

Abstract
History of stress is considered a major risk factor for the development of major depression and posttraumatic stress disorder (PTSD). Elucidating the neurobiological mechanisms of Pavlovian fear conditioning may provide insight into the etiology of PTSD. In the current study, adolescent male Sprague-Dawley rats were exposed to 3 weeks of a chronic-mild-unpredictable stress (CMS) protocol. Immediately following the CMS, the animals were subjected to hippocampal-dependent (trace and contextual) and hippocampal-independent (delay) fear conditioning. CMS exposure enhanced trace freezing behavior compared to non-stress controls. This effect was not observed in contextual or delay conditioned animals. Given that the endocannabinoid system is negatively affected by CMS procedures, separate groups of stressed rats were administered the CB1 receptor agonist, ACEA (0.1 mg/kg), prior to trace fear conditioning or a memory-recall test. Regardless of administration time, ACEA significantly reduced freezing behavior in stressed animals. Furthermore, when administered during the first memory recall test, ACEA enhanced long-term extinction in both stress and non-stress groups. The results demonstrate that chronic unpredictable stress selectively enhances hippocampal-dependent episodic fear memories. Pathologies of the episodic memory and fear response may increase the susceptibility of developing PTSD. Reduction in fear responses via exogenous activation of the CB1 receptor suggests that a deficiency in the endocannabinoid system contributes to this pathology.
Keywords: CB1 receptor, learning, depression, PTSD, hippocampus
Introduction
Life adversities (i.e. stress) are major risk factors for many psychopathologies, including posttraumatic stress disorder (PTSD) and major depression (Kendler et al., 2000; Kessler et al., 2001). In animal models of depression, repeated exposure to mild and unpredictable stress results in behavioral and neurobiological changes similar to those observed in human patients (Krishnan and Nestler, 2010; Yan et al., 2010). For example, anhedonia, a hallmark feature of depression in humans, is often observed as a decreased consumption to a highly-palatable sucrose solution in rodents (Willner, 2005). Chronic mild stress (CMS) also leads to altered glucocorticoid, serotoninergic and neuroimmune activity in animals (Grippo and Johnson, 2009). Endocannabinoid signaling, particularly in the hippocampus, is reported to be exquisitely sensitive to CMS (see Hill and Gorzalka, 2009 for review; Hill et al., 2005; Reich et al, 2009). These stress-induced effects are ameliorated by anti-depressant drugs, providing further evidence that stress and depression share common neural substrates (Grippo and Johnson, 2009; Willner, 2005).
The symptomology of PTSD involves the dysregulation of many emotions and is often comorbid with depression/anxiety disorders (Jovanovic and Ressler, 2010; Rau et al., 2005). In particular, exaggerated responses to fear or impairments in fear controllability are implicated as risk factors for the development of PTSD. As a result, Pavlovian fear conditioning is a widely pre-clinical tool used to explore the etiology of PTSD. A meta-analysis of several fear conditioning experiments reported that humans with anxiety disorders displayed enhanced fear responses compared to healthy, control subjects (Jovanovic and Ressler, 2010). These results suggest that a pre-existing condition, such as anxiety or depression, leads to dysfunction in fear controllability and thus, an increased vulnerability to PTSD.
In related animal studies, a history of stress prior to fear conditioning is capable of modulating fear responses. For example, pre-exposure to restraint stress (acute and chronic), footshocks or exogenous administration of corticosterone facilitates both contextual and delay fear conditioning in male rats (Cordero, et al., 2003a, 2003b; Rau et al., 2005). The effects of chronic stress on fear conditioning have predominantly focused on homotypic (restraint) stress for periods of several weeks (Conrad et al., 1999; Cordero et al., 2003a; Sandi et al., 2001). Homotypic stress protocols induce profound dendritic atrophy and prevent neurogenesis in the hippocampus and other brain regions; effects that are observed to a lesser degree in heterotypic, CMS paradigms (Vyas et al., 2002). Thus, CMS appears to produce non-habituating, although less invasive, effects on hippocampal anatomy and physiology than other chronic stress models (Pêgo et al., 2008; Vyas et al., 2002). These properties of CMS make it an ideal pre-clinical tool for elucidating the neurobiological substrates of stress-induced neuropathologies.
The hippocampus is a medial temporal lobe component of the limbic system that is critically involved in memory formation and in stress regulation (Kim and Diamond, 2002; McEwen, 2005). It functions as a negative feedback control for the hypothalamic-pituitary-adrenal (HPA) axis by assisting in terminating the stress response (Herman et al., 2005). Chronic activation of the HPA axis can lead to stress-induced impairments in hippocampal functions such as learning and memory (Kim and Diamond, 2002; McEwen, 2005). In humans, hippocampal damage is also associated with the symptomologies of depression, PTSD and schizophrenia (Campbell and MacQueen, 2004; Nopoulos et al., 1997; Wong et al., 2000).
The present study investigates the effects of CMS on hippocampal-dependent trace fear conditioning, a model of aversive episodic (autobiographical) memory in adolescent male animals. In this paradigm, the conditioned stimulus (CS; auditory tone) and unconditioned stimulus (US; footshock) are separated by a time gap or trace interval that is several seconds long. The trace interval forms a temporally non-contiguous association between the CS and US by forcing the animal to remember information about the CS during the time gap (Clark et al., 2001). Given that the hippocampus is involved in stress response as well as learning and memory, we hypothesized that animals exposed to 3 weeks of CMS would display enhanced fear acquisition and memory. Other hallmarks of stress-related disorders are deficits in behavioral flexibility and extinction learning. This impairment in behavioral extinction is especially acute in PTSD patients (Jovanovic and Ressler, 2010). Therefore, we also hypothesized that CMS would interfere with extinction of the fear response. Finally, we tested if CMS exposure altered cannabinoid receptor 1 (CB1)-regulation of fear learning and extinction (Marsicano et al., 2002; Reich et al., 2008).
Materials and methods
Subjects
Male Sprague-Dawley rats (Charles River, Boston, MA, USA) were group-caged (three animals per cage) and allowed to acclimate for 5–7 days prior to experimental testing. All animals were 40–45 days old at the beginning of experimental procedures and maintained on a 12-h/12-h light–dark cycle with lights on at 8:00 a.m. Food and water were available ad libitum in the home cages, unless otherwise noted. All experimental procedures were carried out in accordance with protocols established by the Institutional Animal Care and Use Committee of the Ramapo College of New Jersey.
CMS protocol
Animals were subjected to either the CMS protocol or the non-stress protocol (handled daily). Each day, one to three stressors were administered according to a set schedule. The complete regimen lasted 7 days/week for 3 weeks. This protocol is modeled after Willner (2005) in that no individual stressor is considered severe but the unpredictability of the protocol constitutes much of the stress. The stressors were: 1) 5 min swim in 20°C water, 2) cage rotation (social stress), 3) 18-h social isolation with damp bedding, 4) 14-h food deprivation, 14-h water deprivation or 14-h food and water deprivation, 5) 30 min physical restraint, 6) 30 min strobe light exposure and 7) 3-h cage tilt. In a subset of stressed animals, a two-bottle sucrose-to-water preference test was given once a week during a social isolation session. Non-stress control animals were also socially isolated once a week to perform this test. To minimize stress on the control animals, they were acclimated to social isolation prior to testing. Sucrose preference was determined by the equation: 1%-sucrose consumption (mL)/1%-sucrose consumption (mL) + water consumption (mL), where a score of 0.50 indicates equal preference for sucrose and water. In a recent study, we observed that this particular CMS protocol resulted in decreased body weight gain in both male and female animals and reduced sucrose preference in male animals (Reich et al., 2009). These effects are in accordance with the published behavioral effects of CMS protocols (Hill et al, 2005; Willner, 2005).
Apparatus
Experiments were performed using an automated, computerized, fear-conditioning system (Coulbourn Instruments, White Hall, PA, USA). The system consists of three conditioning chambers (30.48 × 25.4 × 30.48 cm) with removable stainless steel grid floors. Footshocks are delivered through the floor grid via a shocker-scrambler unit controlled by custom-designed software (Coulbourn Instruments). Locomotor and freezing activities were monitored through CCD video cameras mounted at the top of each chamber and subsequently analyzed via FreezeFrame software (Coulbourn Instruments). A house light and speaker (1000 Hz tone, 80 dB) is located on the sidewalls of each chamber. In addition, each chamber is contained in a sound-attenuating cubicle equipped with a ventilation fan (60 dB).
Fear conditioning
All fear conditioning procedures were performed in an isolated testing room. Animals were transported in their home cages to the testing room. The cages were covered to blind the animals during transport to and from the colony room.
Acquisition
For trace fear conditioning, each animal had a 120 s baseline acclimation period before receiving three presentations of a tone CS (15 s) followed by a footshock US (2 s, 0.6 mA) that occurred 30 s after the CS offset (i.e. trace period). CS-US presentations were separated by 180 s inter-trial-intervals (ITI). In pilot studies, these trial parameters produced ~40–60% freezing in experimentally-naïve animals. Delay fear conditioning trials were similar to the trace conditioning structure, except the footshock US (2 s, 0.6 mA) co-terminated with the auditory CS (15 s). Two separate protocols were used for contextual fear conditioning: 1) to assess the level of contextual fear conditioning induced by the trace protocol and 2) to assess levels of contextual fear using a more conventional “foreground” contextual conditioning protocol. For the former, footshocks were presented in the same manner as in trace conditioning minus the CS presentations. The “foreground” protocol consisted of placing animals in the chambers for 10 minutes and presenting three shock presentations (2 s, 0.6 mA, 30 s apart) during the last 2 minutes. Immediately after the end of the session, each animal was returned to its home cage in the colony room. Each chamber was then carefully cleaned with Formula 409 cleaner at the end of each session.
Recall/extinction
The strength of the CS-US association was assessed 24 and 48 h after the conditioning sessions. Trace retention/extinction trials consisted of a 60 s baseline acclimation period followed by five trials of CS only presentation at 180 s ITI. These spaced extinction trials were designed to minimize habituation to the altered (smaller, see below) chamber. During Recall Test #1 (24 h after Acquisition), short-term extinction was measured as the difference in freezing between Trial 1 and Trial 5. Long-term extinction was defined as the difference in freezing between Trial 1-Recall Test #2 (48 h after Acquisition) and Trial 1- Recall Test #1. For trace and delay recall/extinction trials, inadvertent contextual conditioning was minimized by altering the chambers during testing sessions. The grid floor was replaced with smooth Plexiglas and another piece of Plexiglas was used to bisect the chambers (Reich et al., 2008). The outer walls were covered in multi-colored construction paper to further eliminate spatial cues. In addition, the chambers were cleaned with orange-scented 409 cleaner and the chamber assignments were shuffled from the previous day. For contextual conditioning recall/extinction, animals were placed in the same unaltered chamber used for conditioning. Once placed in the chamber, freezing was monitored for a 10 minute period.
Pharmacological treatment
Subgroups of stress and non-stress animals were either injected with the CB1 agonist ACEA, N-(2-Chloroethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide (0.1 mg/kg, i.p., Tocris Cookson, St. Louis, MO, USA) or Vehicle (physiological saline and dimethylsulfoxide, 3:1 ratio) solution 30 min prior to experimental procedures. In a preliminary study, we tested the effect of three varying doses of ACEA (0.1 mg/kg, 0.5 mg/kg and 1.0 mg/kg) on the locomotor activity of non-stress control animals. Activity was assessed by placing animals in the conditioning chambers for 15 min. Only the lowest dose of ACEA (0.1 mg/kg) did not significantly affect locomotor activity when compared to vehicle-treated animals; both groups showed ≤1% immobility during the entire test period. A drug-induced increase in immobility may obfuscate changes in freezing behavior; therefore, the 0.1 mg/kg dose was used in subsequent pharmacological experiments.
Behavioral measure and data analysis
Freezing is defined as the absence of all movement except for respiration (Fanselow, 1980), lasting a period ≥ 3 s. For all experimental sessions, time was binned into 15 s intervals and the percentage of time spent freezing was calculated. If an animal had baseline freezing scores ≥ 2.5 standard deviations from the group mean it was removed from further analysis. In this study, no animals were removed from analysis. For the trace and delay recall/extinction trials, freezing scores in 15 s bins were averaged over the first minute following the CS offset. In a previous study, we observed that peak freezing to a CS tone occurs during this 60 s period (Reich et al., 2008). In trace and delay fear recall tests, the first trial was used as an index of recall performance. Contextual recall was analyzed using average freezing during the first minute. According to Rescorla-Wagner learning theory, these trials/time periods reflect the strongest potential for memory recall with later trials/time periods serving as extinction trials (Bouton, 2007). Data analysis (SPSS) was performed by repeated measures, one-way and two-way ANOVAs of group means with Tukey HSD post-hoc, plan-wise comparisons or Student-t-tests (p ≤ 0.05) as indicated.
Results
CMS increases trace but not delay fear memory recall
As mentioned in the Methods section, we observed decreases in body weight gain and reduced sucrose preference in CMS animals (Reich et al., 2009). To ensure our CMS protocol in this study produced similar physiological changes, we measured body weights and sucrose preference in a subset of animals used in the contextual fear experiments. Indeed, as seen in Figure 1, CMS significantly reduced both body weight gain and sucrose preference across the 3-week protocol. These results are consistent with previous studies (Hill et al, 2005; Reich et al., 2009; Willner, 2005).
Figure 1. CMS alters body weight gain and sucrose preference.

(a) CMS significantly reduces body weight gain across the 3-week protocol. Asterisks indicate differences between stress and non-stress animals (p ≤ 0.05).
(b) Stress animals display a significant reduced sucrose preference compared to non-stress controls. * indicate differences between stress and non-stress animals (p ≤ 0.05). Bars represent mean ±SEM.
To assess the effects of CMS on fear acquisition and recall, a memory retention test was performed 24 hours after conditioning. In trace-conditioned animals, a two-way (Group × Trial) repeated measures ANOVA indicated that both stress and non-stress animals displayed significant increases in cued freezing behaviors relative to baseline freezing rates (F (1, 6) = 103.57, p = 0.001, Figure 2(a)). Importantly, CMS significantly increased cued freezing compared to non-stress controls (85.39 ± 4.26% vs 64.31 ± 1.43%, F (1, 6) = 10.30, p = 0.002). No differences in baseline freezing rates were observed between the two groups (stress = 47.93 ± 6.17% vs non-stress = 39.55 ± 3.12%, F (1, 6) = 1.472, p = 0.27).
Figure 2. CMS selectively enhances trace-fear conditioning.

(a) Freezing during the first trial of memory recall (60 s post CS) is significantly greater in stress vs non-stress animals in trace conditioned animals. (b) CMS does not affect freezing in delay fear conditioned animals. Asterisk indicates significant differences between stress and non-stress animals (p ≤ 0.05). Symbols represent mean ±SEM percent freezing.
In trace conditioning procedures, successful memory formation requires hippocampal activation, however amygdala activation is also necessary (LeDoux, 2000). Thus, CMS-induced increases in trace fear conditioning may be attributable to alterations in amygdala activation. To isolate possible effects of CMS on amygdala fear processing, a separate set of stress and non-stress animals underwent delay fear conditioning. Similar to trace-conditioned animals, a 2×2 (Group × Trial) repeated measures ANOVA revealed that both stress and non-stress animals exhibited significant increases in cued freezing compared to baseline freezing (F (1, 6) = 529.48, p = 0.0001, Figure 2(b)). However, no differences were observed between stress and non-stress controls during both cued freezing (62.35 ± 2.05% vs 61.38 ± 90%, F (1, 6) < 1) and baseline freezing (stress = 12.84 ± 3.87% vs. non-stress = 14.81 ± 2.7%, F (1, 6) < 1). Furthermore, no differences were found between the cued freezing rates of both stress and non-stress delay conditioned animals to non-stress trace conditioned animals (F (1, 6) < 1), but significant differences in baseline freezing between delay and trace conditioned animals were observed regardless of stress condition (F (2, 11) = 20.96, p = 0.001). These findings indicate that the trace and delay produced similar levels of cued fear conditioning in non-stress animals, but that trace conditioning produces more generalized (baseline) fear conditioning. It is important to note that all animals received injections of physiological saline prior to conditioning and memory recall protocols. Since injection stress may affect fear conditioning, these injections allow equitable comparisons to the subsequent experiments presented in this paper.
CB1 receptor activation prevents the effects of CMS
CMS produces an approximate 50% reduction in hippocampal CB1 receptors (Hill et al., 2005; Reich et al., 2009). Importantly, Hill et al. (2005) demonstrated that this downregulation of CB1 receptors was accompanied with stressed animals exhibiting difficulty in locating new platform locations (versus the original learned location) in a variant of the Morris water maze. This impairment in behavioral flexibility was rescued by administering the CB1 agonist, HU-210, to the stressed animals. Based on these observations, we hypothesized that exogenous CB1 activation in our stressed animals would rescue the animals from the stress-induced enhancement of fear behavior.
Prior to conditioning, stress animals were split into three groups: those that received the selective CB1 agonist ACEA on acquisition day and vehicle on Recall Test #1 (S_A_V), vehicle on acquisition and ACEA on Recall Test #1 (S_V_A) or vehicle on both acquisition and Recall Test #1 (V_V). Note that neither drug nor vehicle was administered on second Recall Test (see Table 1). A two-way (Group × Trial) repeated-measures ANOVA indicated a significant effect of Trial (F (1, 12) = 237.54, p = 0.001), indicating that all groups froze more during cued freezing compared to baseline levels. No differences were observed during baseline freezing (p > 0.23), signifying that ACEA did not affect basal freezing levels. The ANOVA also yielded a significant interaction effect of the group × trial. Specifically, the S_V_V group froze (75.69 ± 1.24%) more during Trial 1, compared to the S_A_V and S_V_A groups (58.67 ± 1.36%, 65.81 ± 3.63%, respectively; p =0.033, post-hoc analysis; see Figure 3(a)). No differences were observed between these two groups; suggesting that CB1 activation in stress animals during learning itself (S_A_V) or during fear memory recall (S_V_A) weakens the stress-induced fear enhancement. However, only the S_A_V group was comparable to non-stress freezing (55.25 ± 1.08%, p = 0.721), whereas both the S_V_V and S_V_A groups froze significantly more than the non-stress animals (p ≤ 0.01).
Table 1.
ACEA experimental protocol.
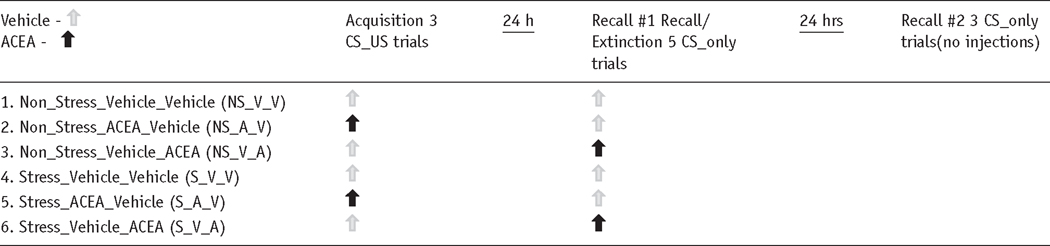
|
Figure 3. The CB1 receptor agonist, ACEA, prevents stress-induced trace-fear enhancement.

(a) In stress animals, administration of ACEA either prior to the Recall Test # 1 (S_V_A) or prior to Acquisition (S_A_V), significantly attenuates the effects of CMS during Trial 1 fear memory recall. * indicates significant higher freezing in the S_V_V group compared to the other three groups (p ≤ 0.05). NS_V_V group is shown for comparison. All groups froze significantly more than baseline but there were no differences among the baseline freezing rates. (b) Conversely, neither baseline nor cued freezing behaviors is significantly affected in ACEA-injected non-stress controls. Symbols represent mean ±SEM percent freezing.
To determine if the ACEA-mediated reduction in freezing is specific to stress animals, we performed a similar experiment in non-stress animals. Three groups of age-matched animals (handled daily): Non-Stress_Vehicle (NS_V_V), Non-Stress_Vehicle_ACEA (NS_V_A) and Non-Stress_ ACEA_Vehicle (NS_A_V) underwent the same fear conditioning acquisition and recall trials with the same dose of ACEA as the stress animals (see Table 1). A one-way ANOVA testing the strength of fear memory during both baseline and first recall trial freezing resulted in no significant differences among the groups (F (2, 9) = 2.81, p = 0.11; Figure 3(b)). These findings suggest that CB1 receptor activation does not affect trace fear acquisition and memory recall in non-stress animals.
Effects of CB1 activation on trace-fear extinction in stress animals
Short-term extinction
During Recall Test #1, effects of CMS and ACEA on short-term extinction were assessed by measuring the difference in freezing behavior between Trial 1 and Trial 5. All groups showed significant short-term extinction (one-way repeated measures ANOVA, F (1, 12) = 91.34, p = .0001). Post-hoc analysis revealed that the S_A_V group froze significantly less than both the S_V_V and NS_V_V groups (35.88 ± 2.98% vs 55.26 ± 3.94% vs 49.52 ± 1.95%, respectively, p ≤ 0.02) (see Figure 4(a)). The S_V_A group displayed freezing levels that were intermediate among the other groups, but freezing was not significantly different from any one group (44.90 ± 4.85%, p = 0.234). Despite the differences between S_A_V and S_V_V animals during Trial 5, the absolute change in freezing from Trial 1 to Trial 5 was similar for both groups (Figure 4(a)). This suggests that CB1 activation in stress animals did not significantly affect short-term extinction rates.
Figure 4. CB1 activation affects long-term, trace-fear extinction.

(a) During Recall Test #1, all groups exhibited significant short-term extinction (less freezing) during Trial 5 compared to Trial 1. S_A_V animals froze significantly less than both the S_V_V and NS_V_V (#). However, extinction rates were similar among groups. * indicate significant differences between trials (p ≤ 0.05). (b) All animals exhibited long-term extinction 24 hours after Recall Test #1. Non-stress animals froze significantly less than S_V_V animals, suggesting CMS results in a more extinction-resistant memory. The S_V_A and NS_V_A groups froze the least compared to the other groups, indicating that ACEA administration during Recall Test 1 may facilitate extinction. * indicate significant differences between trials; # indicates significant difference between S_V_V (and S_A_V) and NS_V_V; ## indicates significant differences among the combined S_V_A and NS_V_A animals and the other groups (p ≤ 0.05). Symbols represent mean ±SEM percent freezing.
Long-term extinction
To test if short-term extinction translated into long-term extinction, we assessed freezing 24 hours following the initial recall testing (48 hours following fear acquisition). A 2×5 (Recall Test × Drug Treatment) repeated-measures MANOVA on first trial freezing resulted in a significant main effect of Recall Test (F(1,15) = 362.49, p = 0.000), indicating that there was a general reduction in freezing from Day 1 (Test #1) to Day 2 (Test #2). Indeed, post-hoc analysis revealed that each group showed significant long-term extinction (see Figure 4(b)). During Recall Test 2, the S_V_V animals froze significantly more than the NS_V_V animals (52.80 ± 2.11% vs.43.27 ± 2.34%, respectively, p = 0.03, independent sample t-test), although the absolute change in freezing rates from Test 1 to Test 2 was ~20% for both groups. Interestingly, both S_V_A and NS_V_A animals that were administered ACEA on Recall Day 1 exhibited the least amount of freezing (27.64 ± 3.56 % and 20.16 ± 3.83, p = 0.000) and the greatest Test 1 to Test 2 change (~40%) compared to the other groups. This indicates that exogenous CB1 activation during extinction learning in both stress and non-stress animals may lead to enhanced performance of future extinction testing.
CMS does not affect contextual fear conditioning
To investigate if CMS modulates other hippocampal-dependent fear memory, contextual fear conditioning was performed in a separate set of experiments. In the first experiment, freezing in the original conditioning chamber (context) was measured 24 hours following trace fear acquisition in stress and non-stress animals. The trace acquisition protocol was used to assess the amount of contextual fear this protocol produces. During the recall test, we measured freezing during the first 60 seconds (trial = 10 minutes) after the animals were placed in the chambers. The CS was never presented during the recall test. As illustrated in Figure 5(a), there were no significant differences between the stress and non-stress animals (19.86 ± 2.29% vs 15.91 ± 3.40%, p = 0.367, paired t-test). Since it was evident that our trace acquisition protocol yielded weak contextual conditioning, we also tested another group of stress and non-stress animals with a more conventional contextual paradigm. In this experiment, acquisition consisted of placing animals in a chamber for 10 minutes with three shocks (0.6 mA, 2 s) spaced 30 seconds apart and occurring during the last 2 minutes. Recall trials were performed 24 hours later and consisted of placing animals into the original conditioning chamber for 5 minutes. Freezing was measured during the initial 60 seconds. Again, we did not observe any significant differences between the stress and non-stress animals (35.42 ± 3.96% vs 36.61 ± 1.27%, p = 0.787, independent-samples t-test; Figure 5(b)), despite the much stronger conditioning. These findings suggest that, in our hands, CMS differentially affects trace and contextual fear conditioning.
Figure 5. CMS does not affect contextual fear conditioning.

(a) Test of contextual memory strength following trace-fear conditioning. (b) Test of contextual memory strength following classical contextual fear conditioning. In either condition, no significant differences were observed between stress and non-stress. Bars represent mean ±SEM percent freezing.
Discussion
The current results demonstrate that chronic mild stress exposure during adolescence increases hippocampal-dependent trace fear conditioning but does not affect contextual or delay fear conditioning. The latter findings are surprising in that chronic restraint stress (CRS) reliably enhances both hippocampal-dependent (contextual) and hippocampal-independent (delay) fear conditioning (Conrad et al., 1999, 2006; Cordero et al., 2003a; Pêgo et al., 2008). Differential effects of CRS and CMS on the hippocampus and the amygdala may explain our observed selective enhancement in trace fear conditioning. For example, CRS (21-days) induces both extensive dendritic atrophy in hippocampal CA3 pyramidal neurons and dendritic hypertrophy in basolateral amygdala (BLA) pyramidal and stellate cells; whereas CMS causes less invasive CA3 dendritic retraction with no effect on BLA pyramidal cells (Pêgo et al., 2008; Vyas et al, 2002). In CRS protocols, the combined atrophy in the hippocampus and hypertrophy in the amygdala may provide a neural substrate for enhanced and more generalized fear conditioning. However, it appears that hippocampal CA3 atrophy, per se, does not lead to enhanced fear conditioning but rather CRS-induced alterations in HPA activity (Conrad, 2006). Specifically, hypertrophy in the amygdala may sensitize BLA pyramidal neurons to increases in HPA-driven glucocorticoid output and this convergence may lead to enhanced fear conditioning (Conrad, 2006). Therefore, CRS appears to be a more invasive stress model than CMS and thus produces a generalized enhancement in aversive learning. It is worth noting that Henningsen et al. (2009) demonstrated that a more intense (7 week) CMS protocol enhances contextual fear conditioning in male rats. Given that our study is the first to report an effect of chronic stress (regardless of the protocol) on trace fear conditioning, it is unknown if CRS or prolonged CMS protocols would also affect freezing in this paradigm. Nonetheless, our findings suggest that a relatively mild CMS protocol (3 weeks) selectively enhances trace fear conditioning. The age of the animals is another important consideration; the previously discussed studies studied adult animals (> 65 days old) while this study investigated effects of CMS exposure during the mid-to-late adolescent period (~40–65 days old) animals. Thus, an alternative hypothesis is that CMS produces varying levels of anatomical and behavioral changes depending on age of exposure.
Evidence from several studies suggests that CMS protocols, similar to the current one, alter CB1 receptor signaling in the hippocampus, hypothalamus, prefrontal cortex, ventral striatum and nucleus accumbens but do not affect CB1 signaling in the amygdala (Hill et al., 2005; 2008; Reich et al., 2009; Wang et al., 2010). These effects are observed in both younger animals and in adult animals. In contrast, CRS protocols affect CB1 signaling in the amygdala (Patel et al., 2009; Sumislawski et al., 2011) in addition to the hippocampus (Hu et al., 2011) and the hypothalamus (Wamsteeker et al., 2010). Interestingly, both CMS and CRS protocols enhance HPA axis activity; however CRS, partially due to enhanced endocannabinoid (eCB) signaling in the amygdala, eventually results in habituation of the stress response (Hill et al., 2010). Conversely, CMS leads to non-habituating activation of the HPA axis (Grippo and Johnson, 2009; Marin et al., 2007). Since the hippocampus acts as an ancillary brake in the HPA axis feedback loop, CB1 downregulation may comprise this hippocampal function; providing the scenario for increased responses to stress, anxiety and fear. CMS-CB1 impairment may preferentially affect circuitry recruited during trace (episodic) conditioning; hence sparing contextual conditioning. Buttressing this idea, pharmacological blockade of CB1 either enhances or reduces tone-signaled contextual fear responses (Arenos et al., 2006; Reich et al., 2009; Sink et al, 2010; although see Pamplona and Takahashi, 2006) but it does not affect unsignaled contextual fear responses (Arenos et al., 2006; Sink et al, 2010). These observations collectively lend further support for dual dichotomies in 1) the neurobehavioral effects of CMS and CRS protocols and 2) CB1 modulation of fear responses.
Based on previous studies that CB1 antagonism/genetic-deletion increases fear and anxiety in both animals and humans (Reich et al., 2008; for review see Moreira and Wotjak, 2010), we hypothesized that CMS-induced downregulation of hippocampal CB1 receptors results in enhanced fear and that exogenous activation of the remaining receptors rescues the stress animals. Indeed, administration of ACEA to stress animals either prior to acquisition or prior to memory recall prevented the stress-enhanced freezing but did not affect freezing in non-stress animals. These results are consistent with data showing that exogenous activation of CB1 receptors in the hippocampus improves behavioral flexibility in the Morris water maze in CMS-exposed animals (Hill et al, 2005). An important caveat is that since ACEA was administered systemically, contributions to the aversive learning and extinction (see below) processes by other fear circuitry nuclei cannot be dismissed. Particularly, the amygdala and ventromedial prefrontal cortex (vmPFC) are considered necessary for fear processing and both contain rich densities of CB1 receptors (LeDoux, 2000; Lisboa et al., 2010; Marsicano et al., 2002; Nieuwenhuis and Takashima, 2011). Isolating and identifying all the CB1-containing nuclei that contribute to stress-induced fear regulation is beyond the scope of the current paper. Future studies will systematically focus on this task.
Endocannabinoids are heavily implicated in modulating the extinction of aversive memories. For example, either pharmacological blockade or genetic deletion of CB1 receptors impairs the ability of animals to extinguish an aversive memory (Chhatwal et al., 2005; Marsicano et al, 2002; Reich et al., 2008; Suzuki et al., 2004). In this study, we observed that CMS, which reduces hippocampal CB1 receptor density (Hill et al., 2005; Reich et al., 2009) appears to impair long-term (24 hour) extinction when comparing raw Recall Test #2 freezing levels. However, comparing the absolute change in freezing rates from Recall Test #1 to Recall Test #2 indicates that both stress and non-stress animals extinguish ~20% of Recall Day 1 rates. This suggests that CMS does not modulate extinction directly, but rather it creates an initial stronger fear memory (i.e. a higher starting level) which may require more extinction training to achieve the same degree of extinction as non-stress animals. Stress and non-stress animals injected with ACEA during extinction learning exhibited the lowest freezing levels and the highest absolute change in freezing between the two recall days (~40%) among the groups. These findings agree with several studies showing that CB1 activation enhances extinction memory (Chhatwal et al., 2005; Marsicano et al., 2002; Pamplona et al., 2006; Suzuki et al., 2004) but also argue that despite CMS enhancing trace fear memory acquisition and recall, it may not alter then neural process of extinction learning. Thus, cannabinoid regulation of extinction is not modulated by stress.
Since ACEA is reported to activate the endovannilloid transient receptor protein (TRPV1) as well as CB1 (Huang et al., 2002; Price et al., 2004; Silveira et al., 2010; Smart et al., 2000; Zygmunt et al., 1999), antagonism of CB1 would be a logical control. Although, the fact that CB1 antagonism increases both fear (Reich et al, 2008) and anxiety (Moreira et al., 2012; Moreira and Wotjak, 2010) it precluded the use of CB1 antagonists in this study. That is, the effects of stress and CB1 antagonism would have occluded each other and thus create a confound. Importantly, several lines of evidence demonstrate that activation of brain TRPV1 increases fear and anxiety (reviewed in Moreira et al., 2012). This action is in stark contrast to the CB1 activation suggesting that the endocannabinoid and endovannilloid regulate diametrically actions on fear and anxiety (Moreira et al., 2012; Moreira and Wotjak, 2010). These behavioral differences are paralleled with differences in synaptic physiology: CB1 activation decreases neurotransmitter release, whereas TRPV1 activation increases neurotransmitter release (Moreira et al., 2012). Therefore, in the current study, if ACEA was acting on TRPV1 rather than CB1, either an increase in freezing in both stress and non-stress animals would have been observed or stress-induced increases in freezing would have been occluded. Moreover, ACEA only activates TRPV1 at higher concentrations. The observation that the current ACEA dose (0.1 mg/kg) only affected fear in stress animals and did not increase fear or immobility in non-stress controls argues against TRPV1 involvement.
In summary, we present three novel findings: 1) CMS, an animal model for major depression, selectively enhances hippocampal-dependent trace fear conditioning, 2) CMS-induced increase in fear learning and memory recall is prevented by exogenous CB1 receptor activation and 3) CB1 activation enhances long-term extinction in both stress and non-stress animals suggesting that cannabinoid modulation of CMS-induced fear enhancement is independent of cannabinoid regulation of fear extinction. These findings suggest that a depressed state induced by uncontrollable stress increases the capacity for aversive learning. Importantly, this stress-induced modulation of fear appears to be gated by the activation of CB1 receptors. Disturbances in autobiographical (episodic) memory are well documented in PTSD (see Dere et al., 2010 for review). The observed selective CMS-induced increase in trace fear conditioning, a model of episodic memory, may serve as a valuable tool in elucidating the neurobiological mechanisms of how stress during adolescence may increase the risk of developing depression and PTSD (Kendler et al., 2000; Kessler et al., 2001).
Acknowledgments
We thank Gregory Mihalik, Cydney Mitchell, Amanda Swanson, Philip Sims, Timur Petrishin, Wendy Levine, Tayla Cacchione and Isabelle Weishaar for their amazing technical assistance. We also thank Michele Reich for proofreading.
Funding
This work was supported by National Institutes of Mental Health: grants RO3 MHO79294-01 and R15 MH085280-01 to Dr. Reich.
Footnotes
Reprints and permissions: sagepub.co.uk/journalsPermissions.nav
Conflict of interest
The authors declare no conflict of interest.
References
- Arenos JD, Musty RE, Bucci DJ. Blockade of cannabinoid CB1 receptors alter contextual learning and memory. Eur J Pharmacol. 2006;539:177–183. doi: 10.1016/j.ejphar.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Bouton M. Learning and Behavior: A Contemporary Synthesis. Sunderland, MA: Sinauer; 2007. [Google Scholar]
- Campbell S, MacQueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatr Neurosci. 2004;29:417–426. [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Davis M, Maguschak KA, et al. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- Clark RE, Manns JR, Squire LR. Trace and delay eyeblink conditioning: Contrasting phenomena of declarative and nondeclarative memory. Psychol Sci. 2001;12:304–308. doi: 10.1111/1467-9280.00356. [DOI] [PubMed] [Google Scholar]
- Conrad CD. What is the functional significance of chronic stress-induced CA3 dendritic retraction within the hippocampus? Behav Cogn Neurosci Rev. 2006;1:41–60. doi: 10.1177/1534582306289043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD, LeDoux JE, Magarinos AM, et al. Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav Neurosci. 1999;113:902–913. doi: 10.1037//0735-7044.113.5.902. [DOI] [PubMed] [Google Scholar]
- Cordero MI, Kruyt ND, Sandi C. Modulation of contextual fear conditioning by chronic stress in rats is related to individual differences in behavioral reactivity to novelty. Brain Res. 2003a;970:242–245. doi: 10.1016/s0006-8993(03)02352-7. [DOI] [PubMed] [Google Scholar]
- Cordero MI, Venero C, Kruyt ND, et al. Prior exposure to a single stress session facilitates subsequent contextual fear conditioning in rats. Evidence for a role of corticosterone. Horm Behav. 2003b;44:338–345. doi: 10.1016/s0018-506x(03)00160-0. [DOI] [PubMed] [Google Scholar]
- Dere E, Pause BM, Pietrowsky R. Emotion and episodic memory in neuropsychiatric disorders. Behav Brain Res. 2010;215:162–171. doi: 10.1016/j.bbr.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15:177–182. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- Grippo AJ, Johnson AK. Stress, depression and cardiovascular dysregulation: A review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress. 2009;12:1–21. doi: 10.1080/10253890802046281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningsen K, Andreasen JT, Bouzinova EV, et al. Cognitive deficits in the rat chronic mild stress model for depression: Relation to anhedonic-like responses. Behav Brain Res. 2009;198:136–141. doi: 10.1016/j.bbr.2008.10.039. [DOI] [PubMed] [Google Scholar]
- Herman JP, Ostrander MM, Mueller NK, et al. Limbic system mechanisms of stress regulation: Hypothalamo-pituitary-adreno-cortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Hill MN, Patel S, Campolongo P, et al. Functional interactions between stress and the endocannabinoid system: From synaptic signaling to behavioral output. J Neurosci. 2010;30:14980–14986. doi: 10.1523/JNEUROSCI.4283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB. The endocannabinoid system and the treatment of mood and anxiety disorders. CNS Neurol Disord Drug Targets. 2009;6:451–458. doi: 10.2174/187152709789824624. [DOI] [PubMed] [Google Scholar]
- Hill MN, Carrier EJ, McLaughlin RJ, et al. Regional alterations in the endocannabinoid system in an animal model of depression: Effects of concurrent antidepressant treatment. J Neurochem. 2008;106:2322–2336. doi: 10.1111/j.1471-4159.2008.05567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Patel S, Carrier EJ, et al. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30:508–515. doi: 10.1038/sj.npp.1300601. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zhang M, Czéh B, et al. Chronic restraint stress impairs endocannabinoid mediated suppression of GABAergic signaling in the hippocampus of adult male rats. Brain Res Bull. 2011;85:374–379. doi: 10.1016/j.brainresbull.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167:648–662. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Thornton LM, Gardner CO. Stressful life events and previous episodes in the etiology of major depression in women: An evaluation of the “kindling” hypothesis. Am J Psychiatry. 2000;157:1243–1251. doi: 10.1176/appi.ajp.157.8.1243. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Avenevoli S, Ries Merikangas K. Mood disorders in children and adolescents: An epidemiologic perspective. Biol Psychiatry. 2001;49:1002–1014. doi: 10.1016/s0006-3223(01)01129-5. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. Linking molecules to mood: New insight into the biology of depression. Am J Psychiatry. 2010;167:1305–1320. doi: 10.1176/appi.ajp.2009.10030434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Lisboa SF, Reis DG, da Silva AL, et al. Cannabinoid CB1 receptors in the medial prefrontal cortex modulate the expression of contextual fear conditioning. Int J Neuropsychopharmacol. 2010;9:1163–1173. doi: 10.1017/S1461145710000684. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Marin MT, Cruz FC, Planeta CS. Chronic restraint or variable stresses differently affect the behavior, corticosterone secretion and body weight in rats. Physiol Behav. 2007;90:29–35. doi: 10.1016/j.physbeh.2006.08.021. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Glucocorticoids, depression, and mood disorders: Structural remodeling in the brain. Metabolism. 2005;54:20–23. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Moreira FA, Aguiar DC, Terzian AL, et al. Cannabinoid type 1 receptors and transient receptor potential vanilloid type 1 channels in fear and anxiety-two sides of one coin? Neuroscience. 2012;204:186–192. doi: 10.1016/j.neuroscience.2011.08.046. [DOI] [PubMed] [Google Scholar]
- Moreira FA, Wotjak CT. Cannabinoids and anxiety. Curr Top Behav Neurosci. 2010;2:429–450. doi: 10.1007/7854_2009_16. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuis IL, Takashima A. The role of the ventromedial prefrontal cortex in memory consolidation. Behav Brain Res. 2011;218:325–334. doi: 10.1016/j.bbr.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Nopoulos P, Flaum M, Andreasen NC. Sex differences in brain morphology in schizophrenia. Am J Psychiatry. 1997;154:1648–1654. doi: 10.1176/ajp.154.12.1648. [DOI] [PubMed] [Google Scholar]
- Pamplona FA, Takahashi RN. WIN 55212–2 impairs contextual fear conditioning through the activation of CB1 cannabinoid receptors. Neurosci Lett. 2006;397:88–92. doi: 10.1016/j.neulet.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Pamplona FA, Prediger RD, Pandolfo P, et al. The cannabinoid receptor agonist WIN 55,212–2 facilitates the extinction of contextual fear memory and spatial memory in rats. Psychopharmacology (Berl) 2006;188:641–649. doi: 10.1007/s00213-006-0514-0. [DOI] [PubMed] [Google Scholar]
- Patel S, Kingsley PJ, Mackie K, et al. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pêgo JM, Morgado P, Pinto LG, et al. Dissociation of the morphological correlates of stress-induced anxiety and fear. Eur Neurosci. 2008;27:1503–1516. doi: 10.1111/j.1460-9568.2008.06112.x. [DOI] [PubMed] [Google Scholar]
- Price TJ, Patwardhan A, Akopian AN, et al. Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyl-dopamine and arachidonyl-2-chloroethylamide. Br J Pharmacol. 2004;141:1118–1130. doi: 10.1038/sj.bjp.0705711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau V, DeCola JP, Fanselow MS. Stress-induced enhancement of fear learning: An animal model of posttraumatic stress disorder. Neurosci Biobehav Rev. 2005;29:1207–1223. doi: 10.1016/j.neubiorev.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Reich CG, Mohammadi M, Alger BE. Endocannabinoid modulation of fear responses: Learning performance and state-dependent effects. J Psychopharmacol. 2008;22:769–777. doi: 10.1177/0269881107083999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich CG, Taylor ME, McCarthy MM. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav Brain Res. 2009;203:264–269. doi: 10.1016/j.bbr.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C, Merino JJ, Cordero MI, et al. Effects of chronic stress on contextual fear conditioning and the hippocampal expression of the neural cell adhesion molecule, its polysialylation, and L1. Neuroscience. 2001;102:329–339. doi: 10.1016/s0306-4522(00)00484-x. [DOI] [PubMed] [Google Scholar]
- Silveira PE, Silveira NA, de Morini VC, et al. Opposing effects of cannabinoids and vanilloids on evoked quantal release at the frog neuromuscular junction. Neurosci Lett. 2010;473:97–101. doi: 10.1016/j.neulet.2010.02.026. [DOI] [PubMed] [Google Scholar]
- Sink KS, Segovia KN, Collins LE, et al. The CB1 inverse agonist AM251, but not the CB1 antagonist AM4113, enhances retention of contextual fear conditioning in rats. Pharmacol Biochem Behav. 2010;95:479–484. doi: 10.1016/j.pbb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Gunthorpe MJ, Jerman JC, et al. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br J Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumislawski JJ, Ramikie TS, Patel S. Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: A potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology. 2011;36:2750–2761. doi: 10.1038/npp.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, et al. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, et al. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–11196. doi: 10.1523/JNEUROSCI.1046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Sun D, Pan B, et al. Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology. 2010;35:2249–2261. doi: 10.1038/npp.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P. Chronic mild stress (CMS) revisited: Consistency and behavioural neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52:90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- Wong ML, Kling MA, Munson PJ, et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: Relation to hypercortisolism and corticotropin-releasing hormone. PNAS. 2000;97:325–330. doi: 10.1073/pnas.97.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HC, Cao X, Das M, et al. Behavioral animal models of depression. Neurosci Bull. 2010;26:327–337. doi: 10.1007/s12264-010-0323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]