

Abstract
Ischemic heart disease (IHD) is one of the leading causes of death worldwide. Unfortunately, current pharmacological treatments for ischemic heart disease do not reliably prevent the remodeling of the left ventricle and the progression to heart failure. Gene therapy offers a novel means to directly treat the pathophysiology underlying the long-term complications of ischemic heart disease. To date, gene therapies directed at single molecular targets have not been successful in the treatment of ischemic heart disease. In this study, we describe a gene therapy combination for inhibiting cardiomyocyte apoptosis under hypoxic conditions. This gene therapy combination utilizes a hypoxia-inducible plasmid expressing both heme oxygenase-1 (HO-1) and the Src homology domain-2 containing tyrosine phosphatase-1 microRNA (miSHP-1): pEpo-SV-miSHP-HO-1. This novel gene therapy construct demonstrated an enhanced expression of HO-1, production of miSHP-1, down-regulation of SHP-1, and inhibition of cardiomyocyte apoptosis under hypoxic compared to normoxic conditions. These results suggest that pEpo-SV-miSHP-HO-1 may be a promising gene therapy combination construct for the clinical treatment of ischemic disease.
Keywords: Myocardial infarction, SHP-1 microRNA, Heme oxygenase-1, Gene therapy, Hypoxia-inducible plasmid
1. Introduction
While current pharmacologic therapy for ischemic heart disease (IHD) can effectively treat symptoms, it cannot prevent the complications of left ventricular remodeling and the progression to heart failure [1]. Gene therapy, by inducing or inhibiting the production of specific proteins, can change the function and fate of the cells in the target tissues. Gene therapy, therefore, is an attractive alternative to current pharmacological therapies because it can potentially reverse the pathophysiology associated with acute myocardial infarction (MI) [2]. To be clinically efficacious, gene therapy for ischemic heart disease should activate and restore the molecular mechanisms responsible for reducing and reversing cardiac injury [3].
To date, single gene therapy has failed to prevent the lethal arrhythmias, acute cardiogenic shock, and chronic end-stage heart failure (HF) that are the potentially fatal complications of ischemic heart disease [4]. A combinatorial gene therapy approach that targets specific cardiac cells and signaling pathways may be clinically more efficacious. Two molecular pathways that are central to the effective treatment of ischemic heart disease are: 1) the inhibition of cardiomyocyte apoptosis and/or necrosis; and 2) the induction of neovascularization. Addressing both pathways simultaneously requires the overexpression of one target gene (knock-in) while at the same time inhibiting the expression of another gene by RNA interference (RNAi) (knockdown). For the present study, we constructed a hypoxia-inducible plasmid for the dual expression of heme oxygenase-1 (HO-1; knock-in, anti-oxidation) and the Src homology domain-2 containing tyrosine phosphatase-1 (SHP-1) microRNA (miSHP-1; knockdown, anti-apoptosis).
HO-1 and miSHP were chosen for their possible synergies in the treatment of ischemic heart disease. In addition to the anti-oxidative and the anti-apoptotic effects of HO-1 and miSHP-1, several studies have reported that the overexpression of HO-1 and the silencing of SHP-1 accelerate angiogenesis in ischemic myocardium [5–7]. HO-1, a stress-inducible anti-oxidant enzyme, exerts potent cardioprotective effects through its anti-inflammatory, anti-apoptotic, and anti-oxidant activity in ischemic tissue [8]. HO-1 gene therapy may protect the heart from ischemia/reperfusion injury by suppressing the early inflammatory response and inhibiting cardiomyocyte apoptosis [9]. Non-specific expression of HO-1, however, may induce unregulated proliferation of cells in normal tissues, which may induce tumor growth.
SHP-1, a key molecular mediator of apoptosis, negatively regulates anti-apoptotic signaling pathways, including extracellular signal-regulated kinase (ERK1/2) and BCL-2. SHP-1 binding to death receptors such as TNFR-1 and FAS-R promotes apoptosis through the regulation of de-phosphorylation in signal transduction pathways [10,11]. Decreased cardiomyocyte apoptosis and increased cardio-protection through Akt activation by the inhibition of the SHP-1 gene suggest that a therapeutic strategy designed to inhibit the expression of SHP-1 by miRNA would be effective in IHD [12,13]. Although miSHP-1 can reduce apoptosis and protect cells in ischemic tissue by inhibiting SHP-1 gene expression, induction of elevated levels of RNAi may be toxic to cells due to the interference with intrinsic cellular RNAi processes. Unlike siRNA, miRNA production can be regulated by promoters/enhancers, which transcribe genes in response to specific intracellular environments or signals, ensuring that RNAi activity occurs in specific tissues or cell conditions [14]. We hypothesized that a hypoxia-inducible plasmid for the expression of HO-1 and the production of miSHP-1 would minimize the side effects caused by unrestricted gene expression and synergistically maximize the survival of cardiomyocytes under hypoxic conditions.
2. Materials and methods
2.1. Materials
Branched poly(ethylenimine) (bPEI, Mw: 25 kDa), antioxidant assay kit, and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma Aldrich (St. Louis, MO). All cell culture products, including fetal bovine serum (FBS), Dulbecco's Phosphate Buffered Saline (DPBS), and Dulbecco's Modified Eagle's Medium (DMEM) were obtained from Invitrogen (GibcoBRL, Carlsbad, CA). Taqman® gene expression assays, Taqman® microRNA assays, and other required reagents for qRT-PCR were obtained from Applied Biosciences (Carlsbad, CA). Carboxy-DCFDA and RNA isolation kits were purchased from Invitrogen (Camarillo, CA). SHP-1, SHP-2 and β-actin antibodies were purchased from Cell Signaling Technology (Danvers, MA). The Amplified Opti-4CN Substrate Kit was obtained from Bio-Rad (Hercules, CA). The LDH Cytotoxicity Assay Kit II was obtained from Abcam Inc. (Cambridge, MA).
2.2. Construction of pEpo-SV-miSHP-HO-1
The HO-1 cDNA was amplified by PCR using pSV-HO-1 as a template and inserted downstream of the Epo enhancer and SV40 promoter of pEpo-SV, resulting in the construction of pEpo-SVHO-1. The SHP-1 siRNA sequence (5′-GGACAUUUCUUGUGCGUGA-3′) was inserted into the miR-30 backbone. The sequence of SHP-1 miRNA was as follows: 5′-GCTCTAGAATTCCAGTGAGCGAGGACATTTCTTGTGCGTGAGTGAAGCCACAGATGTCACGCACAAGAAATGTCCCTGCCTACTGTCTAGAGC-3′. The HinDIII recognition site was inserted for cloning. As a control, luciferase miRNA was also synthesized. The miRNA was inserted into the synthetic intron sequence in the GeneSwitch™ vector (Invitrogen; Carlsbad, CA). The intron containing miRNA was amplified by PCR and inserted into the pEpo-SV-HO-1 at the position of HinDIII, resulting in the construction of pEpo-SV-miSHPHO-1.
2.3. HO-1 quantification and antioxidant activity
The H9C2 cells were maintained in DMEM containing 10% FBS and 1% antibiotics at 37 °C under 5% CO2. At 80% confluence, the cells were seeded on 24-well plates at a density of 2.0×104 cells/well. After 24 h of incubation, the culture media were replaced with plain media containing pDNA/PEI polyplexes prepared by mixing 1 μg pDNA and 1 μg PEI. After 4 h, the cells were washed with PBS and cultured with DMEM. The culture plates were placed in a hypoxia chamber filled with mixed gas composed of 5% CO2, 1% O2, and 94% N2. After 2 days, the cells were lysed with 150 μl of lysis buffer containing a protease inhibitor cocktail (Roche; Indianapolis, IN). The cells were harvested and centrifuged for 30 s at 13,000 rpm. The amount of HO-1 production, antioxidant activity, and total protein was determined using a HO-1 ELISA kit (R&D Systems; Minneapolis, MN), antioxidant assay kit, and BCA assay kit (Pierce, Rockford, IL), respectively, according to the manufacturers' protocols.
2.4. Reactive oxygen species
Cells were prepared and transfected as described above. The culture media were replaced with phenol red-free DMEM containing 5 μmol carboxy-DCFDA at the completion of hypoxic exposure. After 30 min of incubation, the cells were washed with PBS and cultured in DMEM without phenol red. After 2 h, fluorescence intensities were recorded at Ex=495 nm and Em=520 nm using a fluorescence reader. Levels of reactive oxygen species (ROS) were calculated as the percent ROS present relative to normoxic conditions.
2.5. Cell viability and cytotoxicity
An MTT assay was performed to determine both the toxic effect of miRNA production and the cell viability under hypoxic conditions. MTT was added to the cells 48 h after transfection and the cell viability was calculated as a relative value. Cytotoxicity was measured using an LDH cytotoxicity assay kit (Abcam, Cambridge, MA) according to the manufacturer's instructions.
2.6. SHP-1 immunoprecipitation and western blot
Cells were prepared and transfected as described above. For quantitative analysis of SHP-1 protein, the cells were lysed using IP lysis buffer (Pierce; Rockford, IL) containing the protease inhibitor cocktail. The cells were harvested and centrifuged for 30 s at 13,000 rpm. The activity of SHP-1 in the cell lysates was determined using the SHP-1 immunoprecipitation kit (R&D Systems; Minneapolis, MN). The SHP-1 activity was calculated relative to the non-treated control group. For the western blot, the cell lysates were electrophoresed and transferred to a PVDF membrane. The membrane was washed, blocked, and incubated with the SHP-1, SHP-2 or β-actin primary antibody overnight at 4 °C. After washing three times, the membrane was incubated with HRP-conjugated secondary antibody for 1 h at room temperature. The protein present was detected using the Amplified Opti-4CN Substrate Kit.
2.7. SHP-1 microRNA production
Cells were seeded and transfected as described above. Total microRNA was isolated using a mirVana™ miRNA isolation kit, and cDNA was prepared using a TaqMan® microRNA reverse transcription kit according to the manufacturer's protocol (Ambion). The production of SHP-1 microRNA was calculated by the comparative CT (ΔΔCT) method with an endogenous control of 4.5S RNA. No-reverse-transcription controls were run to rule out genomic DNA (gDNA) contamination. A standard reaction was performed using the StepOnePlus™ Real-Time PCR System (Applied Biosystems) following the TaqMan® small RNA assay. The data was analyzed using DataAssist™ Software (Applied Biosystems) for calculating the relative expression of SHP-1.
2.8. SHP-1 mRNA qRT-PCR
Cells were prepared and transfected as described above. Total RNA was isolated using the PureLink™ RNA mini kit (Ambion), and cDNA was prepared using a high capacity RNA to cDNA kit according to the manufacturer's instructions. The level of SHP-1 was determined based on the comparative CT (ΔΔCT) method to calculate the relative expression of SHP-1 to the β-actin control using the TaqMan® gene expression assays (the primer sequence for SHP-1: GGACATTTC TTGTGCGTGA, pre-developed rat actin control for TaqMan® assays). No-reverse-transcription controls were run to rule out gDNA contamination. Fast reaction was performed using the StepOnePlus™ Real-Time PCR System (Applied Biosystems) following the TaqMan® gene expression assay protocol. The data was analyzed using DataAssist™ Software (Applied Biosystems) to calculate the relative expression of SHP-1.
2.9. Caspase activity
Cells were seeded and transfected as described above. The cells were lysed using IP lysis buffer (Pierce; Rockford, IL) containing the protease inhibitor cocktail. The cells were harvested and centrifuged for 30 s at 13,000 rpm, after which the cell lysates were collected. The caspase activity in the cell lysates was measured by Caspase-Glo® 3/7 assay (Promega; Madison, WI) according to the manufacturer's protocol. The caspase activities in the miSHP-1 treatment groups under hypoxic or normoxic conditions were calculated as the decrease in caspase activity compared to the miLuc treatment groups.
3. Results and discussion
3.1. Construction of HO-1 and miSHP-1 dual expression plasmids
The previously validated cDNA sequence [13], contained in the backbone of the primary miR-30 mRNA and encoded the SHP-1 or luciferase (Luc) pre-miRNA, was cloned to express mRNA under the control of the hypoxia-inducible Epo enhancer. The miR-30 stem-loop RNA precursor with the polymerase II-dependent promoter generates miRNA under the precise control of the promoter and produces up to 12 fold higher levels of mature miRNAs than hairpin structures [15–17]. The precursor stem was substituted with the SHP-1- or Luc-specific sequence to yield miRNA, which blocks the expression of endogenous mRNA. The four types of plasmids were constructed with or without the Epo enhancer, SV40 promoter, and miR-30 mRNA for verification of the combinatorial activity of HO-1 and miSHP-1 (Fig. 1). The pEPO-SV-miSHP-HO-1 and pSV-miSHP-HO-1 were prepared to compare the levels of HO-1 expression and miSHP-1 production under hypoxic and normoxic conditions. The pEPO-SV-miLuc-HO-1 and pSV-miLuc-HO-1 were constructed to serve as negative controls.
Fig. 1.
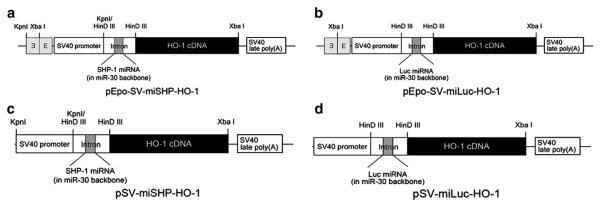
Schematic diagrams of the plasmid constructions. Hypoxia-inducible plasmids were constructed for the expression of HO-1 and the production of (a) miSHP-1 or (b) miLuc. Non-inducible plasmids (c, d) were constructed to serve as negative controls.
3.2. HO-1 expression under hypoxic conditions
HO catalyzes the degradation of heme into carbon monoxide (CO), ferrous iron (Fe2+) and biliverdin [18]. In addition to its role in heme catabolism and erythrocyte turnover, HO-1, the inducible isoenzyme of HO, is a central molecular mediator in vascular biology [8,19,20]. HO-1 plays a role in the anti-oxidative, anti-inflammatory, anti-apoptotic, and pro-angiogenesis pathways [21]. The protective effects of HO-1 make it a promising potential therapy for the treatment of a variety of cardiovascular diseases. Overexpression of HO-1, however, may facilitate tumor growth through its cytoprotective and anti-apoptotic activities [22]. The expression of HO-1, therefore, must to be regulated to inhibit the apoptosis of hypoxic cells, while at the same time not inducing tumorigenesis. The expression of HO-1 in hypoxic and normoxic H9C2 cells, i.e. rat cardiomyocytes, was determined to confirm the Epo enhancer's regulation of gene expression in response to hypoxia. PEI, one of the most powerful cationic polymers for transfection, was used to maximize the transfection efficiency of the plasmids, with the ratio of PEI to DNA fixed at 1:1 (w/w) to minimize cytotoxicity. The polyplex of PEI/pDNA was used to transfect cells incubated under normoxic conditions. 4 h after transfection, the cells were moved to a cell culture chamber filled with mixed gas composed of 94% N2, 5% CO2, and 1% O2. The HO-1 expression in the cell lysates was quantitatively determined. The HO-1 expression in the hypoxic H9C2 cells increased ~3-fold in the pEpo-SV-miSHP-HO-1 and the pEpo-SV-miLuc-HO-1 treatment groups compared to normoxic conditions, while there was no difference in the HO-1 expression following transfection with the pSV-miSHP-HO-1 or the pSV-miLuc-HO-1 plasmids (Fig. 2). The plasmids containing the Epo enhancer consistently produced higher levels of HO-1 in response to hypoxia.
Fig. 2.
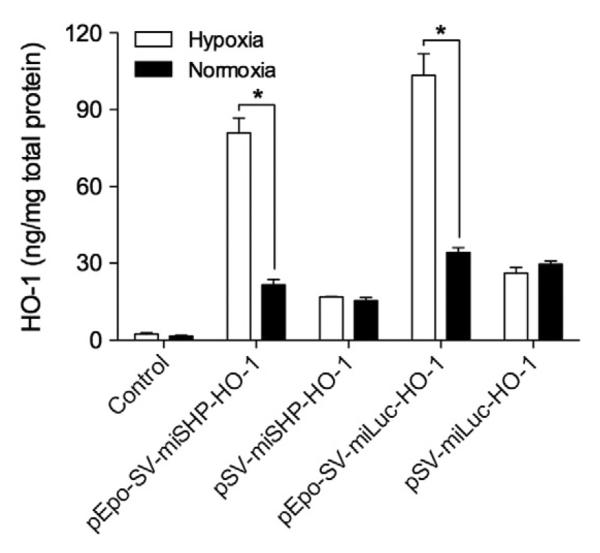
HO-1 expression under hypoxic and normoxic conditions. The H9C2 cells were incubated under hypoxic or normoxic conditions for 48 h following transfection. The level of HO-1 was quantitatively determined in the cell lysates using an ELISA kit, and the total protein was quantified by BCA assay (data presented as mean±SD, n=4 triplicate, *p<0.01).
3.3. Bioactivity of HO-1
The enzymatic activity of HO reduces oxidative stress, the inflammatory response, and apoptosis through the removal of heme and the generation of CO and Fe2+. The final metabolites of heme stimulate a signaling pathway that protects cells against oxidative stress [23]. In addition to the indirect effects of the metabolites, CO is involved in the anti-inflammatory response, while Fe2+ helps to protect cells from oxidative stress [24]. Among the isoforms of HO, HO-1 prevents or moderates a wide variety of diseases associated with elevated levels of reactive oxygen-containing molecules. The anti-oxidative activity of HO-1 occurs indirectly through the interaction between bilirubin and NADPH oxidase [25]. Biliverdin, a metabolite of HO-1, is rapidly reduced to bilirubin. Bilirubin suppresses the enzymatic activity of NADPH oxidase, a major source of ROS, thereby greatly reducing oxidative tissue damage [26,27]. Hence, the levels of ROS in the cells and the anti-oxidant activity present were determined following transfection of the plasmids.
The anti-oxidant activity in H9C2 cell lysates and the level of ROS in intact live H9C2 cells after plasmid transfection were determined under hypoxic and normoxic conditions. The anti-oxidant activity relative to the control group was ~130% in the pEpo-SV-HO-1 group and ~110% in the pSV-HO-1 group under hypoxic conditions. Under normoxic conditions, the anti-oxidant activity relative to the control group was from ~110% to ~115% in the all treatment groups (Fig. 3a). The relative levels of ROS were ~40% in the pEpo-SV-HO-1 group and ~60% in the pSV-HO-1 group under hypoxic conditions, and from ~55% to ~60% in all the treatment groups under normoxic conditions (Fig. 3b). The decrease in ROS was a consequence of the increased anti-oxidant activity following HO-1 transfection. Both biliverdin and bilirubin, metabolites of HO-1, play an important role in scavenging ROS in cells. Bilirubin acts as a chain breaking anti-oxidant to reduce peroxyl radicals, while biliverdin is capable of trapping peroxyl radicals [28]. Since HO-1 is involved in multiple steps of both the anti-oxidative and anti-apoptosis pathways, its mechanism of action is hard to precisely define. The known activities of HO-1 include: 1) a decrease in the level of heme (pro-oxidant); 2) an increase in the level of bilirubin (anti-oxidant); 3) producing CO for anti-oxidation and anti-apoptosis; and 4) an increase in the level of ferritin to remove and detoxify free ferric ion [29].
Fig. 3.

Effects of HO-1 expression as determined by the measurement of anti-oxidant activity and levels of ROS. (a) Total anti-oxidant activity in the cells following transfection with one of four different plasmid constructs expressing HO-1 was determined under hypoxic and normoxic conditions and compared. (b) The levels of ROS were detected under the same experimental conditions as the anti-oxidant activity assay (data presented as the mean±SD, n=4 triplicate, *p<0.01). The anti-oxidant activity and ROS level were calculated as relative values to the non-transfected control. The raw values of non-transfected controls were 1.73±0.04 under hypoxia and 1.96±0.06 under normoxia (anti-oxidant activity/mg total protein) and ~1,210,000±23,000 under hypoxia and ~1,360,000±26,000 under normoxia (fluorescence intensity of ROS/viable cells; data presented as the mean±SD, n=4 triplicate).
3.4. SHP-1 down-regulation
The miRNAs, a class of 21 to 23 nucleotide long noncoding RNAs, provide crucial biological functions by suppressing the translation of target mRNAs. First-generation miRNAs use short hairpin RNA that includes precursor microRNA (pre-miRNA) and are transcribed under the control of RNA polymerase III promoters [30]. Polyadenylation and splicing may occur because ~22 nucleotide long mature miRNA is encoded in the arm of the stem that contains an ~80 nucleotide long hairpin structure [14]. Drosha in the nucleus cleaves the primary transcript for producing pre-miRNA, which is then processed into an siRNA duplex by Dicer in the cytoplasm [31]. The RNA-induced silencing complex (RISC) then binds to the siRNA duplex. Attempts to develop expression-controllable vector-based miRNA have often focused on the human mir-30 miRNA. The mir-30 expresses the primary RNA transcript under the control of RNA polymerase II. The gene-specific duplex for target mRNA silencing can be inserted in the stem of the primary miR-30 transcript without influencing the normal miR-30 maturation and endogenous miRNA processing [17]. The mir-30 precursor is essential to produce mature mir-30 miRNA, and the location of miRNAs present within an ~70 nucleotide precursor RNA stem-loop is a common feature of mir-30 miRNAs [16,32]. We based the plasmids used in this study on mir-30 miRNA control of the expression of miSHP-1 or miLuc under hypoxic conditions.
The anti-SHP-1 or -Luc miRNA sequence was placed in the mir-30 stem loop in the intron region of the plasmid that regulates the gene expression by the Epo enhancer and the SV40 promoter, one of the RNA polymerase II-dependent promoters. The mature miSHP-1 production was first evaluated in H9C2 cells under hypoxic or normoxic conditions (Fig. 4a). The production of miSHP-1 increased in the pEpo-SV-miSHP-1-HO-1 group compared to the pSV-miSHP-1-HO-1, the pEpo-SV-miLuc-HO-1 and the pSV-miLuc-HO-1 groups. The presence of the Epo enhancer significantly increased the production of miSHP-1 in hypoxic cells relative to normoxic cells, while there was no difference in miSHP-1 expression in the pSV-miSHP-HO-1 group between the hypoxic and normoxic states. These results validate our hypothesis that the pEpo-SV-miSHP-1-HO-1 plasmid construct could produce miRNA in response to hypoxia.
Fig. 4.

SHP-1 down-regulation in hypoxic and normoxic cells. Expression profile of: (a) Mature miSHP-1, (b) SHP-1 mRNA, (c) SHP-1 protein expression, (d) SHP-1 and SHP-2 western blot, and (e) cell viability (data presented as the mean±SD, n=4 triplicate, *p<0.01). The production of miSHP-1, level of SHP-1 mRNA, and SHP-1 protein expression were calculated as relative values to the non-transfected control.
The down-regulation of SHP-1 mRNA as a consequence of miSHP-1 production was evaluated under hypoxic and normoxic conditions (Fig. 4b). The level of SHP-1 mRNA in the cells that were transfected with the pEpo-SV-miSHP-HO-1 plasmid was reduced by ~40% under hypoxic conditions and by ~20% under normoxic conditions, whereas the pSV-miSHP-HO-1 decreased the SHP-1 mRNA production by ~20% under both hypoxic and normoxic conditions. The silencing of SHP-1 mRNA led to reduction in the SHP-1 protein expression. The level of SHP-1 protein was determined by SHP-1 immunoprecipitation as shown in Fig. 4c. The relative SHP-1 protein levels in the pEpo-SV-miSHP-HO-1 treatment groups were reduced by ~45% under hypoxic conditions and ~25% under normoxic conditions, while the pEpo-SV-miLuc-HO-1 plasmid decreased SHP-1 expression by only ~5% under both hypoxic and normoxic conditions. The constitutively expressed, non-hypoxia responsive plasmid, pSV-miSHP-HO-1, decreased the expression of SHP-1 protein by ~20%, which approximates the reduction in expression of SHP-1 seen with the pEpo-SV-miSHP-HO-1 plasmid under normoxic conditions. This down-regulation of SHP-1 protein expression was further confirmed by western blot (Fig. 4d). The analysis of cell viability demonstrated that none of the plasmid constructs were associated with cytotoxicity (Fig. 4e). In aggregate, these results indicate that it is possible to construct a miSHP-1 expression vector that specifically produces miSHP-1 and inhibits SHP-1 mRNA, thereby suppressing the expression of SHP-1 in response to hypoxia.
Further, these results demonstrate that miSHP-1 can be produced in hypoxic cells under the control of a hypoxia-inducible promoter/enhancer by substituting the mir-30 stem loop with an anti-SHP-1 miRNA sequence. The hypoxia-inducible miSHP-1 plasmid produced mature miSHP-1 in response to hypoxia in the plasmid-transfected cells. This miSHP-1 induced the specific degradation of SHP-1 mRNA, thereby suppressing the expression of SHP-1 protein. Conventional RNAi based on synthetic dsRNAs have enjoyed only limited success in mammalian systems. Our hypoxia-inducible plasmid-based miRNA offers several advantages over synthetic siRNAs: (1) ease of transfection, (2) cost-effectiveness, (3) controlled miRNA production, and (4) continuous miRNA expression [33]. We believe that the technology described in the present study has the potential to lead to a paradigm change in the application of synthetic RNAi gene therapy.
3.5. Anti-apoptotic effects
Over 100 human protein–tyrosine phosphatases (PTPs) have been reported to play crucial roles in cell signaling transduction, cell physiology, and cellular pathology [10]. The tyrosine-specific PTPs can be classified into the receptor-like PTPs (RPTPs) and the non-receptor-like PTPs (NRPTPs), or cytosolic PTPs. A subfamily of the cytoplasmic PTPs that contains an Src homology-2 N-terminal domain and a C-terminal protein–tyrosine phosphatase domain is denoted as SHP. SHP-1 and SHP-2 are key mediators in cell signaling and cell growth [34,35]. SHP-1 and SHP-2 play important roles in the response to oxidative stress [36,37]. Expression of SHP-1 is increased following a myocardial infarction, suggesting that SHP-1 may contribute to cellular damage [38]. Combining the anti-oxidant activity of overexpression of HO-1 and the anti-apoptotic activity of anti-SHP-1 miRNA may prolong the survival rate of cardiomyocytes under ischemic conditions.
The anti-apoptotic activities of the plasmids were evaluated by measuring caspase activity, a key step in hypoxia-induced apoptosis (Fig. 5a). Caspase activity following transfection of plasmids containing miSHP-1 was calculated as a decrease in caspase activity relative to that of miLuc. The decrease in caspase activity with pEpo-SV-miSHP-HO-1 was ~95,000 RLU/mg protein under hypoxic conditions and ~60,000 RLU/mg protein under normoxic conditions, while the decrease in caspase activity with pSV-miSHP-HO-1 was ~20,000 RLU/mg protein under both hypoxic and normoxic conditions. The decrease in caspase activity associated with pEpo-SV-miSHP-HO-1 suggests that this plasmid may have a role to play in inhibiting ischemia-induced apoptosis. The anti-apoptotic activity of miSHP-1 derives from its negative regulatory role in signal transduction pathways by dephosphorylation of the SHP-1 receptor and by inhibiting the binding of TNFR1 and Fas-R [13,38]. Fas, an important mediator in the setting of acute myocardial ischemia and infarction, induces apoptosis in cardiomyocytes through the stimulation of Fas-R under hypoxic conditions. Our results suggest that the decrease in apoptosis of the hypoxic cells was likely due to the inhibition of Fas binding to Fas-R as a result of silencing the expression of SHP-1. Silencing of SHP-1 mRNA also induces phosphorylation of Akt, which reduces the apoptosis of cardiomyocytes under ischemic conditions.
Fig. 5.

Anti-apoptotic activity of the dual plasmid. (a) Decrease in Caspase-3/7 activity (n=4 triplicate, *p<0.01) and (b) LDH release (*p<0.05, one-way ANOVA) following plasmid transfection (data presented as the mean±SD, n=4 triplicate).
In addition to the determination of caspase activity, the release of LDH as a marker of cell death was measured under the same conditions (Fig. 5b). The percentages of the cell population releasing LDH under hypoxic conditions were ~30% in the control group, ~28% in the pSV-miLuc-HO-1 group, ~25% in the pEpo-SV-miLuc-HO-1 group, ~20% in the pSV-miSHP-HO-1 group, and ~8% in the pEpo-SV-miSHP-HO-1 group. The difference between the pEpo-SV-miSHP-HO-1 group and the pEpo-SV-miLuc-HO-1 group is likely due to the synergistic effects of the down-regulation of SHP-1 and the overexpression of HO-1. It has been reported that both HO-1 overexpression and SHP-1 down-regulation accelerate angiogenesis in ischemic tissues, in addition to the anti-oxidative and the anti-apoptotic activity of HO-1 and miSHP-1 [5–7]. HO-1 and angiogenic factors, including vascular endothelial growth factor (VEGF), activate a positive-feedback cycle to enhance neovascularization in adult tissues. HO-1 expression and its activity in human endothelial cells are prolonged by VEGF, and the angiogenesis induced by VEGF decreases when HO-1 is inhibited [39]. Thus, we assume that HO-1 plays at least two roles in ischemic tissues: 1) as an anti-inflammatory agent that inhibits leukocyte infiltration; and 2) promoting VEGF-mediated angiogenesis to facilitate tissue repair. Similar to HO-1, miSHP-1 is also involved in VEGF-induced angiogenesis. The phosphorylated KDR/flk-1, a substrate for SHP-1, is induced by binding of VEGF to KDR/flk-1, resulting in conformational changes in the receptor and dimerization [40]. The interaction between SHP-1 and the activated VEGF initiates negative regulatory activity in the signal transduction pathways by dephosphorylation of the receptors or their substrates [41,42]. SHP-1 is also associated with the inhibition of endothelial cell proliferation mediated by TNF-alpha and the reduction in angiogenesis by the inactivation of KDR/flk-1 [43]. KDR/flk-1 inactivation by SHP-1 reduces VEGF-induced angiogenesis, suggesting that miSHP-1 may accelerate angiogenesis by silencing SHP-1. We conclude that the synergistic effect of pEpo-SV-miSHP-HO-1 resulting in improved cardiomyocyte survival under hypoxic conditions is due not only of its anti-oxidative and anti-apoptotic activities, but also of enhanced angiogenesis.
4. Conclusion
The hypoxia-inducible plasmid for the dual expression of HO-1 and miSHP-1 demonstrated higher levels of HO-1 expression and mature miSHP-1 production in hypoxic cells compared to normoxic cells. The expression of HO-1 increased total anti-oxidant activity, resulting in a decrease in the level of ROS in cardiomyocytes. The hypoxia-responsive production of miSHP-1 down-regulated SHP-1 mRNA and reduced the production of the SHP-1 protein. The synergistic efficacy of HO-1 and miSHP-1 significantly decreased the apoptosis of cardiomyocytes under hypoxic conditions. This gene expression plasmid combination, pEpo-SV-miSHP-HO-1, is a promising construct for the protection of hypoxic cells. This gene expression plasmid combines the anti-oxidative effects of HO-1 and the anti-apoptotic activity of miSHP-1. In addition, both gene constructs accelerate VEGF-induced angiogenesis. The combination of these knock-in and knockdown genes in one plasmid enhances the expression of HO-1, produces mature miSHP-1, down-regulates SHP-1 mRNA, and reduces the production of SHP-1 protein in hypoxic cells compared to normoxic cells. Examinations of the efficacy of pEpo-SV-miSHP-HO-1 in vivo are warranted to determine its efficacy for the treatment of clinical ischemia.
Acknowledgment
This work was supported by the NIH grants HL065477 (SW Kim) and HL071541 (DA Bull). The work of Minhyung Lee was supported by the Ministry of Education, Science and Technology, Korea (2012K001394).
References
- [1].McGinn AN, Nam HY, Ou M, Hu N, Straub CM, Yockman JW, Bull DA, Kim SW. Bioreducible polymer-transfected skeletal myoblasts for VEGF delivery to acutely ischemic myocardium. Biomaterials. 2011;32:942–949. doi: 10.1016/j.biomaterials.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lavu M, Gundewar S, Lefer DJ. Gene therapy for ischemic heart disease. J. Mol. Cell. Cardiol. 2011;50:742–750. doi: 10.1016/j.yjmcc.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rissanen TT, Yla-Herttuala S. Current status of cardiovascular gene therapy. Mol. Ther. 2007;15:1233–1247. doi: 10.1038/sj.mt.6300175. [DOI] [PubMed] [Google Scholar]
- [4].Vinge LE, Raake PW, Koch WJ. Gene therapy in heart failure. Circ. Res. 2008;102:1458–1470. doi: 10.1161/CIRCRESAHA.108.173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lin HH, Chen YH, Chang PF, Lee YT, Yet SF, Chau LY. Heme oxygenase-1 promotes neovascularization in ischemic heart by coinduction of VEGF and SDF-1. J. Mol. Cell. Cardiol. 2008;45:44–55. doi: 10.1016/j.yjmcc.2008.04.011. [DOI] [PubMed] [Google Scholar]
- [6].Suzuki M, Iso-o N, Takeshita S, Tsukamoto K, Mori I, Sato T, Ohno M, Nagai R, Ishizaka N. Facilitated angiogenesis induced by heme oxygenase-1 gene transfer in a rat model of hindlimb ischemia. Biochem. Biophys. Res. Commun. 2003;302:138–143. doi: 10.1016/s0006-291x(03)00114-1. [DOI] [PubMed] [Google Scholar]
- [7].Sugano M, Tsuchida K, Maeda T, Makino N. SiRNA targeting SHP-1 accelerates angiogenesis in a rat model of hindlimb ischemia. Atherosclerosis. 2007;191:33–39. doi: 10.1016/j.atherosclerosis.2006.04.021. [DOI] [PubMed] [Google Scholar]
- [8].Stocker R, Perrella MA. Heme oxygenase-1: a novel drug target for atherosclerotic diseases? Circulation. 2006;114:2178–2189. doi: 10.1161/CIRCULATIONAHA.105.598698. [DOI] [PubMed] [Google Scholar]
- [9].Tang YL, Tang Y, Zhang YC, Qian K, Shen L, Phillips MI. Protection from ischemic heart injury by a vigilant heme oxygenase-1 plasmid system. Hypertension. 2004;43:746–751. doi: 10.1161/01.HYP.0000120152.27263.87. [DOI] [PubMed] [Google Scholar]
- [10].Chong ZZ, Maiese K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol. Histopathol. 2007;22:1251–1267. doi: 10.14670/hh-22.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Forget G, Gregory DJ, Whitcombe LA, Olivier M. Role of host protein tyrosine phosphatase SHP-1 in Leishmania donovani-induced inhibition of nitric oxide production. Infect. Immun. 2006;74:6272–6279. doi: 10.1128/IAI.00853-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- [13].Nam HY, Kim J, Kim S, Yockman JW, Kim SW, Bull DA. Cell penetrating peptide conjugated bioreducible polymer for siRNA delivery. Biomaterials. 2011;32:5213–5222. doi: 10.1016/j.biomaterials.2011.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zeng Y, Cullen BR. Sequence requirements for micro RNA processing and function in human cells. RNA. 2003;9:112–123. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- [17].Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, Sheth N, Bradshaw J, Burchard J, Kulkarni A, Cavet G, Sachidanandam R, McCombie WR, Cleary MA, Elledge SJ, Hannon GJ. Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- [18].Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. U. S. A. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008;117:231–241. doi: 10.1161/CIRCULATIONAHA.107.698316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Loboda A, Jazwa A, Grochot-Przeczek A, Rutkowski AJ, Cisowski J, Agarwal A, Jozkowicz A, Dulak J. Heme oxygenase-1 and the vascular bed: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2008;10:1767–1812. doi: 10.1089/ars.2008.2043. [DOI] [PubMed] [Google Scholar]
- [21].Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- [22].Jozkowicz A, Was H, Dulak J. Heme oxygenase-1 in tumors: is it a false friend? Antioxid. Redox Signal. 2007;9:2099–2117. doi: 10.1089/ars.2007.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Snyder SH, Baranano DE. Heme oxygenase: a font of multiple messengers. Neuropsychopharmacology. 2001;25:294–298. doi: 10.1016/S0893-133X(01)00275-5. [DOI] [PubMed] [Google Scholar]
- [24].Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- [25].Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16093–16098. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lanone S, Bloc S, Foresti R, Almolki A, Taille C, Callebert J, Conti M, Goven D, Aubier M, Dureuil B, El-Benna J, Motterlini R, Boczkowski J. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J. 2005;19:1890–1892. doi: 10.1096/fj.04-2368fje. [DOI] [PubMed] [Google Scholar]
- [27].Jiang F, Roberts SJ, Datla S, Dusting GJ. NO modulates NADPH oxidase function via heme oxygenase-1 in human endothelial cells. Hypertension. 2006;48:950–957. doi: 10.1161/01.HYP.0000242336.58387.1f. [DOI] [PubMed] [Google Scholar]
- [28].Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- [29].Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009;61:290–302. doi: 10.1016/j.addr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- [30].Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, McCombie WR, Elledge SJ, Hannon GJ. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- [31].Zeng Y, Cai X, Cullen BR. Use of RNA polymerase II to transcribe artificial microRNAs. Methods Enzymol. 2005;392:371–380. doi: 10.1016/S0076-6879(04)92022-8. [DOI] [PubMed] [Google Scholar]
- [32].Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- [33].Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell. 2002;9:1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- [34].Yamauchi K, Milarski KL, Saltiel AR, Pessin JE. Protein–tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc. Natl. Acad. Sci. U. S. A. 1995;92:664–668. doi: 10.1073/pnas.92.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yi T, Mui AL, Krystal G, Ihle JN. Hematopoietic cell phosphatase associates with the interleukin-3 (IL-3) receptor beta chain and down-regulates IL-3-induced tyrosine phosphorylation and mitogenesis. Mol. Cell. Biol. 1993;13:7577–7586. doi: 10.1128/mcb.13.12.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein–tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- [37].Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–252. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- [38].Sugano M, Tsuchida K, Hata T, Makino N. RNA interference targeting SHP-1 attenuates myocardial infarction in rats. FASEB J. 2005;19:2054–2056. doi: 10.1096/fj.05-4020fje. [DOI] [PubMed] [Google Scholar]
- [39].Bussolati B, Ahmed A, Pemberton H, Landis RC, Di Carlo F, Haskard DO, Mason JC. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood. 2004;103:761–766. doi: 10.1182/blood-2003-06-1974. [DOI] [PubMed] [Google Scholar]
- [40].Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- [41].Chen HE, Chang S, Trub T, Neel BG. Regulation of colony-stimulating factor 1 receptor signaling by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol. Cell. Biol. 1996;16:3685–3697. doi: 10.1128/mcb.16.7.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- [43].Guo DQ, Wu LW, Dunbar JD, Ozes ON, Mayo LD, Kessler KM, Gustin JA, Baerwald MR, Jaffe EA, Warren RS, Donner DB. Tumor necrosis factor employs a protein–tyrosine phosphatase to inhibit activation of KDR and vascular endothelial cell growth factor-induced endothelial cell proliferation. J. Biol. Chem. 2000;275:11216–11221. doi: 10.1074/jbc.275.15.11216. [DOI] [PubMed] [Google Scholar]