

Abstract
BACKGROUND
Prostate cancer remains a significant health problem for men in the Western world. Although treatment modalities are available, these do not confer long-term benefit and are accompanied by substantial side effects. Adoptive immunotherapy represents an attractive alternative to conventional treatments as a means to control tumor growth.
METHODS
To selectively target the tumor-expressed form of Muc1 we constructed a retroviral vector encoding a chimeric antigen receptor (CAR) directed against the aberrantly-expressed extracellular portion of Muc1 called the ‘variable number of tandem repeats’.
RESULTS
We now demonstrate that T cells can be genetically engineered to express a CAR targeting the tumor-associated antigen Muc1. CAR-Muc1 T cells were able to selectively kill Muc1-expressing human prostate cancer cells. However, we noted that heterogeneous expression of the Muc1 antigen on tumor cells facilitated immune escape and the outgrowth of target-antigen loss variants of the tumor. Given the importance of androgen ablation therapy in the management of metastatic prostate cancer, we therefore also tested the value of combining conventional (anti-androgen) and experimental (CAR-Muc1 T cells) approaches. We show that CAR-Muc1 T cells were not adversely impacted by anti-androgen therapy and subsequently demonstrate the feasibility of combining the approaches to produce additive anti-tumor effects in vitro.
CONCLUSIONS
Adoptive transfer of CAR-Muc1 T cells alone or in combination with other luteinizing hormone-releasing hormone analogs or antagonists should be tested in human clinical trials.
Keywords: immunotherapy, CAR T cells, tumor immune escape, combination therapy
INTRODUCTION
Prostate cancer is the second highest cause of cancer associated death among men in the United States (Cancer statistics, 2012, DOI: 10.3322/caac.20138) and efforts to develop effective and safe curative therapies for men with advanced/metastatic disease are an area of active investigation. Recently, interest in the use of the immune system to treat advanced prostate cancer has been boosted by the availability of a commercial vaccine to treat men with advanced prostate cancer,1–3 and other immunological approaches are now being tested, including the adoptive transfer of tumor-directed T cells.4,5
We and others have shown that the infusion of T cells, made tumor specific by expression of a transgenic chimeric antigen receptor (CAR), can effectively treat even disseminated malignancies, including neuroblastoma and B-cell tumors.6–8 These CAR-T cells combine the antigen-binding properties of a monoclonal antibody (the CAR element) with the lytic capacity of the T cells by which they are expressed, thus allowing modified (that is, transgenic) T cells to recognize both protein and non-protein antigens on tumor cells and kill targets in an HLA-independent fashion.9
In the current study, we sought to extend the use of such engineered CAR-T-cell therapy to the treatment of prostate cancer. We identified Mucin 1 (Muc1) as a potential target antigen for CAR-T cells.10–13 Muc1 is a type I transmembrane glycoprotein, which is overexpressed in an aberrantly-glycosylated form by many tumors including prostate, breast, colon, lung, gastric and pancreatic cancer (Supplementary Figure 1), while having limited expression on normal tissues.14–18 Moreover, in prostate cancer, Muc1 expression has been correlated with poor prognosis and an increased risk of disease recurrence,19 thus highlighting its relevance as a potential therapeutic target.
To selectively target the tumor-expressed form of Muc1 we constructed a retroviral vector encoding a CAR directed against the aberrantly-expressed extracellular portion of Muc1 called the ‘variable number of tandem repeats’.20,21 In vitro expanded T cells genetically modified to express this Muc1-directed CAR have no detectable activity against non-malignant tissue,22 but efficiently kill Muc1-expressing human prostate cancer cells.22 Unfortunately, expression of Muc1, like that of many other tumor-associated antigens, is heterogeneous and fluctuates, and a common reason for the failure of immunotherapy is the selection of target-antigen loss variants of the tumor. Given the importance of androgen ablation therapy in the management of metastatic prostate cancer, we therefore also tested the value of combining our immunotherapy with Flutamide, an androgen receptor antagonist that spares T cells.23–25 Although CAR-T cells or anti-androgen therapy alone were unable to produce tumor elimination, the combination approach proved additive in our pre-clinical model. This synergy between effector T cells and androgen receptor antagonists should be readily testable in human subjects.
MATERIALS AND METHODS
Donors and cell lines
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers with informed consent on an IRB-approved protocol. The prostate cancer cell lines PC3, LNCaP, DU145 and Human embryonic kidney cell line 293T, were obtained from the American Type Culture Collection (Rockville, MD, USA). Cells were maintained in a humidified atmosphere containing 5% carbon dioxide (CO2) at 37 °C. Tumor cells lines were maintained in complete IMDM (Gibco BRL Life Technologies, Gaithersburg, MD, USA) containing 10% heat-inactivated fetal bovine serum (Hyclone, Waltham, MA, USA), 2 mM L-glutaMAX, 200 IU/ml penicillin and 200 μg/ml streptomycin (all from Gibco BRL Life Technologies).
OKT3/CD28 blast generation
To generate OKT3 blasts, PBMCs were activated with OKT3 (1 mg/ml) (Ortho Biotech, Bridgewater, NJ, USA) and CD28 (1 mg/ml) (Becton Dickinson, Mountain View, CA, USA) antibodies and plated in a non-tissue culture-treated 24-well plate at 1 ×106 PBMCs per 2 ml complete media (RPMI 1640; Gibco BRL Life Technologies) containing 45% Clicks medium (Irvine Scientific, Santa Ana, CA, USA), 10% fetal bovine serum and 2 mM L-glutaMAX. The cells were supplemented with recombinant human interleukin-2 (IL2) (100 U/ml, NIH, Bethesda, VA, USA) on day 1 after activation, and subsequently split and fed with fresh media plus IL2 (50 U/ml).
Generation of retroviral constructs and retroviral transduction
We synthesized (DNA 2.0, Menlo Park, CA, USA) a codon-optimized single-chain variable fragment of Muc1 based on published sequences.22 The scFv fragment was cloned in frame with the human IgG1-ch2ch3 domain and with the ζ-chain of the T-cell receptor (TCR)/CD3 complex in the SFG retroviral backbone.26 We also synthesized (DNA 2.0) the Muc1 antigen based on published sequences.27 The fluorescent marker mOrange was incorporated into the Muc1 antigen construct using an IRES element and a control retroviral vector encoding green fluorescence protein (GFP) was also generated.
Retroviral supernatant was produced using 293T cells, which were co-transfected with the CAR-Muc1, Muc1-mOrange or GFP retroviral vectors, the Peg-Pam-e plasmid containing the sequence for MoMLV gag-pol, and the RDF plasmid containing the sequence for the RD114 envelope, using the Fugene6 transfection reagent (Roche Diagnostics Corporation, Indianapolis, IN, USA), according to the manufacturer’s instructions. Retroviral supernatant was collected at 48 and 72 h post-transfection, filtered (using a 0.45-μm filter) and stored at −80 °C.
T-cell transduction
For T-cell transduction the CAR-Muc1 retroviral supernatant was plated in a non-tissue culture-treated 24-well plate (1 ml per well) pre-coated with a recombinant fibronectin fragment (FN CH-296; Retronectin; Takara Shuzo, Otsu, Japan). OKT3/CD28-activated PBMCs (0.2 ×106 per ml) were resuspended in complete media supplemented with IL2 (100 U/ml) and added to the non-tissue culture-treated 24-well plates (1 ml per well), which was then transferred to the 37 °C, 5% CO2 incubator. Every 3 days cells were fed with complete media supplemented with IL2 (50 U per ml). CAR expression on T cells was measured 72 h post-transduction by flow cytometry.
Tumor cell transduction
For transduction, Muc1-mOrange or GFP viral supernatant was plated in a non-tissue culture-treated 24-well plate (1 ml per well), that had been pre-coated with a recombinant fibronectin fragment. Tumor cells were resuspended at 0.2 ×105 per ml in complete IMDM, 1 ml was added to the supernatant-containing wells, and the plate was transferred to the 37°C, 5% CO2 incubator. Expression of mOrange or GFP was measured 72 h post-transduction by flow cytometry. Cells were maintained or expanded in complete media every 3–4 days.
Cell sorting
293T cells transduced to express either Muc1-mOrange or GFP were harvested, counted, strained through 70 μm-cell strainers and resuspended in phosphate-buffered saline (PBS). The cells were sorted based on GFP and mOrange expression using a Beckman Coulter MoFlo (Brea, CA, USA). Sorted cells were cultured in complete IMDM supplemented with 200 IU/ml penicillin, 200 μg/ml streptomycin and gentamicin (2.5 μg/ml) (Gibco BRL Life Technologies) for 1 week in a six-well plate, then further expanded in a flask T175 using complete media which was replenished every 3–4 days.
Immunohistochemistry (IHC)
Tumor cells were resuspended at a density of 1 ×106 in 1 ml of PBS and 200 μl of the suspension was loaded into appropriate slots in the cytospin. After a 5-min spin at 400 g the filters were removed from their slides and the slides were examined to ensure that cells were dispersed homogenously on a flat layer. The slides were then placed in a steamer for 10 min (high pressure) in Target Retrieval solution (Dako, Carpenteria, CA, USA), washed once with 1x PBS, then immersed in 3% hydrogen peroxide solution for 5–10 min to quench endogenous peroxidase and subsequently blocked with avidin/biotin (Vectastain, Burlingame, CA, USA). The slides were washed with PBS, then incubated in pre-block/diluent for 15 min, after which they were incubated with mouse anti-human Muc1 antibody (no. 4538 VU4H5, Cell Signaling Technology, Danvers, MA, USA) diluted 1:50 in PBS/1% bovine serum albumin for 1 h at room temperature. Cells were washed three times with PBS and then incubated with a biotin-labeled anti-mouse secondary antibody to detect positive cells. After washing with PBS an enzymatic conversion was performed using the chromogenic substrate 3,3 diaminobenzidine. Subsequently we performed a counterstain in hematoxylin for 30 s, dehydrated the slides in 50, 75, 95 and 100% ethanol and then mounted with Permount. Cells were scored by evaluating antigen expression (0 =negative, 1 =up to 10% positive cells, 2 =11–50%, 3 =51–80%, 4 =>80%) and intensity (0 =negative, 1 = weakly positive, 2 =moderately positive, 3 =strongly positive).
Proliferation assay
To evaluate whether Flutamide exposure affected the proliferation of CAR-Muc1 T cells, CAR-T cells were plated at 1 ×105 cells per well in a 96-well U-bottomed plate with serial dilutions of human IL2 ranging from 10–160 U/ml with or without 10 μM of Flutamide. After 72 h, the cells were pulsed with 1 μCi methyl-3[H]thymidine (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and cultured for an additional 15 h. The cells were then harvested onto filters and dried, and cpm measured in a β-scintillation counter (TriCarb 2500TR; Packard BioScience, Meriden, CT, USA). Each condition was tested in triplicate.
Cytotoxicity
To assess the cytolytic specificity and function of non-transduced (NT) and CAR-Muc1 T cells we used either a short-term (6 h) Cr51 release assay or a 3-day co-culture assay.
Chromium release assay
The cytotoxicity specificity of effector T-cell populations was measured in a standard 6 h Cr51 release assay, using E:T ratios ranging from 40:1–5:1. The targets tested include PC3, DU145, CAPAN1, MCF7, LNCaP and 293T cells. Target cells incubated in media alone or in 1% Triton X-100 (Sigma-Aldrich, St Louis, MO, USA) were used to determine spontaneous and maximum Cr51 release, respectively. The mean percentage of specific lysis of triplicate wells was calculated as follows: ((test counts − spontaneous counts)/(maximum counts − spontaneous counts)) ×100%.
Co-culture experiments
The cell lines PC3, DU145, LNCaP, 293T-Muc1-mOrange and 293T-GFP were used as targets. Briefly, PC3 cells or engineered 293T cells were cultured with either NT or CAR-modified T cells at a 1:10 ratio in the presence of IL2 (50 U/ml) in complete media. For our combination therapy approach LNCaP cells were cultured with either NT or CAR-modified T cells at a 1:2 ratio in the presence of IL2 (50 U/ml) in complete media. After 72 h all residual cells were collected, counted, stained with a monoclonal antibody to distinguish T cells (CD3) from tumor cells (GFP/mOrange) and then analyzed by flow cytometry (Gallios; Beckman Coulter).
Immunophenotyping
NT and CAR-modified T cells were surface-stained with monoclonal antibodies to: CD3, CD4, CD8, CD14, CD56, CD27, CD45RO and CD62L (Becton Dickinson BD, Franklin Lakes, NJ, USA). Cells were washed once with PBS (Sigma, St Louis, MO, USA) supplemented with 2% fetal bovine serum, pelleted, and antibodies added in saturating amounts (10 μl). After 15 min incubation at 4 °C in the dark, cells were washed twice and analyzed. To detect CAR-transduced cells, T cells were stained with a monoclonal antibody Fc-specific cyanine-Cy5-conjugated (FcgCy5) antibody (Jackson Immuno Research Laboratories, West Grove, PA, USA), which recognizes the IgG1-ch2ch3 component of the CAR. At least 10 000 live cells from each population were analyzed using a Gallios Flow cytometer and the data analyzed using Kaluza software (Beckman Coulter).
AnnexinV-7-AAD staining
To determine the percentage of apoptotic and necrotic cells in our Flutamide-exposed T-cell cultures we performed Annexin-7-AAD staining, as per manufacturers’ instructions (BD Pharmingentm no. 559763, San Diego, CA, USA). Briefly, CAR-Muc1 T cells were cultured with or without 10 μM of Flutamide for 12, 24 and 48 h, then harvested, washed with cold PBS, resuspended in 1 × binding buffer at a concentration of 1 × 106 cells ml−1, and stained with Annexin V-PE and 7-AAD for 15 min at room temperature (25 °C) in the dark. Subsequently, the cells were analyzed by flow cytometry.
Treatment of T cells and tumor cell lines with Flutamide
LNCaP (5 ×105) cells were plated in complete IMDM and left untreated or were treated with Flutamide (SIGMA F9397) (10 or 20 μM) for three consecutive days. Subsequently, residual tumor cells were quantified by flow cytometry using CountBright microbeads (Invitrogen, Grand Island, NY, USA), according to manufacturers’ instructions. OKT3 blasts from three donors were plated at a cell density of 1 ×106 per well in a 24-well plate and were left untreated or received Flutamide (10 μM) for three consecutive days. Cell expansion was assessed by cell counting using trypan blue exclusion.
Treatment of tumor cell lines with a combination of CAR-Muc1 T cells and Flutamide
LNCaP-mOrange (5 ×105) were plated in complete media on day 0 and either (i) left untreated for 7 days, or (ii) received three consecutive doses of Flutamide (10 μM) on days 1, 2, and 3, or (iii) were treated with CAR-Muc1 T cells on day 4 (2 T cells:1 tumor cell), or (iv) received combination therapy with Flutamide (days 1–3) followed by CAR-Muc1 T-cell treatment on day 4. On day 7 of culture all residual cells were collected, counted, stained with a monoclonal antibody to distinguish T cells (CD3) from tumor cells (mOrange) and then quantified using microbeads. Cells were acquired using a Gallios Flow cytometer and the data analyzed using Kaluza software (Beckman Coulter).
Statistical analysis
All data are presented as mean±s.d. Student’s t-test was used to determine the statistical significance of differences between samples, and P<0.05 was accepted as indicating a significant difference.
RESULTS
T cells can be engineered to recognize and kill prostate cancer cells expressing Muc1
Muc1 is a type I glycoprotein, which is aberrantly overexpressed in human prostate cancer. Expression is associated with more aggressive pathological and clinical features, highlighting its importance as a potential immunotherapeutic target.11,12,15,17–19 To redirect the T-cell response against Muc1-positive prostate cancer cells, we generated a CAR targeting the extracellular portion of Muc1 called the ‘variable number of tandem repeats’. We synthesized and codon-optimized the Muc1 single-chain variable fragment for homosapien expression.22 This was subsequently cloned in frame with the ch2ch3 region of IgG1, the transmembrane domain of CD28, and the ζ-chain of the TCR/ CD3 complex and incorporated into the SFG retroviral vector backbone26,28 (Figure 1a). T cells obtained from four normal donors were activated using anti-CD3/CD28, expanded in the presence of IL2 (50 U/ml), and retrovirally transduced on day 3 of culture with CAR-Muc1. Figure 1b shows a schematic representation of protein expression on the surface of transgenic T cells. Transduction efficiency was assessed by flow cytometric analysis using an antibody directed against the extracellular ch2ch3 portion of the CAR. Sixty six percent of T cells (range 49.5–85.2%) expressed CAR-Muc1; CD4 +(helper) and CD8 +(cytotoxic) T cells were transduced at a similar rate (data not shown). Transduction data from a representative donor are shown in Figure 1c where 85.2% of T cells transgenically expressed CAR-Muc1 after transduction. To evaluate the cytolytic function of CAR-Muc1 T cells, we cultured the prostate cancer cell line, PC3 (Muc1 positive) with control (NT) or CAR-Muc1 transgenic T cells. Figure 1d shows target-specific lysis of PC3 as well as other Muc1-positive tumor cell lines, DU145 (prostate cancer), CAPAN1 (pancreatic cancer), and MCF7 cells (breast cancer), by CAR-Muc1 T cells (55±3, 45±7, 55±8, 39±6% killing, respectively, at an effector:target (E:T) ratio of 40:1) with minimal activity against Muc1-negative control 293T target cells (2±2%, E:T 40:1) in a 6-h Cr51 release assay. As expected, NT T cells had minimal cytotoxicity against the same targets (6±3, 6±3, 11±5, 9±6 and 1±2% killing at E:T 40:1, respectively) (n =3). Hence, CAR-Muc1 T cells specifically kill Muc1-positive tumor cells. However, in this short-term (6 h) in vitro killing assay we did not observe 100% tumor lysis, likely reflecting the fact that not all tumor cells express Muc1 antigen.
Figure 1.

T cells can be engineered to recognize and kill prostate cancer cells expressing Muc1. (a) Shows the retroviral vector map of the CAR-Muc1 construct. (b) Shows a schematic representation of the CAR-Muc1 construct on the surface of transgenic T cells. (c) Shows transduction efficiency of CAR-Muc1 transduced T cells from a representative donor as evaluated by flow cytometry using an antibody directed against the CH2CH3 region of the retroviral construct. (d) Shows that CAR-Muc1 T cells kill the Muc1-expressing prostate cancer cell lines PC3 and DU145, as well as CAPAN1 (pancreatic cancer cell line) and MCF7 (breast cancer cell line), both of which also express Muc1, with negligible activity against Muc1-negative 293T cells. Cytotoxic activity was evaluated in a standard 51Cr-release assay, and results are shown at an E:T ratio of 40:1. Data represent the mean±s.d. of four donors.
Tumor immune escape due to heterogeneous tumor antigen expression
To investigate the cytolytic activity of these CAR-Muc1 T cells in a longer co-culture assay we engineered PC3 tumor cells to express GFP and cultured them with either control (NT) or transgenic T cells (1:10) for 3 days. On day 3, residual GFP-positive tumor cells were quantified by flow cytometric analysis. Figure 2a shows that while CAR-Muc1 T cells killed the majority of PC3 cells (87.7±11.8%), decreasing the number of tumor cells from 26.5±1.7E +05 −3.2±3.1E +05, CAR-Muc1 T-cell treatment again failed to eliminate all tumor cells (n =4). These results were reproduced using a second prostate cancer cell line, DU145. Figure 2c shows that CAR-Muc1 T cells killed the majority of DU145 cells (72%), decreasing the number of tumor cells from 25E + 05 −7E +05, again CAR-Muc1 T-cell treatment failed to eliminate all tumor cells.
Figure 2.
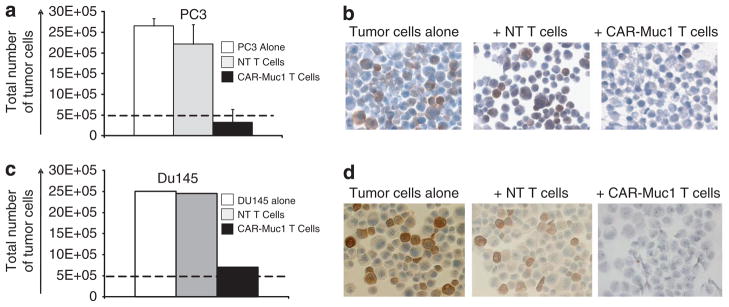
Tumor immune escape due to heterogeneous tumor antigen expression. (a) Shows that CAR-Muc1 T cells (■) kill the Muc1-expressing prostate cancer cell line PC3 while control NT T cells (
 ) have little impact on tumor cell growth. Untreated tumor cells (■) served as an additional control. Cytotoxic activity was evaluated in a 72-h co-culture experiment (ratio 1 tumor cell:10T cells) and results are shown as total number of residual tumor cells. Data represent the mean±s.d. of four donors. (b) Shows IHC analysis for Muc1 tumor antigen expression performed on tumor cells that were untreated (left panel), treated with control NT T cells (middle panel), or treated with CAR-Muc1 T cells (right panel). (c, d) Show similar results for a second prostate cancer cell line, DU145.
) have little impact on tumor cell growth. Untreated tumor cells (■) served as an additional control. Cytotoxic activity was evaluated in a 72-h co-culture experiment (ratio 1 tumor cell:10T cells) and results are shown as total number of residual tumor cells. Data represent the mean±s.d. of four donors. (b) Shows IHC analysis for Muc1 tumor antigen expression performed on tumor cells that were untreated (left panel), treated with control NT T cells (middle panel), or treated with CAR-Muc1 T cells (right panel). (c, d) Show similar results for a second prostate cancer cell line, DU145.
To discover the mechanism of tumor cell escape from immune-mediated destruction we determined the Muc1 antigen expression profile of the GFP-positive tumor cells remaining after co-culture with CAR-Muc1 T cells. Figures 2b and d show that surviving tumor cells lacked expression of the target antigen (Muc1), while tumor cells that were treated with control T cells (that is, without selective pressure) retained Muc1 expression at levels similar to untreated tumor cells. To confirm this mechanism, we engineered the 293T cell line, which is endogenously Muc1 negative, to transgenically express the Muc1 antigen and mOrange as a fluorescent marker. We also generated a control vector encoding GFP (Figure 3a). 293T cells were transduced with either the control GFP or Muc1-mOrange vector and then sorted to ensure that subsequent experiments were performed with pure populations of transgenic target cells. We next mixed GFP-positive and mOrange-positive 293T target cells at a 1:1 ratio and co-cultured these tumor cells with NT or CAR-Muc1 T cells at a 1:10 ratio. After 3 days, the total residual tumor cells (GFP and mOrange) were quantified by flow cytometry and reported as percentage of viable cells relative to the control condition of tumor cells alone. As shown in Figure 3b tumor cells were sensitive to CAR-Muc1 T-cell treatment as evidenced by a 51.1±12% reduction in target cells relative to control NT T cells. We analyzed the composition of these residual tumor cells by sub-fractionating them into GFP +(Muc1 −) and mOrange +(Muc1 +) cells. Figure 3c shows that in cultures treated with NT T cells, residual tumor cells represent a 50/50 mix of mOrange (Muc1 +) and GFP (Muc1 −) cells, while in conditions treated with CAR-Muc1 T cells 95.1±3.1% of cells were GFP +(Muc1 −) (n =6). Figure 3d shows representative results of fluorescent microscope images and flow cytometric analysis of the co-culture experiments that illustrate the changes in the culture composition depending on the effector T-cell treatment and antigen expression profile of the tumor target cells. In this donor, a single treatment with CAR-Muc1 T cells eliminated 96.5% of Muc1 +(mOrange) tumor cells (3.5% residual antigen-expressing cells). After a second treatment with CAR-Muc1 T cells only 0.6% Muc1 antigen-positive cells remained in the culture. Thus, when antigen heterogeneity is eliminated as a variable, T cells display an exquisite ability to eliminate tumor cells based on their expression of the target antigen.
Figure 3.

Engineered 293T tumor model to investigate tumor immune escape due to heterogeneous tumor antigen expression. (a) (Left panel) shows the retroviral vector map of the Muc1-IRES-mOrange construct as well as the control GFP retroviral vector. 293T cells transduced to express either the mOrange (Muc1 +) or GFP (Muc1 −) vectors were sorted to 100% purity and then mixed at a 1:1 ratio, as shown in (a), right panel. (b) (Left panel) shows that CAR-Muc1 T cells (■) were able to kill ~50% of the 1:1 293T tumor mix (GFP/mOrange) while control NT T cells (
) have little impact on tumor cell growth. Cytotoxic activity was evaluated in a 72-h co-culture experiment (1:10 ratio of tumor:T cells) and the presented data represent the mean±s.d. of six donors. (b) (Right panel) shows flow cytometric analysis of residual tumor cells based on expression of GFP (Muc1 −) or mOrange (Muc1 +) (n =6). (c) Shows immunofluorescence images from one representative donor. (d) Shows flow cytometric analysis results from one representative donor where tumor cells were distinguished based on GFP (x axis) and mOrange (y axis) expression. Treatment with control NT T cells had no impact on tumor cell numbers, whereas a single treatment with CAR-Muc1 T cells decreased the mOrange (Muc1 +) population (upper left quadrant) to 3.5%, which was further reduced to 0.6% with a second treatment.
Anti-androgen therapy inhibits prostate cancer cell growth but does not eliminate tumor
To evaluate the effects of anti-androgen therapy on prostate cancer cells, 5 ×105 LNCaP cells (androgen receptor positive) expressing the fluorescent protein mOrange were cultured for 7 days in media alone or media supplemented on days 1–3 with 10 or 20 μM of Flutamide, which blocks androgen binding to its receptor. Flutamide is a competitive inhibitor of testosterone and its metabolites that inhibits the growth of LNCaP and other androgen-sensitive prostate cancer cells.29,30 On day 7 of culture tumor cells were quantified by assessing the frequency of mOrange-positive cells by flow cytometric analysis. The concentrations of Flutamide used were sufficient to inhibit the effects of androgens, as illustrated by characteristic changes in cell morphology and the acquisition of neuroendocrine characteristics in the treated LNCaP cells31 (Figure 4b). As shown in Figure 4a, 10 or 20 μM of Flutamide also significantly reduced the number of tumor cells compared with control cultures, with a decrease from 20±2.5E +05 tumor cells to 1.2±3.2E +05 and 0.24±0.5E +05 cells, respectively, (P =0.00156 and P =0.00028), corresponding with a 40.3±9.1 and 79.2±2.6% reduction, respectively. As a concentration of 10 μM of Flutamide corresponds to levels obtained during clinical administration,29,30 all subsequent experiments were performed using this drug concentration.
Figure 4.

Anti-androgen inhibits prostate cancer cell growth but does not eliminate tumor. (a) LNCaP cells (5 ×105) engineered to express the fluorescent protein mOrange were cultured for 7 days in media alone or media supplemented with 10 or 20 μM of Flutamide on days 1, 2 and 3. On day 7 of culture tumor cells were quantified by assessing the frequency of mOrange-positive cells by flow cytometric analysis. Data represent the mean±s.d. of four donors. (b) Shows that anti-androgen therapy causes changes in cell morphology and results in the acquisition of neuroendocrine characteristics in treated LNCaP cells.
Combination therapy with CAR-Muc1 T cells and Flutamide to improve prostate cancer cell killing
To determine whether immunotherapy could be combined with conventional anti-androgen therapy to produce anti-tumor responses superior to either component alone, we first assessed whether T cells were adversely affected by anti-androgen agents. We compared the proliferative capacity of CAR-T cells alone or CAR-T cells cultured in the presence of 10 μM of Flutamide. As illustrated in Figure 5a, Flutamide exposure did not affect the proliferative capacity of CAR-T cells exposed to increasing doses (ranging from 10–160 U ml −1) of the T-cell growth factor IL2. Similarly, cell viability (Figure 5b) and cytokine production was unaffected (data not shown). Next, we cultured CAR-Muc1 T cells in the absence or presence of 10 μM Flutamide (added 3 × per week) for 2 weeks and assessed expansion, phenotype and function of the cells at the end of culture. As shown in Figure 5c there was no difference in the rate of expansion of T cells alone vs T cells cultured with 10 μM of Flutamide as assessed by cell counting using trypan blue exclusion (18.4-fold increase, range 14–22.3 vs 20.4-fold increase, range 16–26.2, respectively). Transgenic T cells treated with Flutamide retained their ability to kill Muc1-positive prostate cancer cells, as assessed by 6 h Cr51 release assay showing 43±14% specific lysis, E:T 40:1 vs 34±12% killing achieved using untreated CAR-Muc1 T cells (Figure 5d). Finally, the phenotype of the cells was conserved as illustrated by expression of comparable levels of the activation markers CD27, CD28 and CD25 (72.4±7.3 vs 73.1±9.7%) (28.2±13.6 vs 31.6±17.9%) and (5.4±3.4 vs 4.5±2.8%, respectively) or memory markers CD45RA, CD45RO and CD62L (50.6±34.9 vs 46.9±34.9%) (54.9±13.2 vs 59.6±13.8%) and (34.1±21.2 vs 35.1±21.7%, respectively) (Figure 5e). Thus, anti-androgen therapy with Flutamide does not adversely affect T-cell number, phenotype or function.
Figure 5.

Anti-androgen exposure does not adversely affect T-cell growth, phenotype or function. (a) Shows the proliferative capacity of CAR-Muc1 T cells cultured in increasing doses of IL2, with or without Flutamide. (b) Shows cell viability. (c) Shows that CAR-Muc1 T-cell expansion over a 2-week period was unaffected by exposure to Flutamide. (d) Illustrates that untreated or Flutamide-treated CAR-Muc1 T cells killed PC3 cells (■) at similar levels. In contrast, no significant killing of control 293T cells (□) was observed. Cytotoxic activity was evaluated in a standard 51Cr-release assay, and results are shown at an E:T ratio of 40:1. Data represent the mean±s.d. of three donors. (e) Shows the phenotype of control and CAR-Muc1 T cells cultured in the presence or absence of Flutamide for 2 weeks.
To compare the anti-tumor activity of single vs combination therapies, we next cultured Muc1-positive LNCaP cells (mLNCaP—mOrange) (a) alone, (b) with Flutamide (10 μM—added on three consecutive days), (c) with CAR-Muc1 T cells (E:T, 1:2), or (d) with both Flutamide and CAR-Muc1 T cells (Figure 6a). On day 7, we quantified residual tumor cells by flow cytometry (Figure 6b). When LNCaP cells were untreated, we saw an expansion from 5E +05 cells (on day 0)–20.2±2.5E +05 on day 7 (four-fold increase). As expected, treatment with Flutamide alone or CAR-Muc1 T cells alone reduced cancer cell numbers (12±3.2E +05 and 3.2±1.9E +05 residual tumor cells on day 7, respectively). Notably, this effect was enhanced by combining Flutamide and CAR-Muc1 T cells resulting in only 1±0.5E +05 residual viable tumor cells on day 7.
Figure 6.

Combination therapy to overcome tumor heterogeneity. (a) Shows a schematic representation of our experimental design to test the efficacy of combination (Flutamide +CAR-Muc1 T cells) therapy in vitro. (b) To evaluate whether combination therapy would produce superior anti-tumor effects LNCaP cells were left untreated or treated with Flutamide alone, CAR-Muc1 T cells alone or the combination (1:2). After co-culture for 72 h (ratio 1 tumor cell:2 T cells) residual tumor cell numbers were evaluated and total residual cell numbers are reported. Data represent the mean±s.d. of four donors.
DISCUSSION
We have described the use of CAR-modified T cells to selectively target and effectively kill prostate cancer cells expressing the tumor-associated antigen, Muc1, which is expressed in 58% of primary and 90% of lymph node metastases and whose expression appears to be highly related to tumor progression.11,32 We demonstrate that after retroviral transduction 66.2±14.1% of primary T cells stably express the transgene (CAR-Muc1), and this modification enables T cells to kill Muc1-positive prostate cancer cell lines, for example, PC3 and Du145—55±3 and 45±7% specific lysis, respectively, at an E:T of 40:1 as evaluated by a 6 h Cr51 release assay. Furthermore, co-culture of PC3 and D145 with CAR-Muc1 T cells at a 1:10 ratio resulted in an 87±11% and 72% reduction, respectively, in tumor cell numbers. However, after T-cell treatment we observed that the residual tumor population lacked expression of Muc1 antigen, as demonstrated by IHC (Figures 2b and d), making these tumor cells insensitive to CAR-Muc1 T-cell treatment. We confirmed that heterogeneous tumor antigen expression could lead to immune escape by engineering 293T cells to express Muc1 antigen and co-express mOrange. These were then mixed at a 1:1 ratio with Muc1-negative 293T cells transgenically expressing GFP (Figure 3a), and after treatment with CAR-Muc1 T cells we observed a 51.1±12% reduction in the total number of tumor cells (293T Muc1 positive/Muc1 negative), which reflected an almost complete (96.5% reduction after one T-cell treatment and 99.4% reduction after two T-cell treatments) elimination of Muc1/mOrange targets while Muc1-negative targets were unaffected. This suggests that; (i) CAR-Muc1 T cells are highly specific and thus should not produce ‘off target’ toxicity in vivo, (ii) initial treatment with CAR-Muc1 T cells results in a reduction in the number of tumor cells, which is directly proportional to the number of target cells expressing Muc1 antigen, (iii) tumor cells that are resistant to this therapy either express negligible or no Muc1 antigen and (iv) as a monotherapy against tumors with heterogeneous antigen expression T cells with specificity for a single antigen will likely be only partially effective, and may result in the emergence of a new tumor phenotype resistant to the same line of treatment.
Although prostate cancer is amenable to immunotherapeutic intervention, until now the majority of strategies have been restricted to vaccines designed to activate the host cellular immune response to target tumor-associated antigens such as PSA, prostate-specific membrane antigen, Prostatic acid phosphatase and prostate stem cell antigen.25,33–37 However, the clinical responses to some of these vaccines in patients with metastatic disease have not always been successful,38 likely reflecting the fact that vaccines are often administered to patients with bulky or metastatic disease whose immune systems are frequently compromised because of disease burden and prior therapy and whose tumors express or overexpress ‘self antigens’ that are poorly immunogenic and against which circulating T cells are often anergized or tolerized.39
In contrast, CAR therapy is based on the ex vivo generation of immunity after the genetic modification of T cells. CARs combine the binding properties of monoclonal antibodies with the lytic capacity of T cells. Thus, CAR-expressing T cells are able to recognize both protein and non-protein antigens on tumor cells and kill in an MHC-independent fashion—an important consideration as many tumors downregulate MHC or fail to process antigen for presentation. This strategy has already demonstrated clinical efficacy—our group has treated patients with advanced neuro-blastoma using T cells modified with a CAR targeting the tumor-associated antigen GD2 and 6 of 8 patients with no evidence of disease at infusion remain without measurable disease while 6 of 11 with relapsed/resistant disease had tumor responses/necrosis including 3 who achieved complete remissions that were sustained in 2.6,7 Similar encouraging results have been reported by Porter et al. who infused T cells engineered with a CAR targeting CD19, which produced complete clinical responses in 2/3 patients with chronic lymphocytic leukemia.40,41
Irrespective of whether a cellular immune response is activated in vivo or ex vivo there is clear evidence that cancer cells have evolved multiple immune evasion strategies that allow them to escape immune-mediated elimination.39,42 These include (i) the production of immunosuppressive cytokines including TGF-β, interleukin (IL)-10, IL-13 and so on, which inhibit effector T cells, (ii) modulation of MHC and costimulatory molecules to prevent recognition by antigen-specific T cells with native TCR specificity, (iii) recruitment of regulatory T cells (Tregs) which inhibit effector T cells by direct cell-to-cell contact or by the production of soluble factors and (iv) expression of inhibitory cell surface molecules such as PD-L1 (program death ligand), which interacts with PD-1 expressed on activated T cells, and induces T-cell exhaustion.43,44 In addition, heterogeneity in genotype, gene expression, antigen expression profile, provides cancer cells with an evolutionary advantage from an environmental pressure, and confers them with a random fitness to stress.45,46 To our knowledge this is the first report of tumor resistance to CAR-T-cell therapy due to heterogeneity in antigen expression and this phenomenon must be taken into consideration in the design of future clinical studies.
Combination of CAR-T cells with standard treatments for prostate cancer may produce anti-tumor effects that are superior to either approach alone. Anti-androgen therapy, which is the first line standard of care for metastatic prostate cancer, often stunts tumor growth but is not always curative,47,48 though there is now clear evidence demonstrating that even at castrated levels of testosterone, cancer cells continue to rely on androgen signaling for growth. Thus, in coming years new anti-androgens, including abiraterone, TAK-700 and MDV3100, which have produced encouraging results in clinical trials, may emerge as important therapeutics.49–52
Mercader et al.53 reported that androgen ablative therapy-induced infiltration of a mixture of CD4 + and CD8 + T cells into prostate tumors. Similarly, Morse and McNeel54,55 reported the detection of Th1-polarized, prostate-infiltrating cells with an oligoclonal phenotype in patients undergoing neoadjuvant androgen deprivation, while several animal studies have demonstrated that thymic involution can be reversed after castration56 and shown that decreased levels of androgens appear to result in an increase in both the frequency and function of circulating T cells.53–55 This, in addition to reports from Madan et al. demonstrating that combination therapy using vaccines with androgen receptor antagonists or anti-androgens may produce clinical benefit,57,58 led us to investigate whether anti-androgen therapy could be combined with CAR-T cells.
As a proof of concept we tested whether T-cell therapy could be combined with the non-steroidal anti-androgen drug Flutamide. To first confirm that Flutamide would not have any direct adverse effects on CAR-modified T cells we measured a variety of parameters including T-cell proliferation, expansion, viability, phenotype and function (cytokine production and lytic capacity) and found that after 12 days of exposure to this drug at therapeutic levels there was no detectable difference in comparison with cells maintained under normal culture conditions. We subsequently combined the modalities and demonstrated that combination therapy with experimental (CAR-Muc1 T cells) and conventional (Flutamide) agents is feasible and results in superior (additive) anti-tumor effects relative to either therapy alone. Owing to the in vitro nature of the investigations undertaken we were unable to determine whether combination with other luteinizing hormone-releasing hormone analogs or antagonists would produce similar anti-tumor effects but based on the cited literature it appears that irrespective of the mechanism of androgen deprivation, combination with adoptive T-cell transfer should be feasible and result in superior anti-tumor effects in vivo.53–55
Supplementary Material
Acknowledgments
JFV is supported by an Idea Development Award from the Department of Defense Prostate Cancer Research Program (no. W81XWH-11-1-0625). The research work was also supported by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine. MKB is supported by a Fayez Sarofim Chair.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Prostate Cancer and Prostatic Diseases website (http://www.nature.com/pcan)
References
- 1.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 2.Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 3.Higano CS, Schellhammer PF, Small EJ, Burch PA, Nemunaitis J, Yuh L, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- 4.Karan D, Holzbeierlein JM, Van Veldhuizen P, Thrasher JB. Cancer immunotherapy: a paradigm shift for prostate cancer treatment. Nat Rev Urol. 2012;9:376–385. doi: 10.1038/nrurol.2012.106. [DOI] [PubMed] [Google Scholar]
- 5.Longo DL. New therapies for castration-resistant prostate cancer. N Engl J Med. 2010;363:479–481. doi: 10.1056/NEJMe1006300. [DOI] [PubMed] [Google Scholar]
- 6.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cozzi PJ, Wang J, Delprado W, Perkins AC, Allen BJ, Russell PJ, et al. MUC1, MUC2, MUC4, MUC5AC and MUC6 expression in the progression of prostate cancer. Clin Exp Metastasis. 2005;22:565–573. doi: 10.1007/s10585-005-5376-z. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Cozzi PJ. MUC1 is a promising therapeutic target for prostate cancer therapy. Curr Cancer Drug Targets. 2007;7:259–271. doi: 10.2174/156800907780618338. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Cozzi PJ, Russell PJ. Promising tumor-associated antigens for future prostate cancer therapy. Med Res Rev. 2010;30:67–101. doi: 10.1002/med.20165. [DOI] [PubMed] [Google Scholar]
- 14.Beatson RE, Taylor-Papadimitriou J, Burchell JM. MUC1 immunotherapy. Immunotherapy. 2010;2:305–327. doi: 10.2217/imt.10.17. [DOI] [PubMed] [Google Scholar]
- 15.Kufe DW. Functional targeting of the MUC1 oncogene in human cancers. Cancer Biol Ther. 2009;8:1197–1203. doi: 10.4161/cbt.8.13.8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maher J, Wilkie S. CAR mechanics: driving T cells into the MUC of cancer. Cancer Res. 2009;69:4559–4562. doi: 10.1158/0008-5472.CAN-09-0564. [DOI] [PubMed] [Google Scholar]
- 17.Yonezawa S, Goto M, Yamada N, Higashi M, Nomoto M. Expression profiles of MUC1, MUC2, and MUC4 mucins in human neoplasms and their relationship with biological behavior. Proteomics. 2008;8:3329–3341. doi: 10.1002/pmic.200800040. [DOI] [PubMed] [Google Scholar]
- 18.Singh R, Bandyopadhyay D. MUC1: a target molecule for cancer therapy. Cancer Biol Ther. 2007;6:481–486. doi: 10.4161/cbt.6.4.4201. [DOI] [PubMed] [Google Scholar]
- 19.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci USA. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelmann K, Baldus SE, Hanisch FG. Identification and topology of variant sequences within individual repeat domains of the human epithelial tumor mucin MUC1. J Biol Chem. 2001;276:27764–27769. doi: 10.1074/jbc.M103187200. [DOI] [PubMed] [Google Scholar]
- 21.Baldus SE, Engelmann K, Hanisch FG. MUC1 and the MUCs: a family of human mucins with impact in cancer biology. Crit Rev Clin Lab Sci. 2004;41:189–231. doi: 10.1080/10408360490452040. [DOI] [PubMed] [Google Scholar]
- 22.Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901–4909. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 23.Bolla M, de Reijke TM, Van Tienhoven G, Van den Bergh AC, Oddens J, Poortmans PM, et al. Duration of androgen suppression in the treatment of prostate cancer. N Engl J Med. 2009;360:2516–2527. doi: 10.1056/NEJMoa0810095. [DOI] [PubMed] [Google Scholar]
- 24.Jones CU, Hunt D, McGowan DG, Amin MB, Chetner MP, Bruner DW, et al. Radiotherapy and short-term androgen deprivation for localized prostate cancer. N Engl J Med. 2011;365:107–118. doi: 10.1056/NEJMoa1012348. [DOI] [PubMed] [Google Scholar]
- 25.Antonarakis ES, Drake CG. Combining immunological and androgen-directed approaches: an emerging concept in prostate cancer immunotherapy. Curr Opin Oncol. 2012;24:258–265. doi: 10.1097/CCO.0b013e32835205a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanisch FG, Muller S. MUC1: the polymorphic appearance of a human mucin. Glycobiology. 2000;10:439–449. doi: 10.1093/glycob/10.5.439. [DOI] [PubMed] [Google Scholar]
- 28.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Tevell A, Lennernäs H, Jönsson M, Norlin M, Lennernäs B, Bondesson U, et al. Flutamide metabolism in four different species in vitro and identification of flutamide metabolites in human patient urine by high performance liquid chromatography/tandem mass spectrometry. Drug Metab Dispos. 2006;34:984–992. doi: 10.1124/dmd.105.008516. [DOI] [PubMed] [Google Scholar]
- 30.Belanger A, Giasson M, Couture J, Dupont A, Cusan L, Labrie F. Plasma levels of hydroxyflutamide in patients with prostatic cancer receiving the combined hormonal therapy: an LHRH agonist and flutamide. Prostate. 1988;12:79–84. doi: 10.1002/pros.2990120110. [DOI] [PubMed] [Google Scholar]
- 31.Mosca A, Berruti A, Russo L, Torta M, Dogliotti L. The neuroendocrine phenotype in prostate cancer: basic and clinical aspects. J Endocrinol Invest. 2005;28:141–145. [PubMed] [Google Scholar]
- 32.Zellweger T, Ninck C, Bloch M, Mirlacher M, Koivisto PA, Helin HJ, et al. Expression patterns of potential therapeutic targets in prostate cancer. Int J Cancer. 2005;113:619–628. doi: 10.1002/ijc.20615. [DOI] [PubMed] [Google Scholar]
- 33.Frank MO, Kaufman J, Tian S, Suárez-Fariñas M, Parveen S, Blachère NE, et al. Harnessing naturally occurring tumor immunity: a clinical vaccine trial in prostate cancer. PLoS One. 2010;5:e12367. doi: 10.1371/journal.pone.0012367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noguchi M, Uemura H, Naito S, Akaza H, Yamada A, Itoh K. A phase I study of personalized peptide vaccination using 14 kinds of vaccine in combination with low-dose estramustine in HLA-A24-positive patients with castration-resistant prostate cancer. Prostate. 2011;71:470–479. doi: 10.1002/pros.21261. [DOI] [PubMed] [Google Scholar]
- 35.Harrop R, Shingler W, Kelleher M, de Belin J, Treasure P. Cross-trial analysis of immunologic and clinical data resulting from phase I and II trials of MVA-5T4 (TroVax) in colorectal, renal, and prostate cancer patients. J Immunother. 2010;33:999–1005. doi: 10.1097/CJI.0b013e3181f5dac7. [DOI] [PubMed] [Google Scholar]
- 36.Weber JS, Vogelzang NJ, Ernstoff MS, Goodman OB, Cranmer LD, Marshall JL, et al. A phase 1 study of a vaccine targeting preferentially expressed antigen in melanoma and prostate-specific membrane antigen in patients with advanced solid tumors. J Immunother. 2011;34:556–567. doi: 10.1097/CJI.0b013e3182280db1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chudley L, McCann K, Mander A, Tjelle T, Campos-Perez J, Godeseth R, et al. DNA fusion-gene vaccination in patients with prostate cancer induces high-frequency CD8(+) T-cell responses and increases PSA doubling time. Cancer Immunol Immunother. 2012;61:2161–2170. doi: 10.1007/s00262-012-1270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vieweg J, Dannull J. Technology Insight: vaccine therapy for prostate cancer. Nat Clin Pract Urol. 2005;2:44–51. doi: 10.1038/ncpuro0079. [DOI] [PubMed] [Google Scholar]
- 39.Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 40.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vera JF, Brenner MK, Dotti G. Immunotherapy of human cancers using gene modified T lymphocytes. Curr Gene Ther. 2009;9:396–408. doi: 10.2174/156652309789753338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ebelt K, Babaryka G, Frankenberger B, Stief CG, Eisenmenger W, Kirchner T, et al. Prostate cancer lesions are surrounded by FOXP3 +, PD-1 + and B7-H1 + lymphocyte clusters. Eur J Cancer. 2009;45:1664–1672. doi: 10.1016/j.ejca.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 44.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-pd-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durrett R, Foo J, Leder K, Mayberry J, Michor F. Evolutionary dynamics of tumor progression with random fitness values. Theor Popul Biol. 2010;78:54–66. doi: 10.1016/j.tpb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Durrett R, Foo J, Leder K, Mayberry J, Michor F. Intratumor heterogeneity in evolutionary models of tumor progression. Genetics. 2011;188:461–477. doi: 10.1534/genetics.110.125724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim W, Ryan CJ. Androgen receptor directed therapies in castration-resistant metastatic prostate cancer. Curr Treat Options Oncol. 2012;13:189–200. doi: 10.1007/s11864-012-0188-2. [DOI] [PubMed] [Google Scholar]
- 48.Mottet N, Van Damme J, Loulidi S, Russel C, Leitenberger A, Wolff JM. Intermittent hormonal therapy in the treatment of metastatic prostate cancer: a randomized trial. BJU Int. 2012;110:1262–1269. doi: 10.1111/j.1464-410X.2012.11120.x. [DOI] [PubMed] [Google Scholar]
- 49.Dayyani F, Gallick GE, Logothetis CJ, Corn PG. Novel therapies for metastatic castrate-resistant prostate cancer. J Natl Cancer Inst. 2011;103:1665–1675. doi: 10.1093/jnci/djr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reid AH, Attard G, Barrie E, de Bono JS. CYP17 inhibition as a hormonal strategy for prostate cancer. Nat Clin Pract Urol. 2008;5:610–620. doi: 10.1038/ncpuro1237. [DOI] [PubMed] [Google Scholar]
- 51.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mercader M, Bodner BK, Moser MT, Kwon PS, Park ES, Manecke RG, et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc Natl Acad Sci USA. 2001;98:14565–14570. doi: 10.1073/pnas.251140998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morse MD, McNeel DG. T cells localized to the androgen-deprived prostate are T(H) 1 and T(H) 17 biased. Prostate. 2011;72:1239–1247. doi: 10.1002/pros.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morse MD, McNeel DG. Prostate cancer patients on androgen deprivation therapy develop persistent changes in adaptive immune responses. Hum Immunol. 2010;71:496–504. doi: 10.1016/j.humimm.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellis TM, Moser MT, Le PT, Flanigan RC, Kwon ED. Alterations in peripheral B cells and B cell progenitors following androgen ablation in mice. Int Immunol. 2001;13:553–558. doi: 10.1093/intimm/13.4.553. [DOI] [PubMed] [Google Scholar]
- 57.Arlen PM, Gulley JL, Todd N, Lieberman R, Steinberg SM, Morin S, et al. Anti-androgen, vaccine and combination therapy in patients with nonmetastatic hormone refractory prostate cancer. J Urol. 2005;174:539–546. doi: 10.1097/01.ju.0000165159.33772.5b. [DOI] [PubMed] [Google Scholar]
- 58.Madan RA, Gulley JL, Schlom J, Steinberg SM, Liewehr DJ, Dahut WL, et al. Analysis of overall survival in patients with nonmetastatic castration-resistant prostate cancer treated with vaccine, nilutamide, and combination therapy. Clin Cancer Res. 2008;14:4526–4531. doi: 10.1158/1078-0432.CCR-07-5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.