

Abstract
The increasing incidence of Escherichia coli K1 meningitis due to escalating antibiotic resistance warrants alternate treatment options to prevent this deadly disease. We screened a library of small molecules from the National Institutes of Health clinical collection and identified telmisartan, an angiotensin II receptor type 1 (AT1R) blocker, as a potent inhibitor of E. coli invasion into human brain microvascular endothelial cells (HBMECs). Immunoprecipitation studies revealed that AT1R associates with endothelial cell gp96, the receptor in HBMECs for E. coli outer membrane protein A. HBMECs pretreated with telmisartan or transfected with AT1R small interfering RNA were resistant to E. coli invasion because of downregulation of protein kinase C-α phosphorylation. Administration of a soluble derivative of telmisartan to newborn mice before infection with E. coli prevented the onset of meningitis and suppressed neutrophil infiltration and glial cell migration in the brain. Therefore, telmisartan has potential as an alternate treatment option for preventing E. coli meningitis.
Keywords: Angiotensin II receptor type I, protein kinase C-α, human brain endothelial cells, small molecules, brain, invasion, Escherichia coli K1
Escherichia coli continues to be a major healthcare problem, with mortality rates reaching 100% if left untreated. The incidence of infections may further increase because of the recent emergence of antibiotic-resistant E. coli strains. More than 30% of survivors have debilitating neurological sequelae, such as deafness and incoherence, mediated in part by excessive infiltration of immune cells and migration of glial cells and astrocytes into the brain cortex [1–3]. Therefore, alternative strategies to prevent this deadly disease are urgently needed. The expression of outer membrane protein A in E. coli is essential for the bacteria to invade the blood-brain barrier. Previous studies have shown that outer membrane protein A interacts with its receptor, endothelial cell gp96 (Ecgp96), a homologue of Hsp90β, to invade human brain microvascular endothelial cells (HBMECs), an in vitro model of the blood-brain barrier. The interaction of outer membrane protein A with Ecgp96 enhances Toll-like receptor 2 (TLR2) expression in HBMECs, followed by its association with Ecgp96. This interaction further induces phosphorylation of protein kinase C-α (PKC-α) and its recruitment to Ecgp96/TLR2 complex. Phospho–PKC-α then relays signals for nitric oxide (NO) [4, 5]. Phospho–PKC-α also binds vascular endothelial cadherin (VE-cadherin) after dissociation from β-catenin induced by an IQ motif containing GTPase activating-like-protein 1 (IQGAP1) at adherens junctions, which promotes actin polymerization for internalization of E. coli in HBMECs [6]. However, overexpression of C-terminal–truncated Ecgp96 (Ecgp96Δ200) prevented phospho–PKC-α recruitment to the Ecgp96/TLR2 complex and thereby inhibited bacterial invasion, indicating that Ecgp96/PKC-α association is critical for the infection/invasion process [6].
Small-molecule inhibitors of bacterial virulence factors have been explored for pathogens, including staphylococci, enteropathogenic E. coli, Pseudomonas aeruginosa, and Vibrio cholerae [7–10]. However, identification of small-molecule inhibitors of critical host cell signaling pathways that aid bacterial invasion has not been attempted. Here, we sought to identify small molecules that inhibit E. coli invasion of HBMECs, to develop therapeutic strategies against neonatal meningitis. We screened 447 small molecules from the National Institutes of Health Clinical Collection (NCC) and identified telmisartan, which binds angiotensin II receptor type I (AT1R), to be an effective inhibitor of E. coli invasion. AT1R is the G-protein–coupled receptor for angiotensin II, which is an integral part of the renin-angiotensin system that regulates blood pressure and hypertension [11]. The blood-brain barrier expresses high amounts of AT1R and is activated by angiotensin II secreted by the brain or by circulating angiotensin II [12]. Angiotensin II interaction with AT1R stimulates the dissociation of G-protein Gαq11 into the cytoplasm, which activates phospholipase C and increases cytosolic Ca2+ levels. The Ca2+ influx, in turn, triggers the activation of PKC-α. Telmisartan competitively inhibits angiotensin II binding to AT1R and thereby blocks PKC-α activity [13]. We demonstrate that telmisartan treatment of HBMECs inhibited PKC-α phosphorylation, thereby preventing E. coli invasion. Furthermore, treatment with a soluble derivative of telmisartan also inhibited the onset of meningitis in a newborn mouse model. Therefore, telmisartan could be an effective lead molecule for preventing E. coli meningitis.
METHODS
Bacteria, Antibodies, and Other Reagents
E. coli RS218, a rifampin-resistant strain (serotype O18:K1:H7), was isolated from cerebrospinal fluid of a neonate with meningitis [14]. Telmisartan used for initial screening was from the NCC. For the rest of the study, telmisartan and angiotensin II were purchased from Sigma (St. Louis, MO), and valsartan was purchased from AK Scientific (Mountain View, CA). Gp96 (Grp94) antibodies were from Genetex (Irvine, CA), and PKC-α and phospho–PKC-α antibodies were from Cell Signaling (Danvers, MA). AT1R small interfering RNA (siRNA) was from Santa Cruz Biotechnology (Santa Cruz, CA). Lipofectamine was from Life Technologies (Carlsbad, CA), and the PepTag assay kit and Griess reagent for nitric oxide measurement as nitrite were from Promega (Madison, WI). The plasma membrane isolation kit was from Biovision (Mountain View, CA). All other reagents were from Sigma.
Cell Culture and Total Cell-Associated and Invasive Bacteria Assays
HBMECs from previously frozen stocks were cultured and characterized before infection studies as described previously [5]. HBMECs were pretreated with 20 µg/mL of telmisartan or valsartan for 1 hour before performing the assays. Total cell-associated bacteria and invasive bacteria were enumerated as described previously [6].
Plasma Membrane Isolation, Immunoprecipitation, and Western Blotting
Plasma membrane preparations of E. coli–infected HBMECs in the presence or absence of telmisartan, immunoprecipitation, and Western blotting were performed as described previously [5]. Integrative densitometric values were measured using ImageJ software (available at: http://imagej.nih.gov/ij/).
Other General Methods
Flow cytometry, total nitrite production, TEER measurements, and PepTag assays were performed as previously described [5, 15]. HBMECs were transfected with 33 pmol of AT1R siRNA, using lipofectamine and allowed to recover for 18 hours before performing binding/invasion or flow cytometry analysis.
Preparation of a Water-Soluble Telmisartan Ester
To a suspension of telmisartan (25.8 mg, 0.050 mmol, 1.00 equiv) and K2CO3 (13.9 mg, 0.100 mmol, 2.00 equiv) in 0.5 mL dimethylformamide under a N2 atmosphere was added 1,3-propanesultone (8.0 mg, 0.652 mmol, 1.30 equiv), and the resulting mixture was stirred at ambient temperature for 20 hours. The reaction mixture was then diluted with water (5 mL) and 1 M HCl (2.5 mL), basified with aqueous NaHCO3 to a pH of 8, and purified by reverse-phase high-performance liquid chromatography (linear gradient from 10% to 80% ACN in water) to yield the sodium salt of telmisartan ester (26.2 mg, 80.3% yield) as a tan solid. The structural characterization is available in the Supplementary Materials.
Newborn Mouse Model of Meningitis
Animal studies were approved by the Institutional Animal Care and Use Committee of Children's Hospital of Los Angeles and accorded with guidelines for the performance of animal experiments, as mandated by the Office of Laboratory Animal Welfare at the National Institutes of Health. Three-day-old C57BL/6J mouse pups were infected intranasally with 103 colony-forming units of E. coli and monitored as described previously [5]. Pups received an oral dose of 5 mg/kg body weight of telmisartan ester 6 hours before infection. Three additional doses, one during infection, one 6 hours after infection, and one 12 hours after infection, were administered.
RESULTS
Identification of Telmisartan as a Small-Molecule Inhibitor of E. coli Invasion in HBMECs
To identify small-molecule inhibitors, 447 small molecules from the NCC that have high drug-like potential and known safety profiles were tested for their ability to inhibit E. coli entry into HBMECs at 2 different concentrations. Telmisartan was chosen as the active lead, using strategies described in Figure 1. Telmisartan has been reported to exhibit considerable bioavailability in the brain and systemic persistence for about 24 hours and is known to control high blood pressure [16]. Because increased intracranial pressure is a characteristic feature of E. coli meningitis, owing to increased blood flow into the meninges [17], we hypothesized that telmisartan could prevent bacterial entry and inhibit the peripheral effects of meningitis. To assess the activity range of telmisartan, a dose-dependent study of different concentrations of telmisartan was performed. HBMECs were pretreated for 1 hour with different concentrations, ranging from 1 to 50 µg/mL, and then infected with E. coli. Concentrations of 20 µg/mL and 30 µg/mL inhibited the invasion by approximately 50% (Figure 2A). Cytotoxicity analysis revealed that 20 µg/mL of telmisartan was less cytotoxic than 30 µg/mL (17% and 20%, respectively) in HBMECs (Figure 2B). Therefore, 20 µg/mL was used for further analysis.
Figure 1.

Flowchart of steps used in the identification of lead molecules that could potentially prevent Escherichia coli meningitis. Small molecules from the National Institutes of Health Clinical Collection (NCC) were tested for inhibition of total cell association/invasion of E. coli in human brain microvascular endothelial cells (HBMECs). Molecules that inhibited total cell-associated and/or invaded bacteria were referenced with the NCC database files, and molecules that were potential antibiotics were discarded from further studies. Molecules that were noncytotoxic in HBMECs were tested in 3-day-old mouse pups at a dose of 5 mg/kg body weight for their ability to prevent the onset of meningitis by E. coli K1. Finally, the compounds that did not cause deleterious effects in pups were analyzed using Pubchem for log P values, and those with values of >3 were data mined for reports on bioavailability at the blood-brain barrier (BBB). Abbreviation: IC50, median inhibitory concentration.
Figure 2.

Telmisartan (TS) inhibits Escherichia coli invasion of human brain microvascular endothelial cells (HBMECs). A, Confluent monolayers of HBMECs in 24-well plates were treated with different concentrations of TS for 1 hour at 37°C in experimental medium. E. coli at a multiplicity of infection of 100 was added, and cells were subjected to invasion assays. B, HBMEC monolayers were pretreated with different concentrations of TS as mentioned above and subjected to cytotoxicity analysis. C, Confluent HBMECs were pretreated, cotreated with infection, or treated after infection at different time points, and E. coli invasion assays were then performed. D, HBMECs were pretreated for 1 hour with 0.1 µM angiotensin II (Ang-II) at 37°C and then infected with E. coli to analyze total cell-associated and invaded bacteria. E, HBMECs were pretreated with TS or valsartan (VS; 20 µg/mL) and subjected to total cell-associated and invasion assays. Error bars represent mean values ± SD from at least 3 different experiments performed in triplicate. Inhibition of E. coli invasion after treating with inhibitors was statistically significant (* P < .05).
We next examined whether coincubation or postinfection treatment had any effect on invasion. HBMECs were cotreated along with the bacteria or were treated 15 minutes and 30 minutes after infection with telmisartan. Cells pretreated with telmisartan for 30 minutes or 1 hour served as controls. Treatment of HBMECs with telmisartan for 30 minutes and 1 hour before infection inhibited E. coli invasion by 50%, whereas cotreatment of telmisartan along with the bacteria inhibited invasion by 40% (Figure 2C). Telmisartan treatment 15 minutes and 30 minutes after infection inhibited E. coli invasion by 20% and 8%, respectively. Therefore, telmisartan pretreatment or cotreatment inhibits E. coli invasion, whereas postinfection treatment does not affect the invasion process.
Next, we examined the effect of the AT1R natural ligand, angiotensin II, on E. coli invasion. Pretreatment with 0.1 µM and 1 µM concentrations of angiotensin II did not significantly increase invasion or binding (Figure 2D; only data for 0.1 µM are shown). High-performance liquid chromatography of HBMEC supernatants after infection with E. coli was performed to estimate the level of angiotensin II that was secreted and showed no detectable levels of angiotensin II, implying that its secretion is too negligible for detection (data not shown).
Furthermore, we tested whether other angiotensin-receptor blockers, such as valsartan, can also block E. coli invasion of HBMECs. Valsartan (20 µg/mL) also inhibited E. coli invasion by 50%, suggesting that the inhibitory mechanism of invasion is mediated by blocking AT1R signaling. Both telmisartan and valsartan had no effect on bacterial binding to HBMECs (Figure 2E). Both molecules showed no antibacterial activity, even at concentrations 10-fold greater than the amounts used in this study.
AT1R Associates With Ecgp96 in Naive and Infected HBMECs During E. coli Invasion
Previous studies have demonstrated that E. coli invasion of HBMECs requires Ecgp96 association with TLR2 [5]. Pretreatment with blocking antibodies to Ecgp96 or TLR2 prevented invasion, suggesting that both proteins play an important role in the invasion process. Since telmisartan inhibits AT1R and subsequent invasion, we examined whether AT1R also associates with Ecgp96. Immunoprecipitation studies revealed that AT1R associates with Ecgp96 in naive and infected HBMECs. Although Ecgp96 levels increased with infection, AT1R levels were comparable between uninfected control and infected HBMECs. This indicates that E. coli infection does not perturb AT1R levels, even though AT1R remains associated with Ecgp96 (Figure 3A and 3B).
Figure 3.

Angiotensin II receptor type 1 (AT1R) associates with endothelial cell gp96 (Ecgp96) in both naive and infected human brain microvascular endothelial cells (HBMECs). A, HBMECs grown in T75 culture flasks were infected with Escherichia coli at a multiplicity of infection of 100 for various time points, and membrane fractions were isolated. A total of 300 µg of membrane proteins were immunoprecipitated with anti-Ecgp96 antibodies, and the immune complexes were subjected to Western blotting with antibodies to Ecgp96 (96 kDa) or AT1R (43 kDa). Equal loading was verified by the expression of immunoglobulin G (IgG) heavy chain fragment (55 kDa). The blots represent one of 3 experiments with similar results. B, Integrative densitometric values (IDVs) of band intensities of Ecgp96 and AT1R normalized to IgG IDV were graphed, with the IDV of the control considered as 100%. Increase in Ecgp96 expression during infection was statistically significant (*P < .005). C, Total cell lysates from E. coli–infected and TS-treated/E. coli–infected HBMECs were subjected to Western blotting using anti-Ecgp96 or anti-β-actin antibodies. Blots are representative of 3 different experiments. D, The IDV of Ecgp96 expression after normalization with β-actin for different infection time points was plotted as described above. The increase in Ecgp96 expression in HBMECs with or without TS treatment was statistically significant with respect to data for the uninfected control (*P < .01). E, HBMECs grown in 6-well plates were infected with E. coli with or without TS pretreatment for 60 minutes, and the surface expression of Ecgp96 was analyzed by flow cytometry using anti-Ecgp96 antibodies. Data are representative of 3 different experiments. F, Total cell lysates of HBMECs infected with E. coli in the presence or absence of TS were subjected to Western blotting using anti-AT1R antibodies. Equal loading was established using β-actin expression. G, The IDVs of bands from panel F were plotted for AT1R expression as mentioned above.
Because telmisartan inhibits E. coli invasion in HBMECs, we next examined whether telmisartan modulates Ecgp96 expression or its association with AT1R. Western blot analysis of membrane preparations of HBMECs infected with E. coli for various time points in the presence or absence of telmisartan showed no apparent change in Ecgp96 expression when protein levels were normalized to β-actin (Figure 3C and 3D). Flow cytometry also showed that telmisartan did not alter Ecgp96 expression in HBMECs 60 minutes after infection (Figure 3E). To examine whether telmisartan alters the expression of AT1R, HBMECs were treated with E. coli for various time points in the presence or absence of telmisartan, and the membrane fractions were subjected to Western blotting using antibodies to AT1R. E. coli infection alone or in the presence of telmisartan did not alter AT1R expression (Figure 3F and 3G). Treatment of HBMECs with angiotensin II also did not modulate AT1R expression, as determined by flow cytometry (data not shown).
Preincubation of HBMECs With Telmisartan Prevents PKC-α Phosphorylation and Endothelial Cell Permeability
Because telmisartan does not modulate the expression of Ecgp96 and AT1R, the possible role of downstream effectors in inhibiting E. coli invasion was assessed. AT1R signaling induces Ca2+-mediated phospholipase C and PKC-α [17]. We have previously shown that E. coli activates PKC-α 15 minutes after infection during invasion of HBMECs and that inhibition of PKC-α by means of specific inhibitors prevented the invasion [6, 18]. Therefore, we analyzed whether telmisartan or valsartan regulates PKC-α phosphorylation. HBMECs were infected with E. coli for 15 minutes in the presence or absence of telmisartan and subjected to a nonradioactive PepTag assay. Although this assay recognizes all forms of PKC, our previous studies clearly demonstrated that E. coli induces only PKC-α [18]. Phorbol-myristate acetate (PMA) was used as a positive control for PKC phosphorylation. Both E. coli and PMA induced PKC phosphorylation in HBMECs, whereas HBMECs treated with telmisartan or valsartan alone retained the same basal level of phosphorylation exhibited by untreated control HBMECs (Figure 4A). However, telmisartan and valsartan each significantly reduced PKC phosphorylation induced by E. coli in HBMECs. Interestingly, telmisartan did not suppress PMA-induced PKC phosphorylation.
Figure 4.

Telmisartan (TS) inhibits Escherichia coli–induced protein kinase C-α (PKC-α) phosphorylation and thereby endothelial permeability in human brain microvascular endothelial cells (HBMECs). A, HBMECs grown in 6-well plates were treated with TS or valsartan (VS) with or without E. coli infection as mentioned earlier, and PKC phosphorylation was analyzed using a nonradioactive PepTag assay. B, Total phospho–PKC-α was determined by flow cytometry in HBMECs treated with TS with or without E. coli infection. C, Supernatants of HBMECs pretreated with or without TS followed by infection with E. coli were analyzed for total nitrite production, using the Greiss method. D, HBMECs cultured in 3-µm transwell culture inserts were pretreated with TS and infected with E. coli for various periods, and transendothelial electrical resistance (TEER) was measured using a Millicell apparatus. Inhibition of the decrease in TEER values in TS-treated and infected HBMECs was statistically significant, compared with E. coli–infected cells (*P < .02). All experiments were performed at least 3 times in triplicate.
Since PMA activation of PKC-α does not require Ca2+, it is inferred that telmisartan regulates only Ca2+-mediated PKC-α phosphorylation. To confirm the effect of telmisartan on PKC-α phosphorylation, HBMECs infected with E. coli in the presence or absence of telmisartan were subjected to flow cytometry using anti-phospho–PKC-α antibodies. Telmisartan treatment of uninfected HBMECs reduced PKC-α phosphorylation to levels less than that of untreated HBMECs. E. coli infection of HBMECs for 15 minutes showed increased levels of PKC-α phosphorylation (Figure 4B). However, pretreatment with telmisartan prevented E. coli from inducing PKC-α phosphorylation, and the levels were comparable to untreated/uninfected HBMECs.
Since our results establish the role of PKC-α as a critical target in the inhibition of E. coli invasion, we next examined the effect of telmisartan on downstream events dependent on PKC-α phosphorylation. We previously showed that phosphorylated PKC-α might exist as separate pools in HBMECs during E. coli invasion, one regulating NO production and the other regulating endothelial cell permeability [5]. Therefore, E. coli–induced total nitrite production in HBMECs in the presence or absence of telmisartan was determined. The difference in nitrite production between the groups was not statistically significant (Figure 4C). However, telmisartan treatment of HBMECs prevented the decrease in transendothelial cell resistance (TEER) upon E. coli infection, while infection with E. coli alone considerably reduced TEER until 60 minutes after infection (Figure 4D). Previous studies showed that phospho-PKC-α binding to VE-cadherin at adherens junctions in HBMECs induces a decrease in TEER, which releases F-actin to aid E. coli invasion [6]. Therefore telmisartan inhibits a pool of PKC-α, which regulates TEER, thereby preventing E. coli invasion of HBMECs.
Knockdown of AT1R Expression Inhibits E. coli Invasion by Regulating PKC-α Phosphorylation
Since telmisartan is a functional inhibitor of AT1R, we examined whether knockdown of AT1R by siRNA had any effect on bacterial invasion. Suppression of AT1R expression inhibited 80% of E. coli invasion in HBMECs but had no effect on binding (Figure 5A). The knockdown efficiency of AT1R by siRNA was approximately 85% as verified by flow cytometry (Figure 5B).
Figure 5.

Silencing of angiotensin II receptor type 1 (AT1R) prevents Escherichia coli invasion of human brain microvascular endothelial cells (HBMECs) by regulating protein kinase C-α (PKC-α) phosphorylation. A, HBMECs cultured in 24-well plates were transfected with AT1R–small interfering RNA (siRNA), the cells were allowed to recover for 18 hours, and total cell associated/invasion assays were performed. Error bars represent SD from the mean values performed 3 times in triplicate. Inhibition of E. coli invasion in the presence of AT1R-siRNA was statistically significant (*P < .005). B, Efficiency of AT1R silencing in the presence of AT1R siRNA was determined by flow cytometry. AT1R expression was silenced by approximately 85% by siRNA. Flow cytometry analysis for endothelial cell gp96 (C) and phospho–PKC-α expression (D) in AT1R-siRNA/HBMECs with or without E. coli infection was performed. E, Results of flow cytometry analysis for phospho-PKC-α expression in HBMECs treated with phorbol-myristate acetate, TS, and/or AT1R-siRNA. All flow cytometry experiments were performed 3 times individually. F, HBMECs cultured in 3-µm transwell culture inserts were transfected with AT1R-siRNA and infected with E. coli for various times, and transendothelial electrical resistance (TEER) was measured using a Millicell apparatus. Inhibition of the decrease in the TEER in AT1R-siRNA–transfected/infected HBMECs was statistically significant, compared with values exhibited by infected HBMECs (* P < .01). Abbreviations: –, negative; +, positive.
Because AT1R knockdown had no effect on the binding of E. coli K1 to HBMECs, we assume that the lack of AT1R expression did not have any effect on Ecgp96 levels. To confirm this, we also analyzed Ecgp96 expression in AT1R siRNA–transfected HBMECs by flow cytometry. As expected, no significant change in Ecgp96 expression was observed upon E. coli K1 infection (Figure 5C). Thus, it is possible that AT1R suppression affects PKC-α phosphorylation, which is similar to the effect of telmisartan treatment in HBMECs. Therefore, the phosphorylation of PKC-α after treatment with telmisartan or transfection with AT1R siRNA was evaluated. PKC-α phosphorylation was downregulated below the levels observed in untreated cells in both situations (Figure 5D). Telmisartan or AT1R siRNA only partially regulated PKC-α activation by PMA, implying that telmisartan or AT1R siRNA were only able to inhibit Ca2+-mediated PKC-α phosphorylation (Figure 5E). In addition, AT1R knockdown prevented an E. coli–induced decrease in TEER (Figure 5F), suggesting that AT1R-mediated PKC-α activation contributes to the invasion process of E. coli and subsequent tight junction disruption in HBMECs.
A Telmisartan Derivative, Telmisartan Ester, Prevents the Onset of Meningitis in a Newborn Mouse Model
Telmisartan binding to AT1R inhibits downstream signaling and subsequent invasion of E. coli in HBMECs. Therefore, we explored whether telmisartan can prevent bacterial entry into the brain in a newborn mouse model of meningitis. To increase the solubility of telmisartan for use in animal experiments, a derivative of telmisartan containing a sulfate group was prepared by esterification (Figure 6A). The telmisartan ester showed similar inhibitory activity as telmisartan in vitro (data not shown). Oral administration of telmisartan and E. coli infection in newborn mice was performed as described in the Methods section. Cerebrospinal fluid samples from infected pups were positive for E. coli, while cerebrospinal fluid from telmisartan-pretreated and infected pups were not, indicating that telmisartan prevented bacterial entry into the brain (Figure 6B). Brain sections of E. coli–infected pups showed gross morphological changes, such as disruption of cortex and meninges, after hematoxylin-eosin staining, extensive glial cell migration after GFAP staining, and neutrophil infiltration after MPO staining (Figure 6C). However, brain sections of telmisartan-pretreated pups showed no such changes. Uninfected and telmisartan-treated pups showed no adverse reactions to the compound, and the morphologic characteristics of the brains were comparable to those of the brains of control pups.
Figure 6.

Telmisartan ester (TS-S) prevents the onset of meningitis in a newborn mouse model. A, Synthesis of a water-soluble derivative of TS, TS-S, by an esterification process is shown. B, Three-day-old C57BL/6J mouse pups were infected with 103 colony-forming units of Escherichia coli in the presence or absence of TS-S, as mentioned in the Methods section. Cerebrospinal fluid (CSF) samples collected through lumbar puncture were cultured in LB plus rifampicin and incubated overnight. Bacterial growth in CSF cultures was considered positive for meningitis. The asterisk indicates that no bacterial growth was observed in the CSF of any of the pups in all 3 experiments. The experiments were performed 3 times with at least 5 pups per group for each experiment. C, Brain sections of control, TS-S control, E. coli–infected and TS-S–treated/infected pups were subjected to hematoxylin-eosin (H&E) staining for morphological analysis, MPO staining for analysis of neutrophil migration, and GFAP staining for analysis of glial cell migration. Arrows indicate gross morphological changes and neutrophil and glial cell migration in the brains of E. coli–infected pups. Bar, 100 µm.
DISCUSSION
E. coli uses a variety of strategies to evade immune responses to successfully reach and invade the blood-brain barrier. Previous studies show that, although the infection can be cleared using antibiotics, the damage to the brain is permanent [19]. Therefore, in our quest to identify alternate modes of inhibition of E. coli entry into the brain, we screened a small-molecule library from the NCC. We show for the first time that telmisartan can successfully inhibit E. coli invasion of HBMECs and the onset of meningitis in a newborn mouse model. Telmisartan is a Food and Drug Administration–approved drug that has been clinically tested for druglikeness and has a known safety profile. Telmisartan showed no antibacterial activity or cytotoxicity at the dose used in our in vitro and in vivo studies. Angiotensin-receptor blockers like telmisartan exhibit minimal side effects in children aged <18 years, although the caveat remains that the effect of telmisartan was not studied in children <6 years old [20]. Another angiotensin-receptor blocker, valsartan, also had a similar inhibitory effect against invasion in vitro. However, telmisartan has been shown to clear from circulation more rapidly than valsartan (98% through the fecal route), thereby ruling out the possibility of prolonged systemic persistence [16]. Nevertheless, the fraction of telmisartan retained in the system is sufficient to exhibit inhibitory activity on E. coli entry into the blood-brain barrier.
Telmisartan is a competitive inhibitor of angiotensin II binding to AT1R [21]. Our data demonstrate that AT1R associates with the E. coli outer membrane protein A receptor Ecgp96 even in uninfected cells, indicating that E. coli invasion of HBMECs may depend partly on AT1R. We have previously shown that PKC-α phosphorylation and its association with Ecgp96 dictates E. coli invasion into HBMECs [6]. In agreement with this finding, Ecgp96 that lacks its C-terminal domain inhibits PKC-α phosphorylation in HBMECs despite infection with E. coli. Since AT1R also regulates PKC-α phosphorylation, it is possible that AT1R association with Ecgp96 governs PKC-α phosphorylation during E. coli invasion. Confirming this speculation, treatment of HBMECs with telmisartan or knockdown of AT1R expression by specific siRNA significantly reduced PKC-α phosphorylation to levels below that for untreated control cells. TLR2 association also increases with Ecgp96 in HBMECs upon E. coli invasion and recruits phospho–PKC-α to initiate further downstream signaling to induce NO production. Increased NO levels further induce recruitment of more Ecgp96/TLR2 complexes to the membrane to act as receptors for bacteria to bind [5]. Therefore, Ecgp96 forms a complex with multiple proteins at the plasma membrane, and the binding of E. coli outer membrane protein A induces signals via the associated AT1R and TLR2 for PKC-α phosphorylation. Telmisartan inhibits PKC-α phosphorylation without altering the enhanced expression of Ecgp96 that occurs during infection. In agreement, telmisartan only affected invasion, not binding. This is in parallel with our previously reported data that inhibition of PKC-α prevents E. coli invasion of but not binding to HBMECs [18]. NO levels induced by E. coli in the absence or presence of telmisartan were also comparable, indicating that similar NO levels reflected on unchanged Ecgp96 levels. Therefore, it is possible that basal phospho–PKC-α, partly induced by AT1R in HBMECs, is recruited to the Ecgp96/TLR2/AT1R complex during initial bacterial binding. This induces actin rearrangements underneath the bacterial attachment sites by disassembling the tight junctions, which is required for the invasion of E. coli in HBMECs. In parallel, TLR2-mediated activation of PKC-α may contribute to the production of NO, which in turn is responsible for increased expression of Ecgp96 at the plasma membrane. We have also shown previously that E. coli infection increases intracellular Ca2+ levels in HBMECs, leading to PKC-α phosphorylation [18]. Thus, it is assumed that telmisartan binding to AT1R prevents the influx of extracellular Ca2+ into HBMECs and, in turn, hinders PKC-α phosphorylation (Figure 7).
Figure 7.
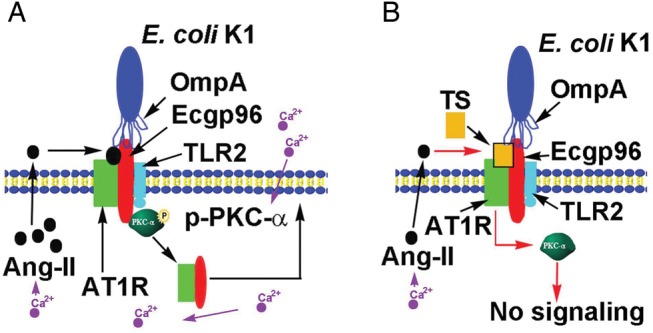
Schematic representation of cellular events during Escherichia coli K1 invasion of human brain microvascular endothelial cells (HBMECs) and the possible mode of its inhibition by telmisartan (TS). A, Interaction of E. coli K1 outer membrane protein A with endothelial cell gp96 (Ecgp96) during invasion mobilizes extracellular Ca2+ into HBMECs, which induces angiotensin II–mediated translocation of angiotensin II receptor type 1 (AT1R) to the plasma membrane and subsequent association with Ecgp96/Toll-like receptor 2 (TLR2) complexes. This interaction also activates phospho–protein kinase C-α (PKC-α), which is recruited to the Ecgp96/AT1R/TLR2 complex and activates downstream signals required for invasion. B, TS pretreatment blocks angiotensin II binding to AT1R and prevents PKC-α phosphorylation, thereby thwarting the downstream signaling required for E. coli K1 invasion.
Telmisartan induces antiinflammatory effects in the brain by suppressing glial cell/astrocyte migration into the brain cortex [22]. We observed that telmisartan treatment in newborn mice completely prevented glial cell migration upon infection with E. coli. Therefore, telmisartan can potentially be an attractive therapeutic molecule for E. coli meningitis because of its bioavailability in the brain, preventing bacterial entry into the brain by inhibiting PKC-α phosphorylation and also preventing potential damage that could result from glial cell migration. Recent studies show that AT1R heterodimerizes with another GPCR, β2-adrenergic receptor (β2AR), and that AT1R also engages β-arrestin for internalization and receptor recycling [23, 24]. Interestingly, Neisseria meningitidis manipulates β2AR/β-arrestin to promote actin-rich cellular protrusions in brain endothelium [25]. A recent report also shows that another angiotensin-receptor blocker, candesartan, binds Hsp90, a homologue of Ecgp96 [26]. Therefore, it is interesting to speculate that telmisartan binds to both Ecgp96/AT1R complexes rather than to AT1R alone, and the concept will be revisited with more experimental data in the future (Figure 7). In addition, if telmisartan binds to the Ecgp96/AT1R/β2AR complex, it can be used to prevent not only E. coli infection, but also infection due to other pathogens, such as meningococci, that cause meningitis. Our future work is aimed at exploring these possibilities.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Financial support. This work was supported by the National Institutes of Health (grants AI40567 and NS73115 to N. V. P.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Khan NA, Goldsworthy GJ. Novel model to study virulence determinants of Escherichia coli K1. Infect Immun. 2007;75:5735–9. doi: 10.1128/IAI.00740-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mittal R, Gonzalez-Gomez I, Goth KA, Prasadarao NV. Inhibition of inducible nitric oxide controls pathogen load and brain damage by enhancing phagocytosis of Escherichia coli K1 in neonatal meningitis. Am J Pathol. 2010;176:1292–305. doi: 10.2353/ajpath.2010.090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubois D, Prasadarao NV, Mittal R, Bret L, Roujou-Gris M, Bonnet R. CTX-M beta-lactamase production and virulence of Escherichia coli K1. Emerg Infect Dis. 2009;15:1988–90. doi: 10.3201/eid1512.090928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prasadarao NV, Srivastava PK, Rudrabhatla RS, Kim KS, Huang SH, Sukumaran SK. Cloning and expression of the Escherichia coli K1 outer membrane protein A receptor, a gp96 homologue. Infect Immun. 2003;71:1680–8. doi: 10.1128/IAI.71.4.1680-1688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishnan S, Chen S, Turcatel G, Arditi M, Prasadarao NV. Regulation of Toll-like receptor 2 interaction with Ecgp96 controls Escherichia coli K1 invasion of brain endothelial cells. Cell Microbiol. 2013;15:63–81. doi: 10.1111/cmi.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krishnan S, Fernandez GE, Sacks DB, Prasadarao NV. IQGAP1 mediates the disruption of adherens junctions to promote Escherichia coli K1 invasion of brain endothelial cells. Cell Microbiol. 2012;14:1415–33. doi: 10.1111/j.1462-5822.2012.01805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banskota AH, McAlpine JB, Sorensen D, et al. Genomic analyses lead to novel secondary metabolites. Part 3. ECO-0501, a novel antibacterial of a new class. J Antibiot (Tokyo) 2006;59:533–42. doi: 10.1038/ja.2006.74. [DOI] [PubMed] [Google Scholar]
- 8.Gauthier A, Robertson ML, Lowden M, Ibarra JA, Puente JL, Finlay BB. Transcriptional inhibitor of virulence factors in enteropathogenic Escherichia coli. Antimicrob Agents Chemother. 2005;49:4101–9. doi: 10.1128/AAC.49.10.4101-4109.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science. 2005;310:670–4. doi: 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- 10.Aiello D, Williams JD, Majgier-Baranowska H, et al. Discovery and characterization of inhibitors of Pseudomonas aeruginosa type III secretion. Antimicrob Agents Chemother. 2010;54:1988–99. doi: 10.1128/AAC.01598-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang SS, Stevenson L, Dzau VJ. Endothelial renin-angiotensin pathway. Adrenergic regulation of angiotensin secretion. Circ Res. 1990;66:103–8. doi: 10.1161/01.res.66.1.103. [DOI] [PubMed] [Google Scholar]
- 12.Ganong WF. Origin of the angiotensin II secreted by cells. Proc Soc Exp Biol Med. 1994;205:213–9. doi: 10.3181/00379727-205-43699a. [DOI] [PubMed] [Google Scholar]
- 13.Arai H, Escobedo JA. Angiotensin II type 1 receptor signals through Raf-1 by a protein kinase C-dependent, Ras-independent mechanism. Mol Pharmacol. 1996;50:522–8. [PubMed] [Google Scholar]
- 14.Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun. 1996;64:146–53. doi: 10.1128/iai.64.1.146-153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shanmuganathan MV, Krishnan S, Fu X, Prasadarao NV. Attenuation of biopterin synthesis prevents Escherichia coli K1 invasion of brain endothelial cells and the development of meningitis in newborn mice. J Infect Dis. 2013;207:61–71. doi: 10.1093/infdis/jis656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Israili ZH. Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J Hum Hypertens. 2000;14(Suppl 1):S73–86. doi: 10.1038/sj.jhh.1000991. [DOI] [PubMed] [Google Scholar]
- 17.Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis. 2004;39:1267–84. doi: 10.1086/425368. [DOI] [PubMed] [Google Scholar]
- 18.Sukumaran SK, Prasadarao NV. Regulation of protein kinase C in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem. 2002;277:12253–62. doi: 10.1074/jbc.M110740200. [DOI] [PubMed] [Google Scholar]
- 19.Mittal R, Gonzalez-Gomez I, Panigrahy A, Goth K, Bonnet R, Prasadarao NV. IL-10 administration reduces PGE-2 levels and promotes CR3-mediated clearance of Escherichia coli K1 by phagocytes in meningitis. J Exp Med. 2010;207:1307–19. doi: 10.1084/jem.20092265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wells TG, Portman R, Norman P, Haertter S, Davidai G, Fei W. Safety, efficacy, and pharmacokinetics of telmisartan in pediatric patients with hypertension. Clin Pediatr (Phila) 2010;49:938–46. doi: 10.1177/0009922810363609. [DOI] [PubMed] [Google Scholar]
- 21.Le MT, Pugsley MK, Vauquelin G, Van Liefde I. Molecular characterisation of the interactions between olmesartan and telmisartan and the human angiotensin II AT1 receptor. Br J Pharmacol. 2007;151:952–62. doi: 10.1038/sj.bjp.0707323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurihara T, Ozawa Y, Shinoda K, et al. Neuroprotective effects of angiotensin II type 1 receptor (AT1R) blocker, telmisartan, via modulating AT1R and AT2R signaling in retinal inflammation. Invest Ophthalmol Vis Sci. 2006;47:5545–52. doi: 10.1167/iovs.06-0478. [DOI] [PubMed] [Google Scholar]
- 23.Hammad MM, Dupre DJ. Chaperones contribute to G protein coupled receptor oligomerization, but do not participate in assembly of the G protein with the receptor signaling complex. J Mol Signal. 2010;5:16. doi: 10.1186/1750-2187-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–12. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 25.Coureuil M, Lecuyer H, Scott MG, et al. Meningococcus hijacks a beta2-adrenoceptor/beta-arrestin pathway to cross brain microvasculature endothelium. Cell. 2010;143:1149–60. doi: 10.1016/j.cell.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 26.Sugawara T, Kokubun K, Ishida R, et al. Angiotensin receptor blocker directly binds to HSP and stimulates its production in the brain. Proceedings of the 11th WSEAS International Conference on Mathematics and Computers in Biology and Chemistry.2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.