

Abstract
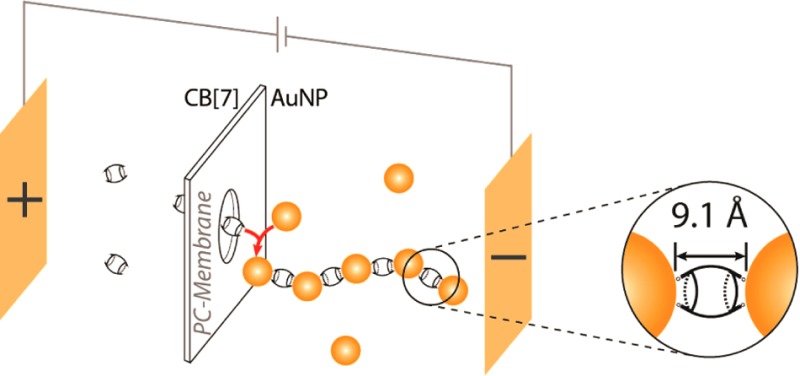
One-dimensional (1D) nanoparticle chains with defined nanojunctions are of strong interest due to their plasmonic and electronic properties. A strategy is presented for the assembly of 1D gold-nanoparticle chains with fixed and rigid cucurbit[n]uril-nanojunctions of 9 Å. The process is electrokinetically accomplished using a nanoporous polycarbonate membrane and controlled by the applied voltage, the nanoparticle/CB[n] concentration ratio, time and temperature. The spatial structure and time-resolved analysis of chain plasmonics confirm a growth mechanism at the membrane nanopores.
Keywords: Nanoparticles, plasmonics, assembly, cucurbit[n]uril, particle chains
One-dimensional (1D) chains of nanoparticles hold great promise as nanoscale connectors in miniaturized electric and optical circuits, because collective electron oscillations along the nanoparticle chains facilitate the transduction of optical and electrical signals.1−4 A precise control of the nanoparticle assembly with defined interparticle distances is essential for tuning the chain properties and minimizing irregularities, such as chain branching or variations in the nanoparticle connection.5 Nanoparticle chains have been synthesized from nanoparticles of different metals including gold,6 silver,7 and palladium,8 as well as semiconducting quantum dots.9 Addressing the general challenge to direct isotropic particles into anisotropic chain assemblies, particle alignment in electric or magnetic fields can be employed for the synthesis of chains of metallic nanoparticles.1,10 Gold nanoparticle (AuNP) chains can be assembled by exploiting the effect of dielectrophoresis (DEP),1,11 which usually leads to larger chain thicknesses, but can be optimized in order to obtain chains with a thickness of a single NP.12 Magnetic cobalt nanoparticles (CoNP) have been assembled by applying an external magnetic field, which guided the assembly of CoNP 1D chains.13 Self-assembly into nanoparticle chains can be furthermore facilitated by the premodification of nanoparticles.14−17 Kuzuya et al. self-assembled DNA-modified AuNPs into 1D chains, which were embedded within a DNA network.18 The modification of AuNPs with an organic polymer facilitated the self-assembly of AuNPs into chains with precise control over the chain thickness.5
The interparticle separation is a critical structural parameter of nanoparticle chains, as the optical coupling and energy loss during the propagation of electromagnetic waves along the chains directly depends on the width of the junctions between the nanoparticles.19 Diminishing the interparticle distance increases the coupling efficiency, enhances the trapped optical field strengths and redshifts the resonance wavelength of the electron oscillations, while also prolonging the wave propagation and reducing the energy loss.20,21 Cucurbit[n]urils (CB[n], known for n = 5, 6, 7, 8, 10) are macrocyclic molecules (Figure 1A),22−24 which efficiently bind to AuNPs via their carbonyl portals.25−27 This dynamic nanojunction generates highly rigid and fixed interparticle separations with a constant value of 9.1 Å (Figure 1B).25−27 Furthermore, CB[n]:NP nanojunctions can serve as SERS hotspots28,29 with the CB[n] being able to incorporate different guests,30,31 thus facilitating chemical or biosensing with CB[n]-bridged nanoparticles.32 AuNPs undergo a specific binding interaction with CB[n], resulting in 3D networks of alternating AuNPs and CB[n], through the monolayer of CB[n], which forms a nanojunction between two nanoparticles. Here, we report a strategy for the assembly of one-dimensional AuNP chains with CB[7] nanojunctions based on the electrokinetic control of the AuNP/CB[7] interaction at the interface of a nanoporous membrane. The chain growth mechanism is studied based on extinction spectroscopy, which allows real-time analysis of the growth mechanism based on the chain plasmonics. A careful study of the impact of different parameters like voltage, AuNP/CB[n] ratio, time and temperature onto the formed chain structures based on transmission electron microscopy (TEM and cryo-TEM) furthermore confirmed the possibility of tuning the chain length and linearity.
Figure 1.
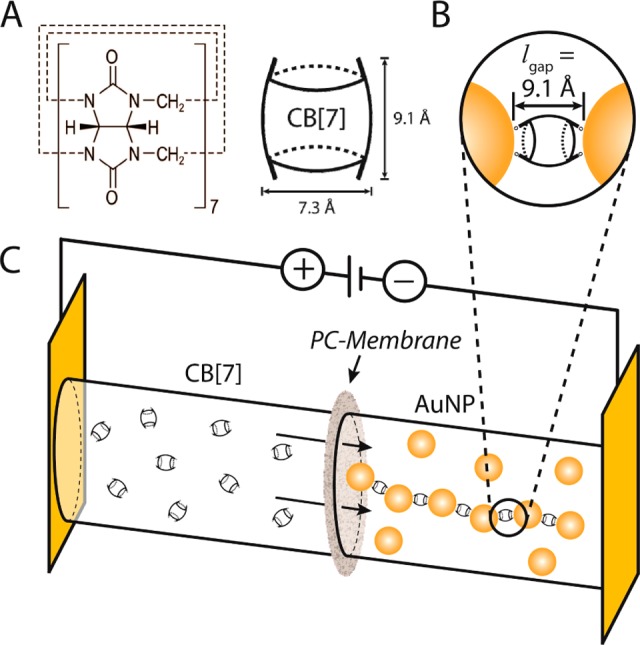
Formation of one-dimensional chains of AuNP and CB[7]. (A) Molecular Structure of cucurbit[7]uril. (B) Magnification of the CB[7]-controlled AuNP–AuNP nanojunction. (C) Electrophoresis cell, where a nanoporous PC membrane separates two compartments containing the AuNP (AuNP: d = 16.5 nm) and CB[7] building blocks, respectively.
Setup
The normally quasi-fractal three-dimensional (3D) growth mechanism of AuNP/CB[n] networks25,33 is redirected into a quasi one-dimensional growth of chains of alternating AuNP and CB[n] building blocks. This is achieved by the electrokinetic control of the AuNP/CB[n] interaction across a nanoporous polycarbonate (PC) membrane, separating two compartments containing the AuNP suspension (AuNP: diameter d = 16.5 nm) and the CB[7] solution, respectively, as shown in Figure 1C.
The application of a small potential difference between two feeder electrodes (E < 2 V), with the negative pole in the AuNP chamber, triggers the formation of chain-type objects in the AuNP compartment, as demonstrated by TEM (see Figure 2). Within the membrane, as well as in the CB[7] chamber, only very few individual AuNP or small AuNP aggregates (dimers/trimers) were observed by TEM. This indicates that only the CB[7] molecules are able to pass through the pores of the membrane, whereas the AuNPs remain in their compartment.
Figure 2.
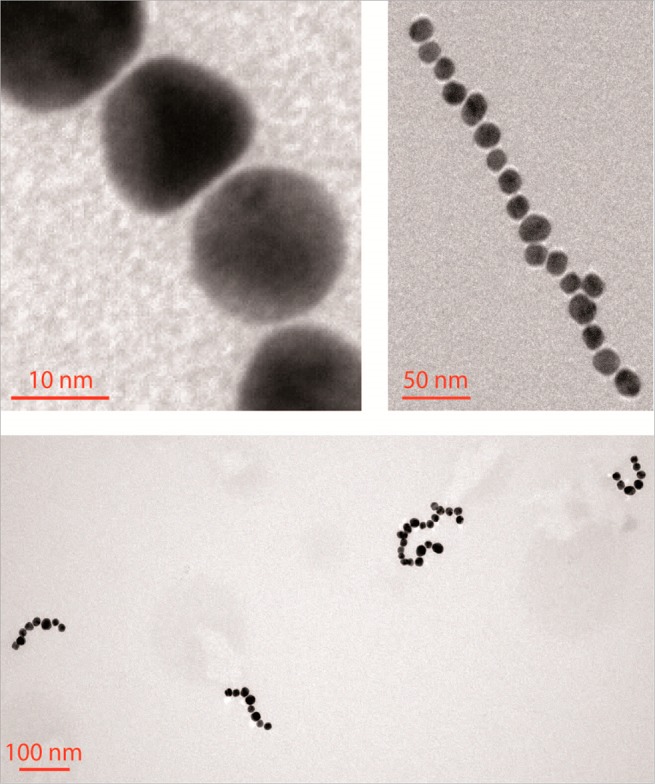
TEM images of AuNP/CB[7] chains at different magnifications.
After transfer of the objects to a TEM grid, the interparticle distances (lgap) were analyzed from TEM images. Figure 2 reveals that the chains exhibit a fixed interparticle distance, which was evaluated as being lgap = 9.13 (±0.41) Å, resulting from an analysis of 100 interparticle distances. This value is in good agreement with the value of 9.1 Å (see Figure 1B), which is theoretically expected for CB[7]-bridging and seen in previous optical experiments.25 This strongly suggests, together with the SERS signal generated by the CBs present in the gap,25 that the generated AuNP chains actually contain CB[7] nanojunctions and consist of alternating AuNP and CB[7] building blocks.
The chain formation is believed to occur at the membrane/solution interface in the AuNP compartment, according to the mechanism proposed in Figure 3, where the movements of AuNP and CB[n] in opposite directions favor the interaction of these two chain building blocks. The application of an electric field induces an electroosmotic flow (EOF) of positive charge carriers present along the negatively charged surface of the nanopores34 (membrane thickness/depth of nanopores = 6 μm) toward the negative electrode,35,36 which transports the neutral CB[7] molecules along the flow direction through the membrane into the AuNP compartment. In contrast, an oppositely directed electrophoretic motion of AuNP, which are stabilized by a negatively charged citrate layer, is induced in direction of the positive electrode. This directs the AuNP toward the membrane, however the opposed electroosmotic flow does not allow AuNP transfer through the nanopores of the membrane. These two electrokinetic phenomena are proportional to the applied electric field and lead to an encounter of the two chain building blocks at the membrane interface in the AuNP compartment, resulting in the formation of anisotropic AuNP/CB chains. The electrophoresis of AuNP toward the membrane interface allows a continuous particle supply for chain prolongation.
Figure 3.
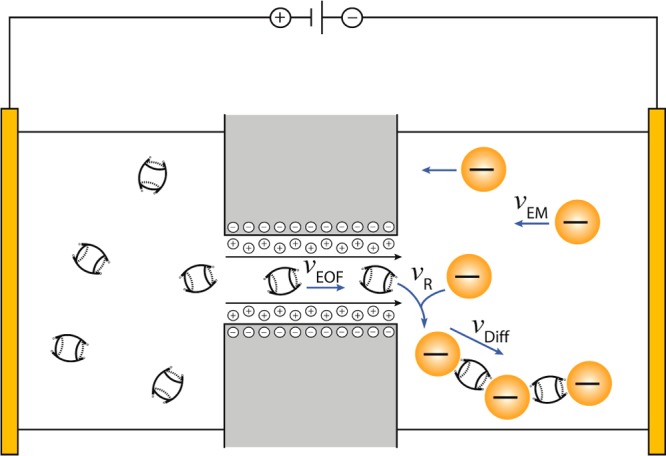
Schematic of one nanopore of the PC membrane to illustrate the synergy between electroosmotic and electrophoretic motion leading to chain formation (vEM, supply rate of the AuNPs by electromigration; vEOF, supply rate of CB[7] by electroosmotic flow; vR, reaction rate of the CB[7]/AuNP assembly; vDiff, evacuation rate of the formed chains).
Tuning of the Chain Assembly
The dependence of the AuNP/CB[7] chain assembly on the applied voltage, the reaction time t, the CB[7]/AuNP ratio (R), as well as the temperature is studied by TEM in order to investigate the possibility of tuning the chain geometry. Figure 4A shows that an increase in the applied DC voltage (at constant t = 30 min, R = 4500:1, and T = 20 °C) first causes an increase in the chain length l with a maximum chain length (l ∼ 48 NP) around 1 V, followed by a significant decrease in chain length. Moreover, the increase in chain length is accompanied by a decrease in chain linearity, until the formation of complex 2D networks with interconnected AuNP chains, as shown in the TEM image in Figure 5C and statistically analyzed in Figure S5 in the Supporting Information. This continuous transition between ideal nonbranched AuNP chains, branched chains, and complex two-dimensional (2D) networks are categorized in Figure 5 into five structural regimes. Ideal nonbranched 1NP chains were observed at small (regime B, 0.5 V < E < 0.75 V) as well as higher voltages (regime D, 1.25 V < E < 2.0 V), whereas a voltage in an intermediate regime C (0.75 V < E < 1.25 V) favors branching with the extreme case of 2D networks of interconnected chains (Figure 5, regime C).
Figure 4.

Statistical analysis of the length of AuNP/CB[7] aggregate chains (based on an analysis of three TEM grids for each set of parameters and two independent sets of experiments), prepared with the setup shown in Figure 1C for (A) different applied DC potentials between 0.25 and 2 V at constant t = 30 min, R = 4500:1 and T = 20 °C; (B) different reaction times t < 75 min at constant E = 0.85 V, R = 4500:1 and T = 20 °C.
Figure 5.
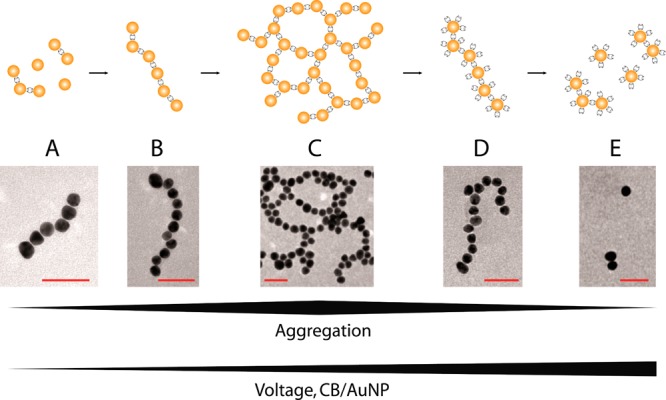
Aggregation regimes during chain formation. The growth regime can be controlled by both the applied voltage and the ratio R of CB/AuNP (TEM images: t = 30 min and R = 4500, varying voltages; the scale bar denotes 50 nm).
An increase in reaction time (at constant E = 0.85 V, R = 4500:1, T = 20 °C) in contrast provokes a clear increase in the chain length (Figure 4B), whereas the chain linearity remains rather constant. An increase in temperature to ∼50 °C under conditions which lead to the branched networks of regime C of Figure 5 (E = 1 V, R = 4500, t = 30 min) increases the chain linearity, whereas a decrease to ∼4 °C increases aggregation. A decrease of R from 4500:1 to 1:1 under regime C conditions (at constant E = 1 V, t = 30 min, T = 20 °C) results in a suppression of the branching and the predominance of 1D chains, corresponding to a shift of the structure spectrum shown in Figure 5 toward the left. A further decrease of R to 0.1:1 leads to shorter chains resembling those of regime A.
The degree of branching of the formed structures between the regimes A–E shown in Figure 5 can be tuned by the applied DC potential, the CB[7]/AuNP ratio R as well as the temperature, whereas an increase in reaction time only provokes an increase in chain length.
The influence of the applied voltage or of R on the formation of the different structural regimes shown in Figure 5 can be explained based on the mechanism depicted in Figure 3. AuNP and CB[7] building blocks reach the membrane interface in the AuNP compartment with an electrophoretic mobility vEM for AuNPs and an electroosmotic flow rate vEOF for CB[7]. The affinity of CBs for gold surfaces suggests that the reaction rate vR should not be a determining kinetic step. However the evacuation rate of the formed chains vDiff, which is composed of an electrophoretic and a diffusional component, has to be considered. This diffusional rate will become smaller with growing chain length.
For successful chain formation in the ideal regime B of Figure 5, the CB[7] supply rate at the nanopore needs to be faster than the rate of AuNP supply due to the need of several CB[7] molecules to be adsorbed for the formation of a successful junction between two gold particles. Furthermore, the supply of AuNPs toward the nanopore should be as fast as the movement of the formed chains away from the nanopore. An excess supply of CB[7] molecules leads on average to more than one junction per AuNP and thus to chain branching, as seen in regime C. A further increase of the local CB[7] concentration can lead to the saturation regimes D and E, where with an increasing CB[7] supply the branched structures of regime C are first cut into individual chains (regime D) in analogy to the dynamics known for 3D AuNP/CB[5] aggregation under an excess of CB[5],26 which then decrease in length with increasing CB[7] concentration (regime E). A deficiency of CB[7] at the nanopore leads to chain termination and thus shortening of the chain lengths according to regime A.
These basic considerations can furthermore explain the impact of the duration of the experiment, as well as the applied temperature on chain formation. Since time does not influence the kinetics of chain assembly, an increase in chain length and no change in the aggregation state is observed when prolonging the experiment. The observed increase in chain linearity at elevated temperatures might be because under these conditions initial alignment errors can be corrected. Individual particles can overcome more easily the activation barrier for disconnection from the aggregate and can then easily rebind at a different position, still under the influence of the electric field. Furthermore the viscosity of the medium is decreasing, allowing faster diffusion and position correction.
Optics
Optical excitation of the chain plasmons facilitates an analysis of the chain structure via extinction spectroscopy. The evolution of a longitudinal surface plasmon resonance (LSPR) mode at longer wavelengths than the plasmon resonance of individual nanoparticles indicates the formation of nanoparticle chains, as it results from the excitation of electron oscillations parallel to the long axis of a nanoparticle chain. Thus, the chain growth can be observed in real time by extinction spectroscopy (λ = 400–800 nm) based on this chain plasmon mode. The extinction measurements were performed in a cell comparable to the one used for the synthesis of the chains for the TEM measurements but in addition a collimated light beam is focused close to the membrane in the AuNP-containing compartment. Figure 6 shows the extinction spectra detected over a time of 2 h when applying parameters optimized to yield regime B-type linear chains.
Figure 6.
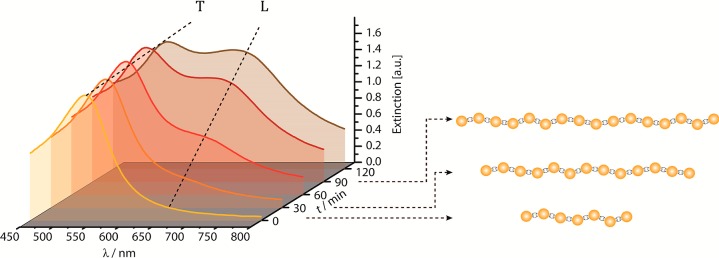
Real-time extinction spectroscopy during the chain growth process (R = 1800, E = 1.5 V).
Figure 6 reveals that over a reaction time of 2 h the LSPR mode grows significantly, as seen by the emergence of the mode at wavelengths λ > 600 nm, next to the transverse surface plasmon resonance (TSPR) mode of the gold nanoparticles at λ = 530 nm. This correlates very well with the observation of an expanding front of gray/blue color in the red AuNP suspension at the membrane interface (see Figure 7).
Figure 7.
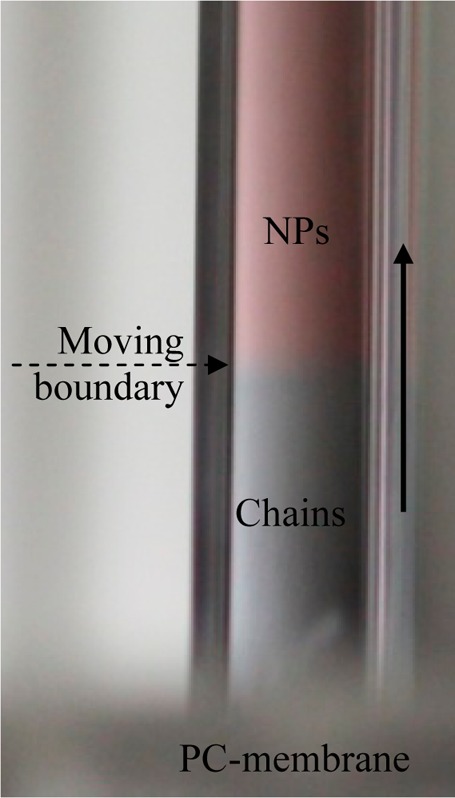
Photo of the nanoparticle compartment at the membrane/solution interface, revealing the development of the AuNP/CB[7] chains as a growing blue front (R = 18720, t = 60 min, E = 1.5 V, d = 16.5 nm).
Figure 8A presents the corresponding analysis of the wavelength of the extinction maximum λagg of the LSPR mode and its peak intensity Amagg after subtraction of the extinction spectrum of individual AuNPs. It shows that Amagg increases over 2 h in a nucleation–growth process that follows a sigmoidal profile (sigmoidal fit: R2 = 0.9978) and λagg increases to values of up to 650 nm during the first 60 min. The increase in Amagg relates to the chain formation, which occurs first with an increasing local chain concentration and stagnates at t > 120 min, when the local chain concentration remains constant as soon as the formation of new chains and the chain transport along the capillary proceed with the same speed. The redshift of λagg can be related to the increase in chain length during the growth process.35
Figure 8.

Analysis of the extinction spectra showing peak intensity Amagg and peak wavelength λagg of the LSPR mode in the extinction spectra. (A) Peak wavelength and amplitude recorded during chain growth over t = 2 h (regime B: R = 1800, E = 1.5 V, d(AuNP) = 16.5 nm). (B) Peak wavelength and amplitude recorded after t = 2 h at increasing distances dm from the membrane with dm = 0.3 mm being the beam position during the experiment in panel A. (C) Extinction difference spectra of regime B-type chains (black line; R = 1800, t = 30 min, E = 1.5 V, d = 16.5 nm) and regime D-type chains (red line; R = 18720, t = 30 min, E = 1.5 V, d = 16.5 nm). (D) Extinction difference spectra of 1D chains (red line) and 3D aggregates (black line; d = 19.5 nm).
Full electromagnetic simulations by Aizpurua et al. for linear chains of alternating AuNP and CB[7] building blocks facilitate the assignment to the extinction spectra of an effective chain length from the peak of the chain plasmon wavelength λagg.37 Thus, the spectra in Figure 6 can be assigned to a chain length l = 7 NPs at t = 15 min, which increases to a length of 16 NPs for t > 60 min. The chain plasmon mode supports nanoparticle coupling over AuNP chain-lengths less than 16 NP, so that no further wavelength shifts are detected even though longer chains can form.38,39 The small concentration of chains in the beginning of the chain growth (t < 15 min) also shows that only chains shorter than 7 NPs are seen during the initial phase of chain growth.
The spatial distribution of the chain concentration profile in the AuNP chamber after 2 h of chain growth was analyzed by extinction spectroscopy in order to gain further insight into the chain growth mechanism and the subsequent chain diffusion. To accomplish this, the cell is scanned by changing the position of the light beam along the long axis of the cell by a distance dm from the membrane. Figure 8B shows the analysis of Amagg and λagg of the chain mode recorded at increasing dm at intervals of 0.5 mm. It reveals that both Amagg, as well as λagg, decrease with increasing dm, indicating that with increasing distance from the membrane the chain concentration decreases and furthermore the predominating chain length gets smaller. According to the theoretical calculations, the longest chains with a length of ∼16 NPs can be found at the membrane interface, while shorter chains with a length of <10 NPs are dominant when moving away from the membrane, on account of their faster diffusion. This profile underscores the proposed mechanism of chain formation at the membrane interface in the AuNP compartment and the subsequent diffusion of the formed chains into the bulk solution, as depicted in Figure 1C and Figure 3.
Extinction spectroscopy provides additional insight into differences in the formation of the two types of 1D chains in regimes B and D (Figure 8C). The spectral analysis of the chain formation under conditions yielding linear chains in regime D (E = 1.5 V, AuNP/CB[7] = 18720) reveals that this process is significantly faster than the regime B-type chain growth, most likely due to the higher CB[7] concentration. At a fixed analysis time of t = 30 min (as in the TEM pictures shown in Figure 5) the faster regime D-type chain growth already yields chains with the maximal detectable length of 16 NPs, whereas the regime B-type chains have only reached a length of ∼12 NPs, which explains the TEM detection of longer regime D-type chains. However, after a reaction time of 2 h, both processes yield LSPR modes related to the maximal detectable chain length of 16 NPs.
Extinction spectroscopy in solution is unable to distinguish 3D fractal aggregates from 1D near-linear chains, because the anisotropy in optical response is washed out by the rotational diffusion of the chains within the optical interrogation volume. However, after both 3D aggregates and 1D chains have been allowed to form over 18 h (by mixing the AuNP and CB in the bulk, or in the electrochemical cell), a different response is clearly observed (Figure 8D). The shorter wavelength peak observed from the 3D aggregates is evidence for less linear, more branched and compact AuNP clusters.
Cryo-TEM
The spatial structure of the formed AuNP/CB[7] chains is studied and visualized by cryo-TEM. This excludes possible misinterpretations about the formed nanoparticle assemblies based on standard TEM measurements. Standard TEM requires the adsorption of the chain assemblies onto the membrane of the TEM grid, which forces the formed 3D structures into adsorbed 2D objects, potentially leading to distortion or destruction of the initially formed structures, as well as the condensation of several objects into new aggregates. Cryo-TEM measurements completely conserve the 3D conformations of the electrokinetically formed nanoparticle assemblies and prevent any interactions between nanoparticle assemblies and the support. The obtained cryo-TEM images reveal one-dimensional linear chains (regimes B and D) as well as branched structures (regime C) similar to what is observed in standard TEM (Figure 9). This confirms the formation and existence of these assemblies in solution, and excludes their accidental formation upon adsorption onto the TEM grid. Furthermore, it can be concluded that the CB[7]-connected nanoparticle chains are sufficiently stable to allow a reliable analysis with standard TEM. However based on cryo-TEM, also full 3D images of the topology of the nanoparticle assemblies can be obtained, which allows further examination of the chain growth mechanism. These images reveal that the detected linear chains, as well as the branched structures, extend mostly in two dimensions and not in three, as would be expected for a random statistical assembly of isotropic particles in solution. This indicates that the chain growth occurs within a two-dimensional plane, which is in agreement with the chain growth mechanism depicted in Figure 3. Although the cryo-TEM images reveal in principle the same structures as the ones observed in standard TEM, they occasionally exhibit AuNP–AuNP distances which are significantly larger than the expected lgap = 9.1 Å of a CB[n]-nanojunction and which are never observed in standard TEM measurements. In addition there is also a constant systematic overestimation of the interparticle distances with respect to the 9.1 Å obtained by normal TEM. Both observations can be explained by the fact that cryo-TEM images are obtained and analyzed by an automatic measurement procedure based on noise-reduced and thresholded images (see Supporting Information), whereas normal TEM images are analyzed manually.
Figure 9.
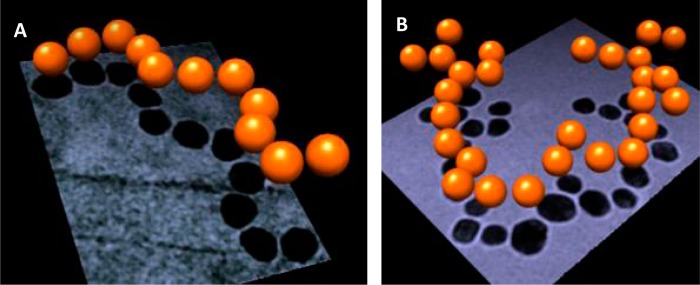
The 3D-images of AuNP/CB[7] chains (orange spheres) of a near-ideal 1D chain (A) and a branched structure (B) calculated from cryo-TEM images, which were recorded at three different tilt angles α = −15°, 0°, and +15° and are shown in the background (method described in the Supporting Information).
The strategy presented in this work allows the synthesis of gold nanoparticle chains with fixed sub-nm gap molecular junctions. The normal spontaneous 3D aggregation of AuNP and CB[7] is redirected into one-dimensional chain growth by the electrokinetic control of the interactions between the AuNP and CB[7] building blocks at the interface of a nanoporous membrane, separating AuNP and CB[7] containing compartments. The nanoparticle assembly can be tuned to yield structures ranging from ideal 1D chains to near-2D networks of interconnected linear chains by adjusting the rate with which the CB[7] molecules enter the AuNP compartment. TEM, cryo-TEM, and spectroscopic analysis of the impact of different experimental parameters on the final aggregate structure allow studying the chain formation mechanism. This synthesis strategy holds promising potential for the controlled assembly of chains of electrostatically interacting building blocks, under the influence of moderate electric fields. No restrictions to conductive, magnetic, or prefunctionalized particles are inherent, which makes this method in principle attractive for generating a large variety of nanoparticle chain systems.
Acknowledgments
The authors thank the French laboratory of excellence AMADEus (Advanced Materials by Design) for a travel grant for N.H. The authors are very grateful to Dr. Tung Chun Lee for fruitful discussions about chain assembly. This work was funded by the CUBiHOLE project in the frame of the European NanoSci-Era+ action under the contract ANR-08-NSCI-008–01, as well as UK EPSRC Grants EP/G060649/1, EP/K028510/1, a CNRS-Royal Society collaboration grant, and ERC Grant 320503 LINASS.
Supporting Information Available
Description of experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hermanson K. D.; Lumsdon S. O.; Williams J. P.; Kaler E. W.; Velev O. D. Science 2001, 294, 1082–1086. [DOI] [PubMed] [Google Scholar]
- Halas N. J.; Lal S.; Chang W.-S.; Link S.; Nordlander P. Chem. Rev. 2011, 111, 3913–3961. [DOI] [PubMed] [Google Scholar]
- Nie Z. H.; Petukhova A.; Kumacheva E. Nat. Nanotechnol. 2010, 5, 15–25. [DOI] [PubMed] [Google Scholar]
- Srivastava S.; Kotov N. A. Soft Matter 2009, 5, 1146–1156. [Google Scholar]
- Wang H.; Chen L.; Shen X.; Zhu L.; He J.; Chen H. Angew. Chem., Int. Ed. 2012, 51, 8021–8025. [DOI] [PubMed] [Google Scholar]
- Li M.; Johnson S.; Guo H.; Dujardin E.; Mann S. Adv. Funct. Mater. 2011, 21, 851–859. [Google Scholar]
- Yan C. C.; Zhang D. H.; Li D. D. J. Appl. Phys. 2011, 109, 063105/1–063105/5. [Google Scholar]
- Feng C.; Guo L.; Shen Z.; Gong J.; Li X.-Y.; Liu C.; Yang S. Solid State Sci. 2008, 10, 1327–1332. [Google Scholar]
- Tang Z.; Ozturk B.; Wang Y.; Kotov N. A. J. Phys. Chem. B 2004, 108, 6972–6931. [Google Scholar]
- Wang H.; Yu Y.; Sun Y.; Chen Q. Nano 2011, 6, 1–17. [Google Scholar]
- Velev O. D.; Gangwal S.; Petsev D. N. Annu. Rep. Prog. Chem., Sect. C: Phys. Chem. 2009, 105, 213–246. [Google Scholar]
- Kretschmer R.; Fritzsche W. Langmuir 2004, 20, 11797–11801. [DOI] [PubMed] [Google Scholar]
- Cheng G.; Romero D.; Fraser G. T.; Hight Walker A. R. Langmuir 2005, 21, 12055–12059. [DOI] [PubMed] [Google Scholar]
- Yamada M.; Shen Z.; Miyake M. Chem. Commun. 2006, 2569–2571. [DOI] [PubMed] [Google Scholar]
- Fernandes R.; Li M.; Dujardin E.; Mann S.; Kanaras A. G. Chem. Commun. 2010, 46, 7602–7604. [DOI] [PubMed] [Google Scholar]
- DeVries G. A; Brunnbauer M.; Hu Y.; Jackson A. M.; Long B.; Neltner B. T.; Uzun O.; Wunsch B. H.; Stellacci F. Science 2007, 315, 358–361. [DOI] [PubMed] [Google Scholar]
- Nakata K.; Hu Y.; Uzun O.; Bakr O.; Stellacci F. Adv. Mater. 2008, 20, 4294–4299. [Google Scholar]
- Ohya Y.; Miyoshi N.; Hashizume M.; Tamaki T.; Uehara T.; Shingubara S.; Kuzuya A. Small 2012, 8, 2335–2340. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Funston A. M.; Gómez D. E.; Davis T. J.; Mulvaney P. Nano Lett. 2011, 11, 4180–4187. [DOI] [PubMed] [Google Scholar]
- Su K.-H.; Wei Q.-H.; Zhang X.; Mock J. J.; Smith D. R.; Schultz S. Nano Lett. 2003, 3, 1087–1090. [Google Scholar]
- Willingham B.; Link S. Opt. Express 2011, 19, 6450–6461. [DOI] [PubMed] [Google Scholar]
- Lagona J.; Mukhopadhyay P.; Chakrabarti S.; Isaacs L. Angew. Chem., Int. Ed. 2005, 44, 4844–4870. [DOI] [PubMed] [Google Scholar]
- Masson E.; Ling X.; Joseph R.; Kyeremeh-Mensah L.; Lu X. RSC Adv. 2012, 2, 1213–1247. [Google Scholar]
- Cheng X.-J.; Liang L.-L.; Chen K.; Ji N.-N.; Xiao X.; Zhang J.-X.; Zhang Y.-Q.; Xue S.-F.; Zhu Q.-J.; Ni X.-L.; Tao Z. Angew. Chem., Int. Ed. 2013, 52, 7252–7255. [DOI] [PubMed] [Google Scholar]
- Taylor R. W.; Lee T. C.; Scherman O. A.; Esteban R.; Aizpurua J.; Huang F. M.; Baumberg J. J.; Mahajan S. ACS Nano 2011, 5, 3878–3887. [DOI] [PubMed] [Google Scholar]
- Lee T.-C.; Scherman O. A. Chem. Commun. 2010, 46, 2438–2440. [DOI] [PubMed] [Google Scholar]
- Lee T.-C.; Scherman O. A. Chem.—Eur. J. 2012, 18, 1628–1633. [DOI] [PubMed] [Google Scholar]
- Tao C.; An Q.; Zhu W.; Yang H.; Li W.; Lin C.; Xu D.; Li G. Chem. Commun. 2011, 47, 9867–9869. [DOI] [PubMed] [Google Scholar]
- Mahajan S.; Lee T.-C.; Biedermann F.; Hugall J. T.; Baumberg J. J.; Scherman O. A. Phys. Chem. Chem. Phys. 2010, 12, 10429–10433. [DOI] [PubMed] [Google Scholar]
- Jiao D. Z.; Biedermann F.; Scherman O. A. Org. Lett. 2011, 13, 3044–3047. [DOI] [PubMed] [Google Scholar]
- Kaifer A. E.; Li W.; Yi S. Isr. J. Chem. 2011, 51, 496–505. [Google Scholar]
- del Pozo M.; Blanco E.; Fatas E.; Hernandez P.; Quintana C. Analyst 2012, 137, 4302–4308. [DOI] [PubMed] [Google Scholar]
- Taylor R. W.; Esteban R.; Mahajan S.; Coulston R.; Scherman O. A.; Aizpurua J.; Baumberg J. J. J. Phys. Chem. C 2012, 116, 25044–25051. [Google Scholar]
- Keesom W. H.; Zelenka R. L.; Radke C. J. J. Colloid Interface Sci. 1988, 125, 575–585. [Google Scholar]
- Ghosal S. Electrophoresis 2004, 25, 214–228. [DOI] [PubMed] [Google Scholar]
- Slater G. W.; Tessier F.; Kopecka K. Methods Mol. Biol. 2010, 583, 121–134. [DOI] [PubMed] [Google Scholar]
- Esteban R.; Taylor R. W.; Baumberg J. J.; Aizpurua J. Langmuir 2012, 28, 8881–8890. [DOI] [PubMed] [Google Scholar]
- Citrin D. S. Nano Lett. 2005, 5, 985–989. [DOI] [PubMed] [Google Scholar]
- Arnold M. D.; Blaber M. G.; Ford M. J.; Harris N. Opt. Express 2010, 18, 7528–7542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.