homozygosity for a variant and an allele frequency < 1% in the NHLBI Exome Sequencing Project (ESP) Exome Variant Server (EVS)19 and the 1000 Genomes Project. These variants were confirmed by Sanger sequence analysis of genomic DNA from family members, and allele frequencies were obtained with the use of ethnically matched control individuals (≥148 chromosomes, Table 1).
Figure 2.
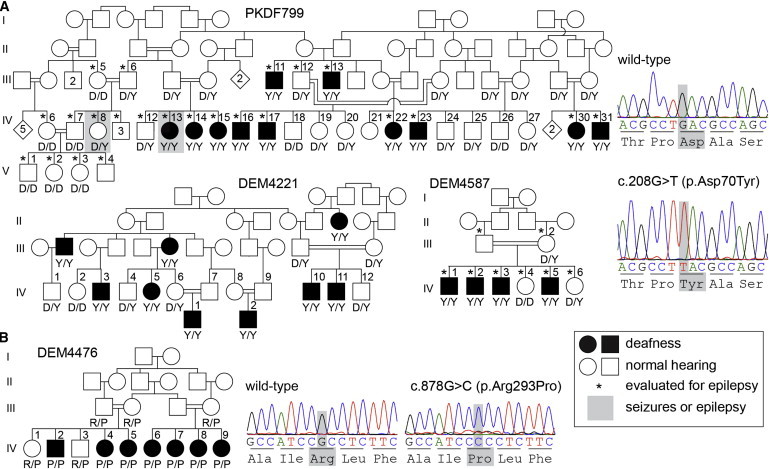
Four Families in which Deafness Cosegregates with Missense Mutations in TBC1D24
(A) Profound deafness in three families cosegregates with the c.208G>T (p.Asp70Tyr) variant in TBC1D24. Representative chromatograms from wild-type and mutant sequences are shown. Altered nucleotides and affected residues are highlighted in gray. Genotypes of the participating family members are shown below each symbol in single-letter amino acid nomenclature. Individuals diagnosed with either seizures or epilepsy are highlighted by a gray rectangle. In family PKDF799, 10 of 11 deaf individuals have no history of seizures. Deaf female IV-13 had febrile seizures at the age of 8 years. In family DEM4587, there is no history of seizures in any of the eight clinically investigated family members, indicated by asterisks. Clinical re-evaluation of families DEM4221 and DEM4476 was not possible.
(B) Deaf individuals from family DEM4476 are homozygous for the c.878G>C (p.Arg293Pro) variant in TBC1D24, whereas normal-hearing parents and siblings of affected family members are heterozygous carriers of this mutation.
Table 1.
Homozygous Variants Identified within the DFNB86 Interval
| Gene | Varianta | mRNA Changeb | Deduced Effect on Proteinb | Family | Mutation Type | Allele Frequency in EVSc | Allele Frequency in Control Chromosomes |
|---|---|---|---|---|---|---|---|
| CACNA1H | g.1257427G>A | c.3060G>A (NM_001005407.1) | p.Ala1020Ala (NP_001005407.1) | PKDF799 | synonymous | 3/12,229 | 2/174 |
| IGFALS | g.1841013C>T | c.1520G>A (NM_001146006.1) | p.Arg507His (NP_001139478.1) | DEM4476 | missense | 0/12,875 | -d |
| TBC1D24 | g.2546357G>T | c.208G>T (NM_001199107.1) | p.Asp70Tyr (NP_001186036.1) | PKDF799 | missense | 0/12,875 | 0/682 |
| TBC1D24 | g.2546357G>T | c.208G>T (NM_001199107.1) | p.Asp70Tyr (NP_001186036.1) | DEM4221 | missense | 0/12,875 | 0/682 |
| TBC1D24 | g.2547027G>C | c.878G>C (NM_001199107.1) | p.Arg293Pro (NP_001186036.1) | DEM4476 | missense | 0/12,875 | 0/634 |
| PRSS27 | g.2762774C>T | c.720G>A (NM_031948.3) | p.Ser240Ser (NP_114154.1) | PKDF799 | synonymous | 61/12,875 | 0/490 |
| PRSS27 | g.2762774C>T | c.720G>A (NM_031948.3) | p.Ser240Ser (NP_114154.1) | DEM4221 | synonymous | 61/12,875 | 0/490 |
| SRRM2 | g.2819161_2819163delTCT | c.7897_7899delTCT (NM_016333.3) | p.Ser2633del (NP_057417.3) | DEM4476 | in-frame deletione | 49/12,396 | 1/290 |
| THOC6 | g.3077605C>T | c.973C>T (NM_024339.3) | p.Arg280Trpf (NP_077315.2) | DEM4221 | missense | 0/12,875 | 0/148 |
Coordinates are based on the hg19 human reference sequence (UCSC Genome Browser).
RefSeq accession numbers are shown in parentheses.
NHLBI Exome Sequencing Project (ESP) Exome Variant Server.
Arg507 is not conserved in mouse, rat, or dog. The substitution of His for Arg507 is predicted to be neutral by MutationTaster, PolyPhen-2, SIFT, LRT, and MutationAssessor.
This deletion of three nucleotides is one of several similar indels previously reported in the EVS as common SNPs resulting in deletion or insertion of one or more serine residues in a stretch of 42 consecutive serines.
Does not cosegregate with deafness in family DEM4221.
On the basis of the criteria listed above, analyses of the WES data sets for the three families revealed a total of seven homozygous DNA variants within the refined DFNB86 interval (Table 1). Five of the seven variants were not considered to be pathogenic. A synonymous variant, c.3060G>A (p.Ala1020Ala), in CACNA1H (MIM 607904) is a polymorphism in the Pakistani population. The c.1520G>A (p.Arg507His) variant in IGFALS (MIM 601489) was not studied further because the affected amino acid is not evolutionarily conserved and the substitution is predicted to not be detrimental by five in silico programs, including MutationTaster, PolyPhen-2, and SIFT. The synonymous variant c.720G>A (p.Ser240Ser) in PRSS27 (MIM 608018) was not detected in 490 ethnically matched control chromosomes but has an allele frequency of 0.47% in the EVS (Table 1). A transition mutation, c.973C>T (p.Arg280Trp) in THOC6 (MIM 615403), identified in family DEM4221 does not cosegregate with deafness, further refining the linkage interval of this family (Figure 1).
TBC1D24 was the only gene in which homozygous pathogenic mutations were found in the exome data sets of all three families (Table 1). Deaf members of families PKDF799 and DEM4221 are homozygous for a transversion mutation, c.208G>T, predicted to cause a p.Asp70Tyr substitution. Deaf members of family DEM4476 are homozygous for c.878G>C (p.Arg293Pro) in TBC1D24. We also Sanger sequenced the coding exons of TBC1D24 in affected individuals from the fourth family, DEM4587, and found the c.208G>T (p.Asp70Tyr) mutation. The c.208G>T mutation occurs on a haplotype spanning 477 kb shared among three families (Table S3). The two missense alleles of TBC1D24 cosegregate with deafness in these families, are absent from the EVS and 1000 Genomes Project, and were not observed in at least 634 control chromosomes from ethnically matched individuals (Table 1). The substitutions p.Asp70Tyr and p.Arg293Pro affect residues conserved in orthologs of TBC1D24 from Drosophila melanogaster to Homo sapiens (Figure 3A). The p.Asp70Tyr substitution is predicted to be “damaging” to TBC1D24 function by MutationTaster, Mutation Assessor, LRT, SIFT, and PolyPhen-2. The p.Arg293Pro variant was assessed by SIFT as “tolerated” but was predicted to be “damaging” by the other four algorithms and is not reported in the EVS or 1000 Genomes Project. However, the EVS does contain two variants for the Arg293 codon: c.877C>T (p.Arg293Cys) and c.878G>A (p.Arg293His). In the ∼6,500 EVS exomes, a heterozygous p.Arg293Cys substitution was detected once among 12,838 chromosomes, whereas heterozygosity for p.Arg293His was detected seven times among 12,828 chromosomes. Both of these rare variants are predicted by in silico programs to be damaging to TBC1D24 function, suggesting a crucial role for Arg293 in the normal function of TBC1D24.
Figure 3.
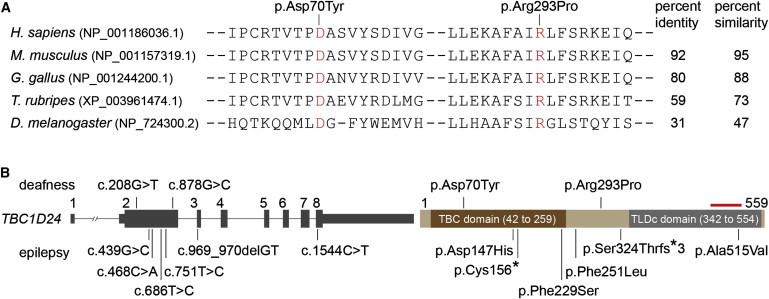
Structure of TBC1D24 and Amino Acid Conservation of the Affected TBC1D24 Residues
(A) ClustalW2 alignment of TBC1D24 orthologs shows that the two DFNB86-associated mutations affect conserved residues (red font). Amino acid residue identity and similarity percentages were calculated for full-length TBC1D24.
(B) Graphic representation of TBC1D24 structure (left) and its encoded protein (right). The red bar above the TLDc domain represents the epitope for the commercial antibody (Abcam, ab101933) that we used in Figure 4. DFNB86-associated mutations are shown above the diagram, and epilepsy-associated mutations are shown below. Tall and short gray boxes represent coding exons and UTRs of the transcribed gene, respectively. Horizontal lines joining the gray boxes denote introns. The longest isoform of TBC1D24 was used as the reference sequence for mutation nomenclature (RefSeq NM_001199107.1 and NP_001186036.1). c. 1544C>T (p.Ala515Val) is renamed here according to the longest isoform but was originally reported as c.1526C>T (p.Ala509Val) on the basis of a shorter isoform of TBC1D24 (Falace et al.24).
Human TBC1D24 has eight exons, and the longest mRNA encodes a protein of 559 amino acids (Figure 3B). In mammals, TBC1D24 is one of 42 TBC (Tre-2, Bub2, Cdc16)-domain-containing family members.20 TBC domains have approximately 200 residues that are conserved in amino acid sequence from yeast to human.21 The p.Asp70Tyr substitution is located within the TBC domain of TBC1D24 (Figure 3B). Although the crystal structure of TBC1D24 is not known, the sequence can be compared with that of other TBC domains that have X-ray crystal structures available. Comparison with the sequence of TBC1D4 suggests that the altered residue Asp70 is analogous to residue Asp950 of TBC1D4 (3QYB).22 This residue is located in a loop between two helices, termed α3 and α4, in the TBC1D4 structure. The boundaries of this loop vary significantly between different TBC domains, and high B-factors indicate a relatively large degree of flexibility or disorder in this region. Although it is possible that the loop might be the site of interaction with a different region of TBC1D24 or a binding region for another protein, no specific roles for the loop have yet been identified.
Some TBC-domain-containing proteins have been shown to function as GTPase-activating proteins (GAPs), which accelerate the intrinsic rate of GTP hydrolysis of specific Rab-GTPases.23 When the GTP of a Rab-GTPase is hydrolyzed to GDP by a GAP, the protein becomes inactive until the GDP is exchanged for GTP by another class of regulatory proteins termed guanine nucleotide exchange factors.20 Thus, GAPs are involved in the regulation of numerous membrane-trafficking and sorting processes of vesicles by modulating the activity of Rab-GTPases.20 A direct Rab-GTPase target of TBC1D24 has not been reported and might not exist. In yeast, the TBC-domain-containing protein Gyp1p (RefSeq accession number NP_014713.1) has been demonstrated to require an arginine at residue 343 for its Rab-GAP-stimulating activity.23 Clustal Omega alignment of the TBC domains of yeast Gyp1p and human TBC1D24 shows that there is a glutamine rather than a catalytic arginine at residue 100 of TBC1D24, suggesting that TBC1D24 lacks Rab-GAP GTPase-stimulating activity.20 However, ARF6 (ADP-ribosylation factor 6) was reported to be a partner of TBC1D24 and could provide GTPase activity.24
To begin to understand wild-type TBC1D24 function in normal hearing, we examined its pattern of expression, alternative transcript splicing, and immunolocalization of TBC1D24 in the mouse inner ear. Although not previously reported in the auditory system, human TBC1D24 is expressed in a variety of tissues, including the heart, liver, kidney, stomach, lungs, and brain.24 The NCBI Gene database lists eight splice variants composing two protein isoforms (a and b) of mouse Tbc1d24 (NCBI Gene ID 224617). Isoforms a and b are predicted to encode 561 amino acids (63.2 kDa) and 555 amino acids (62.6 kDa), respectively. We investigated mouse Tbc1d24 mRNA expression after preparing a cDNA library from P12 mouse inner-ear tissue. We used forward and reverse PCR primers complementary to the 5′ UTR and 3′ UTR of the full-length transcript to amplify and characterize splice isoforms of this gene (Figure S1). Agarose gel electrophoresis separated the PCR product into five bands of distinct sizes (data not shown) that were individually isolated, cloned, and Sanger sequenced. We detected nine alternatively spliced transcripts, including the previously reported isoforms a and b, of protein-coding exons of Tbc1d24 (Figure S1). Isoforms a–e include large exon 5, which encodes 57% of the full-length protein, including the translation initiation codon. However, exon 5 is not included in isoforms f–i. If translated, these shorter transcripts would utilize in-frame ATG codons that do not satisfy the consensus (−3A and +4G) for a Kozak start site,25 which is not an absolute requirement for translation initiation. If these short isoforms are also expressed in humans, they would not be affected by the two TBC1D24 missense mutations associated with DFNB86 deafness.
Immunoblot analysis of P12 mouse brain and cochlea lysates with the use of a commercially available TBC1D24 antibody (Abcam, ab101933) revealed a signal at 60 kDa, consistent with the expected size for the two previously reported TBC1D24 isoforms. There was also a signal at 20 kDa in untransfected COS-7 cells and in mouse brain and cochlear lysates. The 20 kDa signal might be the short isoform f, which includes the epitope recognized by TBC1D24 antibody ab101933 (Figure S1). To determine whether this antibody recognizes TBC1D24, we included a lysate from COS-7 cells transfected with an expression vector encoding pEGFP-TBC1D24 (full length) in the immunoblot analysis, and the expected product corresponding to EGFP-TBC1D24 (26.9 + 63.2 kDa) was detected at approximately 90 kDa (Figure S1).
To investigate TBC1D24 localization in P30 mouse inner-ear, we immunolabeled26 cryosections with the TBC1D24 antibody and counterstained them with DAPI and rhodamine phalloidin. The strong signal for TBC1D24 was observed in spiral ganglion cells, a collection of neurons critical for hearing and balance (Figure 4).
Figure 4.
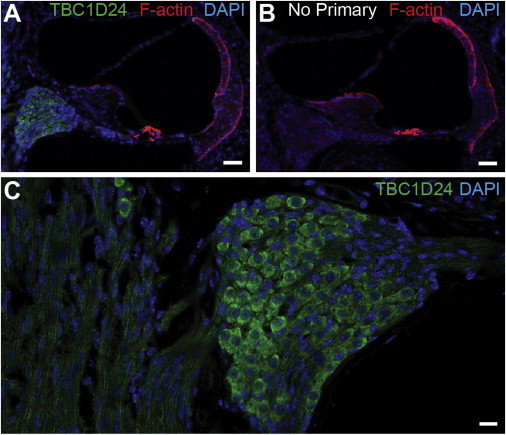
TBC1D24 Is Found in Spiral Ganglion Neurons of the Mouse Inner Ear
(A) TBC1D24 antibody binds to spiral ganglion cells in the P30 cochlea.
(B) The corresponding signal is absent in a control experiment with no added primary antibody.
(C) A higher-magnification image reveals TBC1D24 antibody binding to the cell body and axonal projections of spiral ganglion. Scale bars in (A) and (B) represent 40 μm, whereas that in (C) represents 10 μm.
In addition to a TBC domain, the only other predicted domain in TBC1D24 is a C-terminal TLDc domain (TBC, LysM, domain catalytic). Neither of the two TBC1D24 missense mutations associated with deafness alters the TLDc domain. There are four other human TLDc-domain-containing proteins, including NCOA7 and OXR1, both of which have been demonstrated to defend cells against oxidative stress.27,28 Interestingly, the mouse OXR1 TLDc domain alone is sufficient to protect granule cells of the cerebellum against oxidative stress.27 Clustal Omega alignment of the 163 residues of the TLDc domain of mouse OXR1 shows 66% similarity to the TLDc domain of human TBC1D24. Given the reported role of TLDc domains, an additional wild-type function of TBC1D24 might be to help safeguard spiral ganglion neurons against oxidative stress.
Assuming that p.Asp70Tyr and p.Arg293Pro cause deafness as a result of an altered function of TBC1D24 in the spiral ganglion neurons of the inner ear, DFNB86 deafness might be an auditory neuropathy spectrum disorder.29–31 These disorders are audiologically defined by abnormal auditory brainstem response (ABR) waveforms and interpeak intervals in combination with normal otoacoustic emissions (OAEs).32 These findings reflect an underlying lesion in the afferent auditory pathway, which includes the auditory nerve, the afferent synapse with the inner hair cell, or the inner hair cell itself. In the absence of afferent stimulation, the remaining hair cells and organ of Corti can eventually degenerate secondarily. Unfortunately, we were not able to obtain OAE and ABR data from any of our subjects. However, mouse models of DFNB86 might allow testing of the hypothesis that the primary lesion in TBC1D24 affects spiral ganglion neurons.
Four independent publications have described families in which epilepsy, with varying degrees of severity, segregates with recessive mutations in TBC1D24 (Figure 3B and Table 2). Individuals from an Italian family affected by familial infantile myoclonic epilepsy (FIME [MIM 605021]) were found to be compound heterozygous for c.439G>C (p.Asp147His) and c.1544C>T (p.Ala515Val) in TBC1D24.24 In another family (ethnicity not reported), two siblings presented with malignant migrating partial seizures of infancy (MIM 605021) as a result of compound heterozygosity for c.468C>A (p.Cys156∗) and c.686T>C (p.Phe229Ser) in TBC1D24.35 In an Arab family, four siblings with a focal epilepsy and intellectual disability syndrome (MIM 605021) were homozygous for a c.751T>C (p.Phe251Leu) allele of TBC1D24.34 Finally, in a family from Turkey, a homozygous 2-nucleotide deletion located in cassette exon 3 of TBC1D24 (Figure 3B) caused myoclonic epilepsy and severe neurodegeneration culminating in childhood death between the ages of 1.5 and 7 years.33
Table 2.
TBC1D24 Mutations and Associated Phenotypes
| Origin | Phenotype | Mutationa | Zygosity in Affected Individuals | Reference |
|---|---|---|---|---|
| Pakistan | nonsyndromic deafness | c.208G>T (p.Asp70Tyr) | homozygous | this study |
| Pakistan | nonsyndromic deafness | c.878G>C (p.Arg293Pro) | homozygous | this study |
| Turkey | myoclonic epilepsy, dystonia, developmental and neurological disability, childhood lethality | c.969_970delGT (p.Ser324Thrfs∗3) | homozygous | Guven et al.33 |
| Israel | focal epilepsy and intellectual disability | c.751T>C (p.Phe251Leu) | homozygous | Corbett et al.34 |
| Italy | myoclonic and generalized tonic-clonic seizures, photosensitivity | c.439G>C (p.Asp147His) | compound heterozygous | Falace et al.24 |
| c.1544C>T (p.Ala515Val)b | ||||
| Not reported | malignant migrating partial seizures of infancy | c.468C>A (p.Cys156∗) | compound heterozygous | Milh et al.35 |
| c.686T>C (p.Phe229Ser) |
Nomenclature in this table is based on the longest isoform (RefSeq NM_001199107.1 and NP_001186036.1) of TBC1D24.
Reported as p.Ala509Val on the basis of a shorter isoform (RefSeq NP_065756.1) of TBC1D24.
Do mutations in TBC1D24 cause either epilepsy without hearing loss or hearing loss without epilepsy? The four reports documenting seizure disorders do not mention hearing loss in epileptic individuals.24,33–35 In the four DFNB86-affected families, epilepsy might initially have gone undetected given that a family history of seizures was not an element of ascertainment. Therefore, we reassessed 15 deaf subjects in our study for a history of epilepsy. We were able to obtain clinical data for only two of the four families (DEM4587 and PKDF799, Figure 2). In family DEM4587, no history of seizures was reported for the eight individuals indicated by asterisks in Figure 2A. Brain MRI and electroencephalography (EEG) of deaf individuals IV-1 and IV-2 were normal. In family PKDF799, a history regarding seizures and epilepsy was obtained from 25 individuals, and EEG was performed for individuals III-12, IV-8, IV-12, IV-14, IV-15, and IV-17 (Figure 2A). EEG of a 16-year old individual with normal hearing (IV-12) was consistent with potential seizure activity, but he reported not ever having a seizure. Individual IV-8 (18 years old), who has normal hearing, has seizures that began when she was approximately 3 years of age and now occur two to three times a month. At the age 8 years, a third family member, an 18-year-old deaf individual (IV-13) had seizures that, according to her parents, were associated with a fever, which is common in children with hyperpyrexia. She has not had a seizure since then. The remaining 22 evaluated family members have no history of seizures. Individuals IV-8 and IV-12 are heterozygous for c.208G>T (p.Asp70Tyr) in TBC1D24, and their audiograms showed normal hearing thresholds (data not shown). For p.Asp70Tyr carrier IV-8, all eight exons of TBC1D24 were sequenced and no other variants of this gene were found. To date, only recessive mutations in TBC1D24 have been associated with epilepsy.24,33–35 Only one of two individuals with epilepsy in family PKDF799 is both deaf and homozygous for a mutation in TBC1D24. The World Health Organization reports that the world prevalence of epilepsy is 0.4%–1%. Given the high prevalence of epilepsy, especially in developing countries, we suspect that this association is a coincidence.
The localization of TBC1D24 in neurons links two distinct phenotypes, epilepsy and deafness. In the inner ear, TBC1D24 was immunolocalized predominantly in spiral ganglion neurons (Figure 4), whereas in the brain, TBC1D24 mRNA was localized by in situ hybridization to the hippocampus and cortex.24 Falace and coauthors also reported that ARF6, a member of a family with GTPase activity, is a partner of TBC1D24 and is involved in neurite outgrowth.24 ARF6 is also a component of a complex with scaffold protein GAB1 (GRB2-associated binding protein 1), GGA3 (golgi-associated, gamma adaptin ear-containing Arf-binding protein 3), and CRK (v-crk avian sarcoma virus CT10 oncogene homolog). This complex mediates the MET (receptor tyrosine kinase) internalization and recycling pathway36,37 only after MET is activated by its ligand HGF. We previously reported that non-protein-coding recessive mutant alleles of HGF cause human nonsyndromic deafness DFNB39.14 The p.Asp147His epilepsy-associated substitution, located in the TBC domain of TBC1D24, reduces its binding to ARF6.24 It remains to be determined whether ARF6 is also localized to spiral ganglion neurons and whether or not the deafness-associated substitution p.Asp70Tyr alters ARF6 regulatory activity.
Assuming that there is normal hearing in epileptic individuals with recessive mutations in TBC1D24,24,33–35 it is intriguing that different recessive alleles of TBC1D24 cause two distinct phenotypes, deafness and epilepsy. One possibility is that genetic background modifies the phenotypic outcome of TBC1D24 mutations. Alternatively, differences in mutant alleles of TBC1D24 might directly account for the different phenotypic outcomes, epilepsy and deafness. Mouse Tbc1d24 knockin alleles engineered to model the human mutations could address these questions.
Acknowledgments
We thank the subjects who participated in this study, as well as Dennis Drayna, Katie S. Kindt, and Julie Schultz for their critiques of our manuscript. We also thank Barbara Zwiesler, Elizabeth Wilson, Roger Murayi, and Hashim Raza for technical assistance. This study was supported by National Institutes of Health (NIH) National Institute on Deafness and Other Communication Disorders (NIDCD) grants R01 DC011651 and R01 DC003594 to S.M.L., the Higher Education Commission, the Ministry of Science and Technology of Pakistan (Sh.R. and W.A.), the International Center for Genetic Engineering and Biotechnology in Trieste (CRP/PAK08-01 contract 08/009 to Sh.R.), and the NIH National Human Genome Research Institute and National Heart, Lung, and Blood Institute (NHLBI) (U54 HG006493 to the University of Washington Center for Mendelian Genomics). Genotyping for S.M.L. was performed at the Center for Inherited Disease Research, which is funded by the NIH (contract number N01 HG65403 to Johns Hopkins University). N.B. was supported by NHLBI intramural funds HL004232 to James Sellers. Work at the NIH NIDCD was supported by intramural funds DC000060-12 to A.J.G. and DC000039-16 to T.B.F.
Contributor Information
Suzanne M. Leal, Email: sleal@bcm.edu.
Thomas B. Friedman, Email: friedman@nidcd.nih.gov.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
Clustal Omega, http://www.ebi.ac.uk/Tools/msa/clustalo/
Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org/
MutationAssessor, http://mutationassessor.org/
MutationTaster, http://www.mutationtaster.org/
NCBI Gene, http://www.ncbi.nlm.nih.gov/gene
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer-BLAST, http://www.ncbi.nlm.nih.gov/tools/primer-blast/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Hilgert N., Smith R.J., Van Camp G. Function and expression pattern of nonsyndromic deafness genes. Curr. Mol. Med. 2009;9:546–564. doi: 10.2174/156652409788488775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duman D., Tekin M. Autosomal recessive nonsyndromic deafness genes: a review. Front Biosci (Landmark Ed) 2012;17:2213–2236. doi: 10.2741/4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richardson G.P., de Monvel J.B., Petit C. How the genetics of deafness illuminates auditory physiology. Annu. Rev. Physiol. 2011;73:311–334. doi: 10.1146/annurev-physiol-012110-142228. [DOI] [PubMed] [Google Scholar]
- 4.Drummond M.C., Belyantseva I.A., Friderici K.H., Friedman T.B. Actin in hair cells and hearing loss. Hear. Res. 2012;288:89–99. doi: 10.1016/j.heares.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kazmierczak P., Müller U. Sensing sound: molecules that orchestrate mechanotransduction by hair cells. Trends Neurosci. 2012;35:220–229. doi: 10.1016/j.tins.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuknecht H.F. Lea & Febiger; Malvern: 1993. Pathology of the Ear, Second Edition. [Google Scholar]
- 7.Vollrath M.A., Kwan K.Y., Corey D.P. The micromachinery of mechanotransduction in hair cells. Annu. Rev. Neurosci. 2007;30:339–365. doi: 10.1146/annurev.neuro.29.051605.112917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verhoeven K., Van Laer L., Kirschhofer K., Legan P.K., Hughes D.C., Schatteman I., Verstreken M., Van Hauwe P., Coucke P., Chen A. Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat. Genet. 1998;19:60–62. doi: 10.1038/ng0598-60. [DOI] [PubMed] [Google Scholar]
- 9.Verpy E., Masmoudi S., Zwaenepoel I., Leibovici M., Hutchin T.P., Del Castillo I., Nouaille S., Blanchard S., Lainé S., Popot J.L. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat. Genet. 2001;29:345–349. doi: 10.1038/ng726. [DOI] [PubMed] [Google Scholar]
- 10.Scott H.S., Kudoh J., Wattenhofer M., Shibuya K., Berry A., Chrast R., Guipponi M., Wang J., Kawasaki K., Asakawa S. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed Z.M., Masmoudi S., Kalay E., Belyantseva I.A., Mosrati M.A., Collin R.W., Riazuddin S., Hmani-Aifa M., Venselaar H., Kawar M.N. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat. Genet. 2008;40:1335–1340. doi: 10.1038/ng.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehman A.U., Morell R.J., Belyantseva I.A., Khan S.Y., Boger E.T., Shahzad M., Ahmed Z.M., Riazuddin S., Khan S.N., Riazuddin S., Friedman T.B. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am. J. Hum. Genet. 2010;86:378–388. doi: 10.1016/j.ajhg.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu M., Yang T., Wei S., DeWan A.T., Morell R.J., Elfenbein J.L., Fisher R.A., Leal S.M., Smith R.J., Friderici K.H. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26) Am. J. Hum. Genet. 2003;73:1082–1091. doi: 10.1086/379286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schultz J.M., Khan S.N., Ahmed Z.M., Riazuddin S., Waryah A.M., Chhatre D., Starost M.F., Ploplis B., Buckley S., Velásquez D. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am. J. Hum. Genet. 2009;85:25–39. doi: 10.1016/j.ajhg.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolze A., Mahlaoui N., Byun M., Turner B., Trede N., Ellis S.R., Abhyankar A., Itan Y., Patin E., Brebner S. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340:976–978. doi: 10.1126/science.1234864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCann K.L., Baserga S.J. Genetics. Mysterious ribosomopathies. Science. 2013;341:849–850. doi: 10.1126/science.1244156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali R.A., Rehman A.U., Khan S.N., Husnain T., Riazuddin S., Friedman T.B., Ahmed Z.M., Riazuddin S. DFNB86, a novel autosomal recessive non-syndromic deafness locus on chromosome 16p13.3. Clin. Genet. 2012;81:498–500. doi: 10.1111/j.1399-0004.2011.01729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukuda M. TBC proteins: GAPs for mammalian small GTPase Rab? Biosci. Rep. 2011;31:159–168. doi: 10.1042/BSR20100112. [DOI] [PubMed] [Google Scholar]
- 21.Gao X., Jin C., Xue Y., Yao X. Computational analyses of TBC protein family in eukaryotes. Protein Pept. Lett. 2008;15:505–509. doi: 10.2174/092986608784567483. [DOI] [PubMed] [Google Scholar]
- 22.Park S.Y., Jin W., Woo J.R., Shoelson S.E. Crystal structures of human TBC1D1 and TBC1D4 (AS160) RabGTPase-activating protein (RabGAP) domains reveal critical elements for GLUT4 translocation. J. Biol. Chem. 2011;286:18130–18138. doi: 10.1074/jbc.M110.217323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan X., Eathiraj S., Munson M., Lambright D.G. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442:303–306. doi: 10.1038/nature04847. [DOI] [PubMed] [Google Scholar]
- 24.Falace A., Filipello F., La Padula V., Vanni N., Madia F., De Pietri Tonelli D., de Falco F.A., Striano P., Dagna Bricarelli F., Minetti C. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am. J. Hum. Genet. 2010;87:365–370. doi: 10.1016/j.ajhg.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kozak M. The scanning model for translation: an update. J. Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi B.Y., Kim H.M., Ito T., Lee K.Y., Li X., Monahan K., Wen Y., Wilson E., Kurima K., Saunders T.L. Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J. Clin. Invest. 2011;121:4516–4525. doi: 10.1172/JCI59353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliver P.L., Finelli M.J., Edwards B., Bitoun E., Butts D.L., Becker E.B., Cheeseman M.T., Davies B., Davies K.E. Oxr1 is essential for protection against oxidative stress-induced neurodegeneration. PLoS Genet. 2011;7:e1002338. doi: 10.1371/journal.pgen.1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durand M., Kolpak A., Farrell T., Elliott N.A., Shao W., Brown M., Volkert M.R. The OXR domain defines a conserved family of eukaryotic oxidation resistance proteins. BMC Cell Biol. 2007;8:13. doi: 10.1186/1471-2121-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moser T., Predoehl F., Starr A. Review of hair cell synapse defects in sensorineural hearing impairment. Otol. Neurotol. 2013;34:995–1004. doi: 10.1097/MAO.0b013e3182814d4a. [DOI] [PubMed] [Google Scholar]
- 30.Schoen C.J., Emery S.B., Thorne M.C., Ammana H.R., Sliwerska E., Arnett J., Hortsch M., Hannan F., Burmeister M., Lesperance M.M. Increased activity of Diaphanous homolog 3 (DIAPH3)/diaphanous causes hearing defects in humans with auditory neuropathy and in Drosophila. Proc. Natl. Acad. Sci. USA. 2010;107:13396–13401. doi: 10.1073/pnas.1003027107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Starr A., Picton T.W., Sininger Y., Hood L.J., Berlin C.I. Auditory neuropathy. Brain. 1996;119:741–753. doi: 10.1093/brain/119.3.741. [DOI] [PubMed] [Google Scholar]
- 32.Madden C., Rutter M., Hilbert L., Greinwald J.H., Jr., Choo D.I. Clinical and audiological features in auditory neuropathy. Arch. Otolaryngol. Head Neck Surg. 2002;128:1026–1030. doi: 10.1001/archotol.128.9.1026. [DOI] [PubMed] [Google Scholar]
- 33.Guven A., Tolun A. TBC1D24 truncating mutation resulting in severe neurodegeneration. J. Med. Genet. 2013;50:199–202. doi: 10.1136/jmedgenet-2012-101313. [DOI] [PubMed] [Google Scholar]
- 34.Corbett M.A., Bahlo M., Jolly L., Afawi Z., Gardner A.E., Oliver K.L., Tan S., Coffey A., Mulley J.C., Dibbens L.M. A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24. Am. J. Hum. Genet. 2010;87:371–375. doi: 10.1016/j.ajhg.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milh M., Falace A., Villeneuve N., Vanni N., Cacciagli P., Assereto S., Nabbout R., Benfenati F., Zara F., Chabrol B. Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum. Mutat. 2013;34:869–872. doi: 10.1002/humu.22318. [DOI] [PubMed] [Google Scholar]
- 36.Clague M.J. Met receptor: a moving target. Sci. Signal. 2011;4:pe40. doi: 10.1126/scisignal.2002422. [DOI] [PubMed] [Google Scholar]
- 37.Parachoniak C.A., Luo Y., Abella J.V., Keen J.H., Park M. GGA3 functions as a switch to promote Met receptor recycling, essential for sustained ERK and cell migration. Dev. Cell. 2011;20:751–763. doi: 10.1016/j.devcel.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.