

Abstract
This Letter describes the continued optimization of an MLPCN probe molecule M1 antagonist (ML012) through an iterative parallel synthesis approach. After several rounds of modifications of the parent compound, we arrived at a new azetidine scaffold that displayed improved potency while maintaining a desirable level of selectivity over other muscarinic receptor subtypes. Data for representative molecules 7w (VU0452865) and 12a (VU0455691) are presented.
Keywords: Muscarinic acetylcholine receptor 1, M1 Antagonist, ML012, VU0455691, VU0452865
Acetylcholine (ACh) is a critical neurotransmitter with diverse functions both within the central nervous system (CNS) and in peripheral signaling pathways.1-4 ACh operates by interacting with two very distinct groups of receptors; a set of ligand-gated ion channels - the nicotinic acetylcholine receptors (nAChRs) - and a set of family A, G protein-coupled receptors (GPCRs) - the muscarinic acetylcholine receptors (mAChRs). The muscarinic family of acetylcholine receptors is divided into five subtypes (M1-5).5 These subtypes can be further classified into two subsets based on their G protein-coupling partners, with the M1, 3, 5 receptors preferentially coupling to Gq (stimulating PLC and intracellular calcium mobilization) and the M2, 4 receptors preferentially coupling to Gi/o (inhibiting adenylate cyclase (AC), thereby decreasing cAMP production).4 The specific subtypes of mAChRs are expressed throughout the body with varying degrees of expression levels based on the particular site or organ.6 As a result, mAChRs play significant roles in a wide range of physiological functions such as memory and attention, motor control, nociception, regulation of sleep-wake cycles, cardiovascular function, secretory functions, and mediators of inflammation, renal and gastrointestinal (GI) function, among many others.6,7
It has been postulated that M1, 4, 5 receptors are the relevant subtypes for CNS therapies; however, specific functions for each receptor subtype are still being investigated.7 This is a direct result of the highly conserved orthosteric binding site for the endogenous ligand (ACh) that is shared across all five subtypes of mAChRs. This similarity has stymied the discovery and development of muscarinic ligands with high selectivity for a particular subtype.8 Yet, this lack of selectivity has not precluded the development of pharmaceuticals with activity at mAChRs for a range of indications. Many of these non-selective compounds have undesirable side-effects that are attributed to activity at the other mAChRs (often M2 and M3), limiting their clinical impact. For example, xanomeline, a reported M1- and M4-selective agonist, showed robust clinical efficacy in Phase II trials for Alzheimer’s disease and schizophrenia,9,10 but also has nearly equivalent agonist activity at M3.8 Even in the absence of offtarget mAChR activity, the debate still remains whether a single mAChR subtype (M1 or M4) is responsible for the positive outcomes in these trials; although, recent studies using mAChR genetic knockout (KO) mice have shed additional light on this topic.11 More highly selective mAChR ligands would allow for more direct pharmacological insight and a better understanding of the individual roles for each of the five mAChRs. We envision two ways to obtain mAChR subtype selectivity with synthetic ligands: 1) simultaneous binding to the orthosteric site and into adjacent areas which may be less structurally conserved among the other mAChRs12 or 2) binding to a completely distinct region of the mAChR at an allosteric site, imparting a level of selectivity to the ligand not found relative to the other four mAChRs. This allosteric approach has been highly successful for a number of the individual mAChRs: M1,13,14,15 M4,16 and M5.17
We have previously reported on the selective M1 antagonist, ML012 (VU0255035, Fig. 1), and progress on optimization of the ML012 scaffold. ML012 showed 45- to 159-fold selectivity for M1 over the other subtypes.12 ML012 also reduced pilocarpine-induced seizures in rodents at doses that had no negative impact on contextual fear conditioning, a behavioral model of hippocampal-dependent cognitive function. These findings demonstrated that selective M1 antagonists have therapeutic potential over non-selective muscarinic antagonists. Given the potential for M1 antagonists in such indications as Parkinson’s disease, movement disorders, and Fragile X syndrome,18,19 we engaged in an optimization campaign of ML012. Our efforts yielded compound 1 (VU0415248, Fig. 1), a more potent antagonist with better selectivity for M1.20 These efforts also expanded the structure-activity relationship (SAR) of ML012 and other compounds in this series. Herein, we report further modifications which provided a panel of compounds with improved potency and good selectivity for the M1 muscarinic receptor, and more importantly, divergent SAR from the ML012 series.
Figure 1.
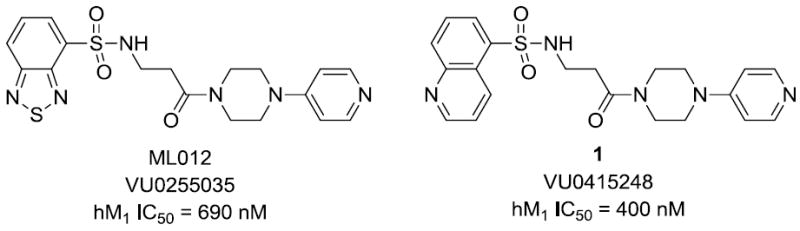
MLPCN Probe ML012 and VU0415248, a selective M1 antagonist.
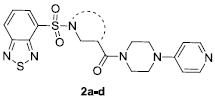
In our previous work on ML012 optimization, the central linker was modified through the introduction of methyl substitution and fluorination at the alpha position of the beta-alanine moiety. Of these modifications, none provided a desirable increase in potency or selectivity and many abolished activity altogether.20 Concurrently, we prepared a limited series of cyclic constrained analogs and screened these compounds for antagonism at M1 (Table 1). For compounds 2a-c, potency was significantly decreased while some slight activity at M1 remained (for 2a, 33% activity and for 2c, 47% activity). We were encouraged that an azetidine analog, 2d, was tolerated, albeit 3-fold less active than ML012. This provided an opportunity to enter into new chemical space and investigate compounds for improved potency and selectivity over ML012. Previous modifications to the Western thiadiazole of ML012 led us to determine that an oxadiazole was a suitable replacement and generally maintained potency.20 We procured 3 (Fig. 2), which contained the desired oxadiazole and a central azetidine linker, and found that it was equipotent to 1 (vida supra), our improved M1 antagonist.
Table 1.
Structures and potencies of M1 antagonist analogs 2a-d with cyclic constraints.
| ||||
|---|---|---|---|---|
| Cmpd | aza ring constraintsa | pIC50 ± SEMb | hM1 IC50 (μM)b | %EC min ± SEMb |
| 2a |
|
>10 | 33.4±3.9 | |
| 2b |
|
inactive | ||
| 2c |
|
>10 | 47.1±4.0 | |
| 2d |
|
5.68±0.07 | 2.2 | 10.1±3.3 |
Examples 2a and 2c were prepared and screened as racemic mixtures.
Values represent the mean ± standard error mean of at least three independent determinations performed in triplicate.
Figure 2.
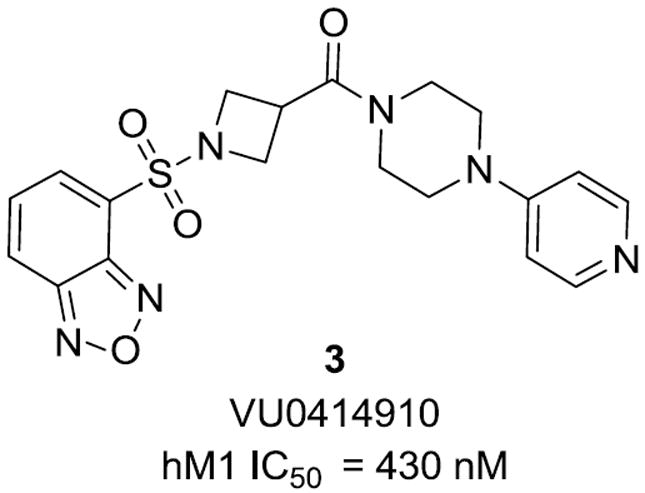
VU0414910, an M1 antagonist.
With 3 in hand, we explored the SAR at both termini of the molecule through an iterative parallel synthesis approach. These routes, illustrated in Scheme 1, made use of the commercially available azetidine central linkers 4 and 8. For the Eastern SAR, azetidine 4 and aryl sulfonyl chlorides were reacted to provide sulfonamide 5, followed by saponification which yielded acid 6. Amide coupling with substituted piperazines provided the target compound (7). As with our previous findings from the optimization of ML012, Eastern SAR around the pyridine ring was unforgivingly steep.20 Indeed, only compound 3 (hM1 IC50 = 430 nM) or substitution with a (5-bromopyridin-2-yl)piperazine moiety (hM1 IC50 = 280 nM, structure not shown) maintained activity.21 Focusing on the Western SAR and starting with azetidine 8, amide coupling to give 9 was followed by deprotection of the N-Boc with TFA in DCM to provide bis-TFA salt 10. Sulfonation with aryl sulfonyl chlorides generated sulfonamides (7), shown in Table 2 with the requisite (pyridin-4-yl)-piperazine and a variety of Ar groups.
Scheme 1.
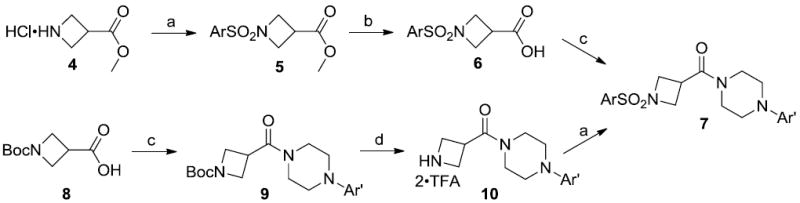
Reagents: (a) ArSO2Cl, NEt3, DCM; (b) NaOH (aq), MeOH; (c) amine, EDCI, HOBt, DIEA, DMF; (d) TFA/DCM (1:1).
Table 2.
Structures and activities of M1 antagonist analogs 7a-7aa.
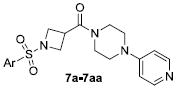
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | Ar | pIC50 ± SEMa | hM1 IC50 (μM)a | %EC min ± SEMa | Cmpd | Ar | pIC50 ± SEMa | hM1 IC50 (μM)a | %EC min ± SEMa | Cmpd | Ar | pIC50 ± SEMa | hM1 IC50 (μM)a | %EC min ± SEMa |
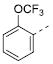
| 7a |
|
5.60±0.07 | 2.5 | 8.4±1.0 | 7j |
|
5.91±0.09 | 1.2 | 12.3±1.0 | 7s |
|
5.88±0.06 | 1.3 | 8.2±1.0 |
| 7b |
|
5.77±0.04 | 1.7 | 9.5±0.8 | 7k |
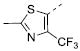
|
5.60±0.11 | 2.5 | 12.7±1.6 | 7t |
|
>10 | 20.6±1.6 | |
| 7c |
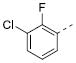
|
5.55±0.04 | 2.8 | 11.4±1.0 | 7l |
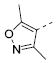
|
5.64±0.02 | 2.3 | 15.3±1.5 | 7u |
|
>10 | 40.7±0.8 | |
| 7d |
|
5.39±0.05 | 4.1 | 20.7±2.3 | 7m |
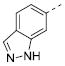
|
>10 | 33.0±0.9 | 7v |
|
6.90±0.07 | 0.13 | 3.6±0.6 | |
| 7e |
|
>10 | 41.3±1.6 | 7n |
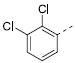
|
6.08±0.08 | 0.84 | 3.5±0.2 | 7w |
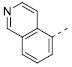
|
6.94±0.14 | 0.11 | 2.7±0.3 | |
| 7f |
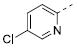
|
>10 | 38.5±0.7 | 7o |
|
6.08±0.11 | 0.83 | 5.3±0.8 | 7x |
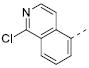
|
6.13±0.01 | 0.74 | 3.5±0.4 | |
| 7g |
|
5.75±0.07 | 1.8 | 8.8±1.1 | 7p |
|
6.43±0.07 | 0.37 | 4.9±0.2 | 7y |
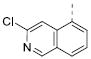
|
5.81±0.02 | 1.6 | 6.0±0.9 |
| 7h |
|
5.77±0.02 | 1.7 | 8.0±0.8 | 7q |
|
6.30±0.04 | 0.50 | 5.7±0.8 | 7z |
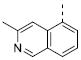
|
5.67±0.11 | 2.2 | 9.8±1.6 |
| 7i |
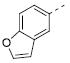
|
5.64±0.01 | 2.3 | 10.5±1.2 | 7r |
|
6.51±0.05 | 0.31 | 4.9±0.8 | 7aa |
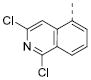
|
5.26±0.01 | 5.5 | 12.7±0.7 |
Values represent the mean ± standard error mean of at least three independent determinations performed in triplicate.
Table 2 illustrates the Western SAR of azetidine-linked aryl piperazines. While not performed extensively, we did investigate other ways to functionalize the N-H of the azetidine, including sulfonamide and amide bond formation. Amides were not a fruitful endeavor, resulting in all compounds with >10 μM potency at M1 (data not shown). Aryl sulfonamides were tolerated in a much broader sense than seen previously with the ML012 scaffold,19 with many compounds displaying IC50 values in the low to mid-micromolar range. Simple benzene sulfonamides 7a-d were 2- to 5-fold less potent than ML012 (used as our standard in this context, hM1 IC50 = 0.81 μM). 2-Pyridyl sulfonamides 7e and 7f were weak antagonists with both compounds displaying hM1 IC50 > 10 μM, 38% and 41% ACh activity remaining at the highest concentration tested (30 μM). Other heterocyclic aromatics were modest antagonists with low to mid-micromolar potencies (see 7g-m, Table 2). 2,3-Dichlorination of the benzene ring (7n) gave a compound that was nearly equipotent to ML012 as did piperonyl sulfonamide, 7o. In the beta-alanine-linked ML012 series, 3-pyridyl sulfonamides were inactive,19 yet a small subset of 3-pyridyl sulfonamides, including compound 7p, were more potent than ML012 (hM1 IC50 = 0.37 μM). Substituted pyridine replacements such as 4-chloropyridin-3-yl sulfonamide 7q and 4-chloro-5-methylpyridin-3-yl sulfonamide 7r were potent (hM1 IC50 = 0.50 μM and hM1 IC50 = 0.31 μM, respectively), as was 2-chloropyridin-3-yl sulfonamide 7s, albeit to a lesser extent. 4-methoxypyridin-3-yl and 4-trifluoromethylpyridin-3-yl sulfonamides 7t and 7u were weak antagonists (hM1 IC50 > 10 μM, 20% and 40% ACh activity remaining at the highest concentration tested (30 μM)). This loss of potency could be attributed to the increased steric bulk at the 4-position of the pyridine. The largest improvements in potency were seen with compounds 7v and 7w (hM1 IC50 = 0.13 μM and hM1 IC50 = 0.11 μM, respectively, Table 2) with an 8-fold increase. It seemed that many of these azetidine analogs of ML012 began to show a divergent SAR profile from the parent molecule. Finally, compounds 7x-aa exhibited a steric intolerance at the α-position of the isoquinoline and were less potent antagonists of M1.
Isoquinoline sulfonamide 7w represented a new and attractive scaffold on which to work. The strategic introduction of a critical nitrogen atom improved potency over benzothiadiazole and benzoxadiazole sulfonamides 2d and 3 and likely mimics an interaction at the N-1 nitrogen of both structures. Previously, this trend was also observed in the optimization of ML012 to arrive at VU0451248.20 With this structure in hand, we moved to probe the Eastern SAR once more, given the breadth of changes made to the original scaffold. To ensure that we had found the optimal Eastern heterocycle, we held the Western isoquinoline sulfonamide and the azetidine linker constant. Following our iterative parallel synthesis approach described in Scheme 1 (compounds 4-7, vida supra), we generated a small set of Eastern replacements to the pyridin-4-yl piperazine (Table 3). We anticipated that only basic moieties would be tolerated in Ar substitutions on compound 11 from the SAR profiles previously described. Indeed, 11a proved to be a modest antagonist of M1 (hM1 IC50 = 1.5 μM). Improved potencies were observed for a more basic set of substituents (11b-e).
Table 3.
Structures and activities of M1 antagonist analogs 11a-e with Eastern ring replacements.
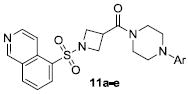
| ||||
|---|---|---|---|---|
| Cmpd | Ar | pIC50 ± SEMa | hM1 IC50 (μM)a | %EC min ± SEMa |
| 11a |
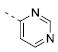
|
5.83±0.14 | 1.5 | 7.3±1.4 |
| 11b |
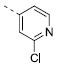
|
6.74±0.18 | 0.18 | 3.6±0.7 |
| 11c |
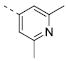
|
6.16±0.09 | 0.69 | 4.7±0.5 |
| 11d |
|
6.29±0.01 | 0.51 | 7.3±1.4 |
| 11e |
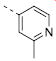
|
6.97±0.08 | 0.11 | 3.1±0.1 |
Values represent the mean ± standard error mean of at least three independent determinations performed in triplicate.
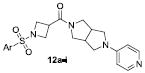
Next, replacements for the piperazine were explored. We utilized common isosteres for this aza-heterocycle, shown in Figure 3. Many of these variants did not provide antagonists with any appreciable activity. The one notable exception was the 3,7-diazabicyclo[3.3.0]octane 12a. This isostere was remarkably potent (hM1 IC50 = 0.23 μM, Table 4).
Figure 3.
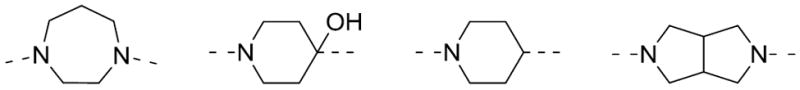
Piperazine replacements for M1 antagonists.
Table 4.
Structures and activities of M1 antagonist analogs 12a-i with Western ring replacements.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | Ar | pIC50 ± SEMa | hM1 IC50 (μM)a | %EC min ± SEMa | Cmpd | Ar | pIC50 ± SEMa | hM1 IC50 (μM)a | %EC min ± SEMa |
| 12a |
|
6.64±0.15 | 0.23 | 2.8±0.4 | 12f |
|
5.72±0.04 | 1.9 | 13.5±1.8 |
| 12b |
|
>10 | 25.8±0.9 | 12g |
|
5.81±0.06 | 1.5 | 6.6±1.1 | |
| 12c |
|
5.57±0.05 | 2.7 | 14.8±1.5 | 12h |
|
5.50±0.07 | 3.2 | 18.9±1.1 |
| 12d |
|
5.55±0.02 | 2.8 | 16.2±0.6 | 12i |
|
5.83±0.02 | 1.5 | 9.9±0.8 |
| 12e |
|
5.71±0.01 | 2.0 | 12.6±1.0 | |||||
Values represent the mean ± standard error mean of at least three independent determinations performed in triplicate.
Probing the ML012 scaffold for SAR revealed that rather stark modifications to the central linker and piperazine would afford a new SAR paradigm to be explored. Holding the central azetidine and the newly discovered 3,7-diazabicyclo[3.3.0]octane substitutions constant, we performed another round of explorations on the Western side of compound 12a (VU0455691, Table 4). Revisiting many of the same aryl sulfonamides as described in Table 3, 5-quinoline sulfonamide 12b was a weak antagonist (hM1 IC50 > 10 μM, 26% activity remaining) yet 6-methylisoquinoline-5-sulfonamide 12c was a micromolar antagonist (hM1 IC50 = 2.7 μM). Unfortunately, many of these aryl sulfonamides that were potent in the piperazine series were less so in the 3,7-diazabicyclo-[3.3.0]octane series. This was exemplified by compounds 12d-i which had modest antagonist activity at M1 (Table 4).
The development of selective allosteric compounds for mAChRs is well documented in our laboratories.13-17 Selective orthosteric compounds are more difficult to achieve due to a high conservation of the binding site between the five mAChR subtypes. In the case of ML012 and VU0415248, both compounds were selective orthosteric antagonists of M1 mAChR.20 These new series of antagonists merited a reexamination of the nature of their interaction with the M1 mAChR due to the changes made to the original ML012 scaffold. With this in mind, equilibrium radioligand binding studies using 1-[N-methyl-3H]scopolamine ([3H]NMS) were performed using two representative compounds, one from the azetidine linked piperazine series (VU0456704, 7o) and one from the [3.3.0] series (VU0455691, 12a). Both compounds showed complete displacement of the orthosteric ligand [3H]NMS, Figure 4A, consistent with a competitive interaction. Additionally, functional Schild analyses of compounds 7o and 12a were also performed.12 Both 7o and 12a displayed a parallel rightward shift in the ACh concentration response curves as the concentration of 7o or 12a was increased, as shown in Figure 4B and Figure 4D (analysis of Schild data showed a linear regression of essentially unity, Fig. 4C and Fig. 4E). These data further support an orthosteric binding mode as well as a competitive orthosteric ligand in functional assays.
Figure 4.
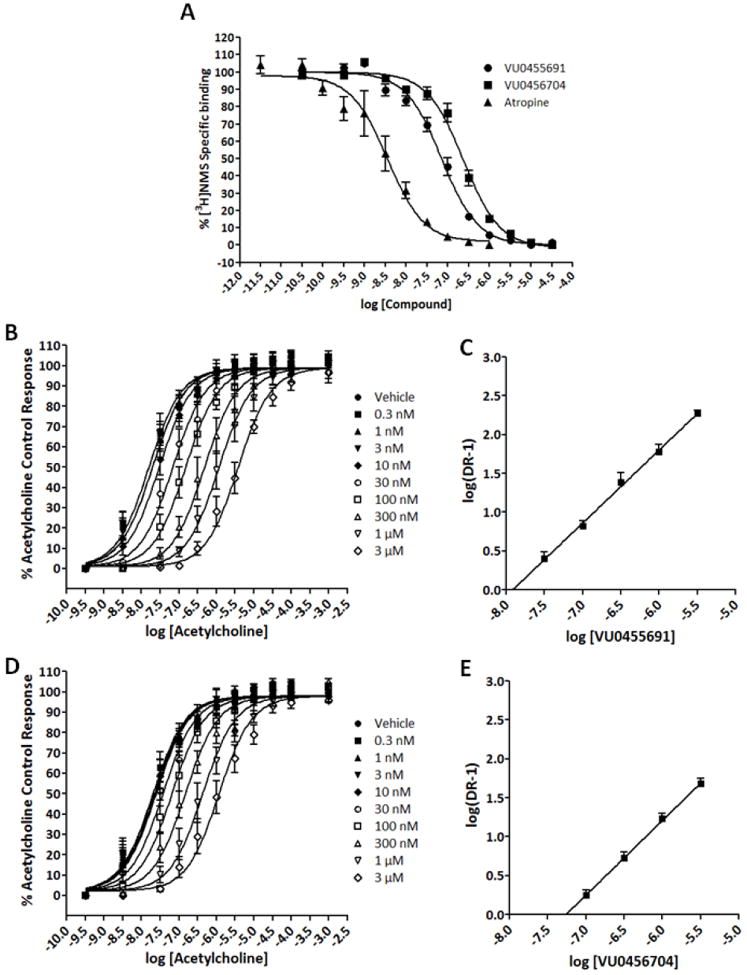
A, Compound 7o (VU0456704), 12a (VU0455691), and atropine compete with [3H]NMS binding at M1. B, 12a (VU0455691) competitively antagonizes M1 response to ACh in a concentration-dependent manner in a calcium mobilization assay. C, Schild regression of the concentration ratios derived from 12a (VU0455691) antagonism of ACh (slope of this regression is 0.94 ± 0.01. Kd = 12 ± 2.5 nM. R2 = 0.993). D, 7o (VU0456704) competitively antagonizes the M1 response to ACh in a concentration-dependent manner in a calcium mobilization assay. E, Schild regression of the concentration ratios derived from 7o (VU0456704) antagonism of ACh (slope of this regression is 0.96 ± 0.02. Kd = 54 ± 8.5 nM. R2 = 0.999). Values represent the mean ± S.E.M. of three experiments conducted in triplicate.
Our most potent compounds from the piperazine and the 3,7-diazabicyclo[3.3.0]octane series (7w and 12a, respectively) were chosen for profiling against both human and rat M1-5 receptors in a calcium mobilization assay. Both compounds compounds were highly selective for human M1 over the other human muscarinic subtypes (hM2-5 IC50s > 10 μM, data not shown), despite their orthosteric behavior observed in binding and functional assays. Figure 5 shows these data for the rat (r) M1-5 receptors. One would anticipate that ML012 and 1 would have more structural flexibility in the linker region relative to 7w and 12a. Indeed, when the same quinoline sulfonamide of 1 is made in the piperazine and the 3,7-diazabicyclo[3.3.0]octane series (7h and 12b, Tables 2 and 4, respectively) resulting compounds are inactive or weak antagonists at best. The structural rigidity provided by the azetidine linker seems to require a regioisomeric quinoline sulfonamide relative to 1 to maintain potency at M1. Additionally, we suspect this key nitrogen interaction is vital for selectivity at M1 over M2-5. We proceeded to evaluate our most divergent compound from ML012 and 1, compound 12a, for its pharmacokinetic properties. This compound was extremely hydrophilic which is not surprising, given its remarkably low cLogP (cLogP = 0.78) and, consequently, exhibited a high % unbound in plasma protein binding assays (human PPB fu = 0.97, rat PPB fu = 0.60). Unfortunately, 12a also displayed an IV plasma clearance value in the rat of 68.6 mL/min/kg, which correlated well with its moderate to high in vitro hepatic microsome intrinsic clearance (CLINT: 107 mL/min/kg) and predicted hepatic clearance (CLHEP: 42.3 mL/min/kg). 12a was also measured for its ability to inhibit the more common cytochrome P450 enzymes. Three of the four P450 enzymes tested were inhibited at low micromolar concentrations of 12a (IC50 < 2.5 μM, isoforms 2C9, 2D6, 3A4; IC50 > 30 μM for 1A2). This compound was also measured in a rat brain homogenate binding experiment and exhibited desirable levels of % unbound (fu = 0.08).
Figure 5.
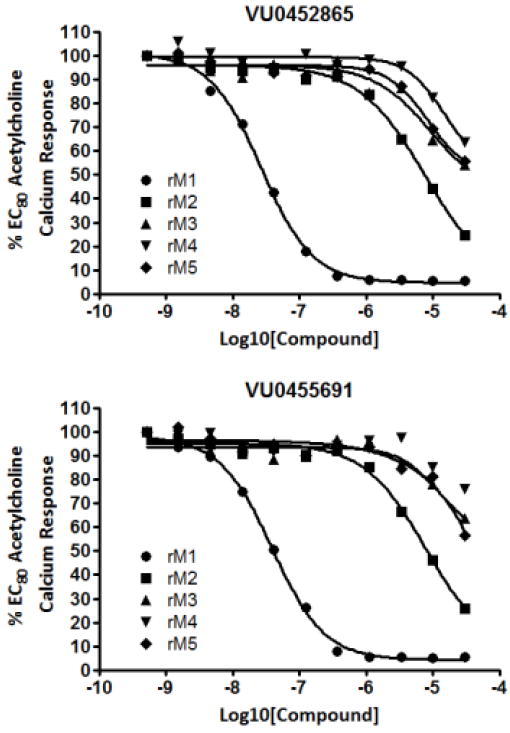
Compound 7w (VU0452865) and compound 12a (VU0455691) selectively antagonize M1 when compared to M2-5 receptors. CRCs were obtained in the presence of an EC80 concentration of ACh for each receptor in calcium mobilization assays. Data were normalized to the maximum response of 30 μM ACh and are presented as a percentage of the EC80 ACh response.
In conclusion, we have further expanded the SAR surrounding ML012 which has culminated in the development of selective orthosteric M1 antagonists 7w (VU0452865) and 12a (VU0455691). These antagonists utilized a novel scaffold relative to ML012 and clearly displayed a unique and separate SAR from the previous series. These compounds represent valuable in vitro tools with improved selectivity over ML012. Continuing work on the SAR described here may yet improve the DMPK properties of these classes of antagonists. This work will be reported in due course. ML012 is an MLPCN probe and is freely available upon request.22
Acknowledgments
The authors thank Seaside Therapeutics, NIMH (RO1MH082867), NIH (U54MH084659) and NINDS (P50NS071669) for support of our Center in the development of subtype selective mAChR antagonists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 2.Bonner TI, Young AC, Brann MR, Buckley NJ. Neuron. 1988;1:403. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 3.Wess J. Annu Rev Pharmacol Toxicol. 2004;44:423–450. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- 4.Langmead CJ, Watson J, Reavill C. Pharmacol Ther. 2008;117:232. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Wess J. Crit Rev Neurobiol. 1996;10:69. doi: 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- 6.Wess J, Eglen RM, Gautam D. Nat Rev Drug Discov. 2007;6:721. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 7.Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones JK, Conn PJ, Lindsley CW. Drug News Perspect. 2010;23:229. doi: 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinrich JN, Butera JA, Carrick T, Kramer A, Kowal D, Lock T, Marquis KL, Pausch MH, Popiolek M, Sun S-C, Tseng E, Uveges AJ, Mayer SC. Eur J Pharmacol. 2009;605:53. doi: 10.1016/j.ejphar.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 9.Bymaster FP, Whitesitt CA, Channon HE, DeLapp N, Ward JS, Calligaro DO, Shipley LA, Buelke-Sam JL, Bodick NC, Farde L, Sheardown MJ, Olesen PH, Hansen KT, Suzdak PD, Swedberg MDB, Sauerberg P, Mitch CH. Drug Dev Res. 1997;40:158. [Google Scholar]
- 10.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Am J Psychiatry. 2008;165:1033. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 11.Woolley ML, Carter HJ, Gartlon JE, Watson JM, Dawson LA. Eur J Pharmacol. 2009;603:147. doi: 10.1016/j.ejphar.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Sheffler DJ, Williams R, Bridges TM, Lewis LM, Xiang Z, Kane AS, Byun NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Lindsley CW, Conn PJ. Mol Pharmacol. 2009;76:356. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bridges TM, Kennedy JP, Noetzel MJ, Breininger ML, Gentry PR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:1972. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reid PR, Bridges TM, Sheffler DJ, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Bioorg Med Chem Lett. 2011;21:2697. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuduk SD, Chang RK, Di Marco CN, Ray WJ, Ma L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. ACS Med Chem Lett. 2010;1:263. doi: 10.1021/ml100095k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. J Pharmacol Exp Ther. 2008;327:941. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bridges TM, Kennedy JP, Hopkins CR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:5617. doi: 10.1016/j.bmcl.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veeraragavan S, Bui N, Perkins JR, Yuva-Paylor LA, Carpenter RL, Paylor R. Psychopharmacology. 2011;217:143. doi: 10.1007/s00213-011-2276-6. [DOI] [PubMed] [Google Scholar]
- 19.Healy A, Rush R, Ocain T. ACS Chem Neuro. 2011;2:402. doi: 10.1021/cn200019z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melancon BJ, Bridges TM, Lamers AP, Sulikowski GA, Sheffler DJ, Niswender CM, Noetzel MJ, Utley TJ, Daniels JS, Morrison RD, Jones CK, Conn PJ, Lindsley CW, Wood MR. Bioorg Med Chem Lett. 2012;22:1044. doi: 10.1016/j.bmcl.2011.11.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(1-(Benzo[c][1,2,5]oxadiazol-4-ylsulfonyl)azetidin-3-yl)(4-(pyridin-2-yl)piperazin-1-yl)methanone had an IC50 = 5 μM.
- 22.For information on the MLPCN and information on how to request probe compounds, such as ML012, see: http://mli.nih.gov/mli/mlpcn/.