

SUMMARY
Previous studies have indicated that NAD(P)H:quinone oxidoreductase [DT-diaphorase (NQO1)] plays an important role in the bioreductive activation of quinone-containing antitumor agents. Although these studies demonstrated that purified NQO1 can reduce these compounds in vitro, the importance NQO1 in the intracellular activation of quinone-containing antitumor agents remains controversial. In our study, we transfected human NQO1 into Chinese hamster ovary cells that do not normally express NQO1 activity and obtained stable clones that expressed NQO1 activity of 19–3527 nmol of 2,6-dichlorophenolindophenol reduced/min/mg of protein. The level of NQO1 expression correlated with an increased killing by streptonigrin, EO9 (3-hydroxymethyl-5-aziridinyl-1-methyl-2-(1H-indole-4,7-dione)-propenol), and 2,5-diaziridinyl-3,6-dimethyl-1,4-benzoquinone, but mitomycin C sensitivity was independent of this activity. NQO1 expression also led to a slight decrease in the sensitivity of cells to menadione. Our data demonstrate that compounds that are efficient substrates for NQO1 in vitro are also bioactivated in cultured mammalian cells when they are transfected with human NQO1. These results are consistent with the relative abilities of mitomycin C, streptonigrin, EO9, and 2,5-diaziridinyl-3,6-dimethyl-1,4-benzoquinone to serve as substrates for bioreduction by human NQO1, and show that NQO1 levels are not necessarily predictive in terms of sensitivity to mitomycin C.
NQO1 (DT-diaphorase; E.C. 1.6.99.2) is an obligate two-electron reductase that can reduce a variety of diverse compounds and is thought to serve a protective function in cells by reducing potentially damaging compounds to less reactive metabolites. Conversely, NQO1 has been shown to reduce antitumor quinones to reactive intermediates. This bioactivation is thought to play an important role in the cytotoxicity of these agents (1, 2). Elevated levels of NQO1 in tumor compared with nontumor tissues have been reported (3–6), so NQO1 has become a target for the selective activation of bioreductive agents in tumors (7, 8).
MMC is considered to be the prototype bioreductive alkylating agent. Most information regarding the role of NQO1 in the activation of MMC has come from studies using the NQO1 inhibitor dicumarol (9,10) or studies in which NQO1 activity levels in cell lines were correlated to sensitivity to MMC (2, 11, 12). A complication with studies using dicumarol is that dicumarol is not specific for NQO1 (13), and it affects other enzymes that are capable of MMC bioactivation (14, 15). MMC can be metabolized to a reactive intermediate by many enzymes, including NQO1 (2, 16, 17), NADPH: cytochrome P-450 reductase (16, 18), NADH:cytochrome b5; reductase (15), xanthine oxidase (18, 19), and xanthine dehydrogenase (19). The relative contribution of these enzyme systems in activation of MMC in vivo is difficult to delineate because these reductive enzymes are present in most cells.
Although it is clear that MMC can be metabolized by NQO1 (2) and can be bioactivated by NQO1 to DNA cross-linking species in a cell-free system at physiological pH (17), the role of NQO1 in the toxicity of MMC in a cellular system remains controversial. The ability of MMC to function as a substrate for numerous reductases makes the contribution and relative roles of individual reductases difficult to elucidate. Many studies have associated NQO1 activity with MMC sensitivity in tumor cell systems (2, 11, 20) and xenografts (12). Other studies brought the role of NQO1 in MMC toxicity into question because of a demonstrated lack of a correlation between MMC sensitivity and NQO1 activity in a panel of 15 tumor cell lines (21). In the same study, a correlation was observed between NQO1 activity and EO9 cytotoxicity (21). More recently, a correlation was observed between MMC cytotoxicity and NQO1 activity in a series of human lung and breast cancer cell lines (22). Studies using the NCI panel of tumor cell lines have also shown that MMC and porfiromycin are among the top four agents whose toxicity correlates with NQO1 expression (23). Although useful in suggesting a direction for further research, the major problem with these data is that, by definition, they are correlative rather than causal.
The only direct mechanistic evidence that NQO1 plays a role in MMC toxicity in cells was obtained by Hodnick et al. (24), who observed an increase in MMC and porfiromycin cytotoxicity in CHO cells after transfection with rat NQO1 cDNA. There are major differences, however, in the catalytic efficiencies of rat and human NQO1 with both antitumor quinones (25) and nitroaziridines such as CB1954 [5-(aziridin-1-yl)-2,4-dinitrobenzamide] (26). In addition, there have been no attempts to address the role of NQO1 in the cytotoxicity of quinones other than MMC and porfiromycin using transfection studies. To this end, we focused our research on the use of CHO cells transfected with the human NQO1 cDNA to address the role of NQO1 in bioactivation of MMC and other antitumor quinones.
Experimental Procedures
Materials
Ampicillin, aprotinin, bovine serum albumin (fraction V), cytochrome c, DCPIP, dicumarol, glutamine, menadione (2-methyl-1,4-napthoquinone), MTT formazan, NADH, NADPH, phenylmethylsulfonyl fluoride, Triton X-100, Tween 20, and xanthine were purchased from Sigma Chemical (St. Louis, MO). G418, Lipofectin reagent, Puck’s Saline G, and subcloning efficiency DH5α Escherichia coli were purchased from Life Technologies (Grand Island, NY). Plasmid DNA isolation kits and gel extraction kits were purchased from Qiagen (Studio City, CA). Restriction endonucleases and DNA modifying enzymes were purchased from Boehringer-Mannheim (Indianapolis, IN). PCDNA3 plasmid was purchased from In-Vitrogen (San Diego, CA). Ham’s F12 media and penicillin/streptomycin were purchased from Irvine Scientific (Santa Ana, CA). Fetal calf serum was purchased from Atlanta Biologicals (Norcross, GA), and newborn calf serum was from Gemini (Irvine, CA). sodium dodecyl sulfate-polyacrylamide gel electrophoresis reagents were purchased from BioRad (Richmond, CA). Anti-mouse IgG-horseradish peroxidase conjugate and ECL detection kits were purchased from Amersham (Arlington Heights, IL). Bicinchoninic acid total protein assay reagent was purchased from Pierce Chemical (Rockford, IL). Bacto-tryptone, yeast extract and Bacto-Agar for LB were purchased from Difco (Detroit, MI). All other reagents were of analytical grade.
Cell culture
CHO cells used for these studies were a glyA− auxotroph derived from CHO-K1 cells. Cells were grown in Ham’s F12 medium supplemented with 4% fetal calf serum, 3% newborn calf serum, 20 mm HEPES, pH 7.4, 1 mm glutamine, and 50 U/ml penicillin/50 μg/ml streptomycin in a humidified atmosphere of 5% CO2/95% air at 37° in tissue culture plates.
Construction of mammalian cell expression plasmid and transfection of NQO1 into CHO cells
The human NQO1 cDNA was contained in the pKKRDTD bacterial expression plasmid. The construction of this plasmid has been described elsewhere (25). The cDNA for human NQO1 was isolated from the pKKRDTD plasmid by digestion with NcoI/HindIII, and the resulting cDNA was isolated from a preparative agarose gel using a gel extraction kit. The cDNA fragment was blunt-ended using T4 DNA polymerase and ligated into the mammalian expression vector PCDNA3, which had been digested with EcoRV and treated with shrimp alkaline phosphatase. The products from the ligation reaction were transfected into subcloning efficiency DH5α E. coli and plated on LB agar plates containing 50 μg/ml ampicillin. Resulting ampicillin-resistant clones were picked and grown up in a volume of 5 ml of LB broth containing 50 μg/ml ampicillin, and plasmid DNA was isolated from the individual clones. The plasmids were then digested with HindIII/XhoI and analyzed by agarose gel to verify ligation of the cDNA into PCDNA3. Plasmids that had incorporated human NQO1 cDNA were transfected into CHO cells using Lipofectin reagent, and the resulting G418-resistant cells were screened for NQO1 activity. NQO1 expression verified that the cDNA had been cloned with the proper orientation. Populations of CHO cells expressing human NQO1 were plated out at 300 cells/100-mm tissue culture plate, and individual clones were picked 10 days later using cloning cylinders. Individual clones were screened for NQO1 activity.
Enzyme activity measurements
Cells were grown to ~75% confluency in 100-mm tissue culture dishes (~2 × 106 cells/plate) and harvested by scraping the cells off the plate with a rubber policeman in phosphate-buffered saline. The cells were pelleted by centrifugation for 5 min in a microcentrifuge and then resuspended in cold 25 mm Tris·HCl, pH 7.4, or 100 mm phosphate buffer, pH 7.5. Cells were disrupted by three cycles of freeze/thawing using an ethanol/dry ice bath and centrifuged for 5 min in a microcentrifuge. The supernatant was collected and stored on ice until assay for enzyme activity. Whole-cell lysates were used for determining NADPH:cytochrome P-450 reductase and NADH:cytochrome b5 reductase activities. NQO1 activity was measured as described by Ernster (27) and modified by Benson et al. (28) using DCPIP as a substrate. NADPH:cytochrome P-450 reductase was assayed using the cytochrome c reduction method of Strobel and Dignam (29). The same assay was used for NADH:cytochrome b5 reductase determination except that NADH was substituted for NADPH. Xanthine dehydrogenase/oxidase was measured as described by Stirpe and Della Corte (30).
Western blot analysis of NQO1 protein
Cells were grown in 100-mm tissue culture dishes and lysed by the addition of 50 mm Tria·HCl, pH 8, 150 mm NaCl, 1 mm EDTA, 50 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, and 1% Triton X-100. The lysate was then centrifuged for 10 min at 13,000 × g, and the supernatant was collected. Twenty-five micrograms of total protein was then separated on a 15% polyacrylamide gel electrophoresis gel and transferred to nitrocellulose filters by electrophoretic transfer. Filters were then blocked overnight at 4° with TBST (10 mm Tria·HCl, pH 7.5, 100 mm NaCl, 0.1% Tween 20) containing 5% nonfat dried milk. The filter was then incubated with mouse anti-human NQO1 hybridoma medium in TBST containing 5% nonfat dry milk for 1 hr at room temperature. After the addition of primary antibody, the filter was washed three times with TBST. The secondary antibody was anti-mouse IgG-horseradish peroxidase conjugate and was added to TBST with 5% nonfat dry milk and incubated at room temperature for 1 hr. After 2° antibody addition, the filter was washed three times with TBST. The blot was then developed using ECL detection.
Toxicity assays
Cells were grown in 60-mm tissue culture dishes as described above and harvested by trypsinization. Trypsinized cells were counted using a hemacytometer and plated out at an appropriate cell density. After a 3-hr period for cell attachment, cell were treated with graded concentrations of MeDZQ, STPN, EO9, menadione, or MMC for 2 hr. After drug exposure, cells were washed with Puck’s Saline G, and complete Ham’s F12 medium was added. Colony formation was assessed 7–10 days later by fixing, staining, and counting. Surviving fractions were determined by standard techniques.
MTT assay
Cells were plated in 96-well plates at a density of 2 × 104 cells/ml and incubated overnight. Drug solutions were added in medium containing no serum, and the cells were treated for 2 hr. After treatment, cells were washed with Saline G, growth medium was added, and cells were incubated for 5–7 days. MTT (50 μg) was added, and the cells were incubated for an additional 4 hr. Medium/MTT solutions were removed carefully by aspiration, the MTT formazan crystals were dissolved in 100 μl of dimethylsulfoxide, and the absorbance was determined on a plate reader at 550 nm 31). IC50 (drug concentration resulting in 50% inhibition of absorbance at 550 nm) values were determined from semilog plots of percentage of control versus concentration of drug.
Protein assay
Total protein was determined using the bicinchoninic acid total protein assay reagent with bovine serum albumin as a standard.
Results
Expression of human NQO1 in CHO cells
The parental CHO cell line that was used for these transfections had no detectable NQO1 activity. Four transfected CHO cell lines with large differences in human NQO1 expression were selected for comparative study. NQO1 activity in the four transfected clones (DTD1, DTD812, DTD815, and DTD818) ranged from 19 ± 2 to 3527 ± 194 nmol of DCPIP reduced/min/mg of protein (Table 1). The NQO1-transfected cell lines were screened throughout the study period for NQO1 activity, and NQO1 activity remained stable in the absence of G418 selection. Western blot analysis of the NQO1-expressing clones showed similar results to the activity measurements in terms of levels of NQO1 (Fig. 1A). NQO1 activity versus NQO1 protein content in the transfected cell lines was linear, showing that expressed protein was active (Fig. 1B). Southern blot analysis of the DNA from the transfected cell lines indicated that the levels of NQO1 activity in the four clones were a function of the number of plasmids incorporated (data not shown).
TABLE 1. NQO1 activity in CHO cell lines transfected with human NQO1 cDNA.
Values are mean ± standard deviations of at least three independent determinations. NQO1 activity was measured in cell extracts by monitoring the dicumarol-inhibitable reduction of DCPIP at 600 nm using NADH as the electron donor substrate. NQO1 activity was stable in the transfected cell lines, and expression was maintained in the absence of G418 selection.
NQO1 activity is expressed in nmol of DCPIP reduced/min/mg of protein.
n.d. = not detectable.
Fig. 1.

A, Immunoblot of NQO1 protein in CHO and CHO cells transfected with human NQO1 cDNA. Twenty-five micrograms of cellular protein was separated on a 12.5% sodium dodecyl sulfate-polyacrylamide gel and transferred to a nitrocellulose membrane. NQO1 protein was detected with mouse anti-human NQO1 hybridoma medium, anti-mouse IgG-horseradish peroxidase conjugate, and ECL detection (Amersham). The mouse anti-human NQO1 hybridoma cell line was isolated at the Monoclonal Core Facility, Cancer Center, University of Colorado Health Sciences Center using recombinant human NQO1 protein. This mouse anti-human NQO1 antibody cross-reacts with rat NQO1. Lane 1, CHO; lane 2, DTD1; lane 3, DTD812; lane 4, DTD815; lane 5, DTD818; lane 6, H460 human lung carcinoma. H460 cellular protein was used as a positive control because the human NQO1 cDNA used in this study was originally cloned from this cell line. B, NQO1 protein content versus NQO1 activity in CHO cells expressing human NQO1. NQO1 protein content was quantified using an imaging densitometer (BioRad).
Effect of human NQO1 expression on antitumor quinone cytotoxicity
Clonogenic dose-response survival curves for CHO and the transfected cell lines exposed to MMC are presented in Fig. 2A. Toxicity data were obtained for the parent line and the transfectants indicating that MMC cytotoxicity is not dependent on the levels of NQO1 activity in this series of CHO cells. These data were confirmed using MTT assays shown in Table 2. Clonogenic dose-response curves for CHO and the four human NQO1 expressing cell lines exposed to EO9, STPN, and MeDZQ are shown in Fig. 2, B–D, respectively. For these three agents, the parental cell line, which has no detectable NQO1 activity, was the most resistant of the cell lines tested. Interestingly, there was no significant differences in the toxicity of EO9, STPN, and MeDZQ to DTD812 and DTD818 despite a ~5-6-fold difference in NQO1 activity, suggesting that there may be a threshold level of NQO1 activity above which cytotoxicity does not correlate with NQO1 activity. A threshold model for the relationship of NQO1 activity to cytotoxicity of agents that are activated by NQO1 has been proposed (32).
Fig. 2.

Toxicity of MMC (A), EO9 (B), STPN (C), and MeOZQ (D) to CHO and human NQO1-expressing CHO cells. Cells were plated out at appropriate cell densities on 60-mm tissue culture plates and exposed to graded drug concentrations for 2 hr. Drugs were dissolved in media before addition. After the 2-hr period, plates were rinsed twice with Puck’s Saline G, and complete medium was added to allow colony formation. Colonies were fixed and stained after 10 days, and surviving fractions were determined from the number of colonies formed. Plating efficiency for untreated control cells was 50–90%. Points, mean ± standard deviation of at least three independent determinations. ○, CHO; □, DTD1; △, DTD812; ▽, DTD815; ◇, DTD818.
TABLE 2. Toxicity of MMC, MeDZQ, STPN, and EO9 to CHO, DTD812, and DTD818 as measured with the MTT assay.
IC50 ((dose resulting in a 50% reduction in absorbance at 550 nm) values are mean ± standard deviations of three independent determinations. Cells (~5 × 104) were treated for 2 hr with graded concentrations of MMC, MeDZQ, STPN, or EO9, and MTT formazan crystal staining was determined 5–7 days after treatment. IC50 values were determined from semilog plots of percentage of control absorbance at 550 nm versus concentration of drug.
| Cell line | IC50 |
|||
|---|---|---|---|---|
| MMC | MeDZQ | STPN | EO9 | |
| nm | ||||
| CHO | 1360 ± 140 | 289 ± 2 | 98 ± 19 | 625 ± 9 |
| DTD812 | 1560 ± 230 | 71 ± 26 | 58 ± 9 | 215 ± 81 |
| DTD818 | 1900 ± 340 | 73 ± 3 | 65 ± 9 | 223 ± 50 |
Sensitivity to MMC, EO9, STPN, and MeDZQ versus NQO1 levels
The D50 values (drug concentrations resulting in 50% survival) and D10 values (drug concentrations resulting in 10% survival) of MMC, EO9, STPN, and MeDZQ to CHO and human NQO1 expressing CHO cells were plotted versus NQO1 levels (Fig. 3). MMC toxicity showed no dependence on NQO1 activity, whereas EO9, STPN, and MeDZQ showed a decrease in D50 and D10 values with respect to NQO1 activity.
Fig. 3.

D50 and D10 values for MMC (A), EO9 (B), STPN (C), and MeDZQ (D) to CHO and human NQO1-expressing CHO cells plotted versus NQO1 activity in each cell line. Values were calculated from semilog plots of percent survival versus drug concentration. D10 values were estimated by extrapolation for survival curves not extending below 10% survival. Filled symbols, D50; open symbols, D10.
Toxicity of menadione to CHO and NQO1 transfectants
The expression of human NQO1 acted to reduce the toxicity of menadione in CHO cells (Fig. 4). Menadione sensitivity in human NQO1 expressing CHO cells was ~50% less than that seen to parental CHO cells. The D50 for menadione to CHO cells was ~2 μg/ml, and it was ~2.8 μg/ml in NQO1-expressing CHO cells. The decreased sensitivity of NQO1-expressing cells to menadione is consistent with previous studies with menadione-resistant Chinese hamster V79 cells that had elevated NQO1 activity (33) and with the proposed protective role of NQO1 in menadione toxicity (34).
Fig. 4.
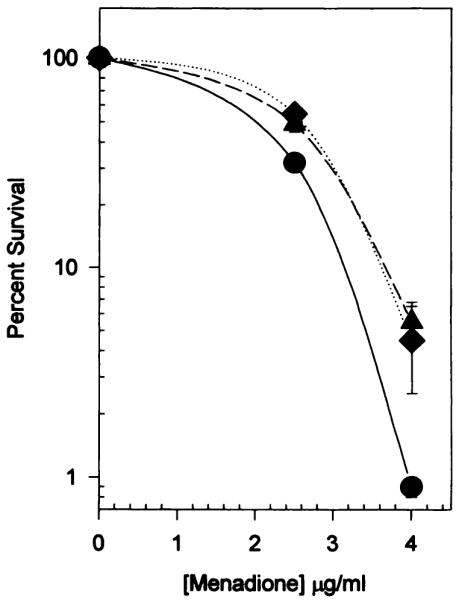
Effect of human NQO1 expression on the toxicity of menadione to CHO cells. Cells were plated out at the appropriate cell densities and exposed to menadione for 2 hr, followed by rinsing twice with Puck’s Saline G and the addition of complete growth medium. Colonies were fixed and stained after 10 days, and surviving fractions were determined. The D50 for CHO cells to menadione was ~2 μg/ml and to human NQO1-expressing CHO cells was ~2.8 μg/ml. Only the two CHO clones expressing the highest levels of human NQO1 were tested for differences in sensitivity to menadione. ●, CHO: ▲, DTD812; ◆, DTD818.
Effect of dicumarol on the toxicity of EO9 to CHO and NQO1-expressing CHO cells
The effect of dicumarol on the toxicity of EO9 to CHO cells and the highest expressing human NQO1-transfected cell line (DTD812) was assessed, and the results are shown in Fig. 5. The inclusion of 20 μm dicumarol in EO9 treatment lead to a large increase in survival in both CHO and DTD812 cells. Because protection was observed in both cells that had no detectable NQO1 activity and in those expressing human NQO1, these data indicate that dicumarol-mediated protection is not necessarily NQO1 dependent.
Fig. 5.
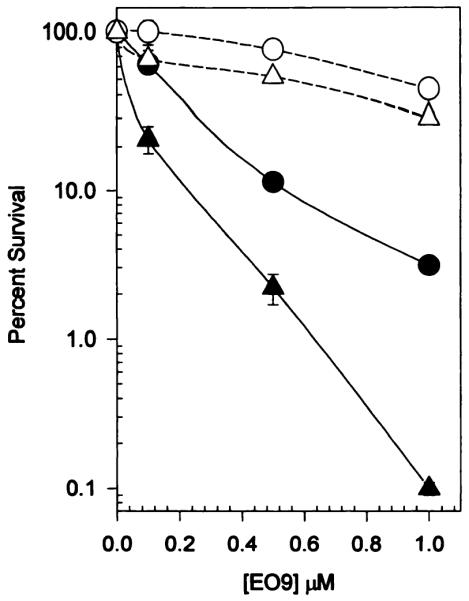
Protective effect of dicumarol against EO9 toxicity in CHO and in CHO cells expressing human NQO1. EO9 toxicity was measured as previously described in the presence (open symbols) and absence (filled symbols) of 20 μm dicumarol. Dicumarol was present throughout the 2-hr EO9 exposure period and was removed when the cells were rinsed with Puck’s Saline G. The protective effect of dicumarol to EO9 toxicity was also observed in other human NQO1 expressing CHO clones (data not shown). Points, mean ± standard deviation of three determinations. ● and ○, CHO; ▲ and △, DTD812.
Activities of NADPH:cytochrome P-450 reductase, NADH cytochrome b5 reductase, and xanthine dehydrogenase/oxidase
The activities of other enzymes that may play a role in determining the sensitivity of cells to quinone antitumor agents were measured, and the results are shown in Table 3. NADPH:cytochrome P-450 reductase levels in CHO, DTD812, DTD815, and DTD818 ranged from 3.3 ± 1.2 to 9.6 ± 4.0 nmol of cytochrome c reduced/min/mg of protein. Analysis of variance of the results showed that only the CHO and DTD815 cell lines had significant differences (p < 0.05) in activity. NADH:cytochrome b5 reductase levels ranged from 33.7 ± 12.0 to 55.4 ± 9.0 nmol of cytochrome c reduced/min/mg of protein. Analysis of variance showed no significant differences among any of the cell lines. Xanthine dehydrogenase/oxidase activity ranged from 5.7 ± 2.3 to 1.3 ± 1.2 nmol of uric acid formed/min/mg of protein. All of the xanthine dehydrogenase/oxidase activity was NAD+ dependent, showing that the dehydrogenase form of the enzyme predominates in these cells. Analysis of variance showed no significant difference in xanthine dehydrogenase/oxidase activity among the cell lines.
TABLE 3. Activity of NADPH:cytochrome P-450 reductase, NADH:cytochrome b5 reductase, and xanthine dehydrogenase/oxidase in parental CHO cells and transfectants expressing human NQO1.
Values are mean ± standard deviations of at least four independent determinations. NADPH:cytochrome P-450 reductase and NADH:cytochrome b5 reductase were measured in whole-cell lysates by monitoring the reduction of cytochrome c in the presence of NADPH or NADH, respectively. Xanthine dehydrogenase/oxidase activity was measured by monitoring the conversion of xanthine to uric acid in the presence and absence of NAD+. Assays were done on whole-cell lysates for NADPH:cytochrome P-450 reductase and NADH:cytochrome b5 reductase and the supernatant after centrifugation at 15,000 × g for xanthine dehydrogenase/oxidase. Lysate from ~106 cells was used for each assay, and total protein was determined in the samples using the BCA total protein assay.
| Cell line | P-450Ra | b5Rb | XDH/XOc |
|---|---|---|---|
| CHO | 3.3 ± 1.2 | 33.7 ± 12.0 | 5.7 ± 2.3 |
| DTD1 | 5.0 ± 2.1 | ||
| DTD812 | 4.5 ± 1.1 | 47.9 ± 4.0 | 3.4 ± 1.4 |
| DTD815 | 9.6 ± 4.0 | 55.4 ± 9.0 | 1.3 ± 1.2 |
| DTD818 | 7.4 ± 1.7 | 51.0 ± 6.2 | 4.9 ± 1.0 |
NADPH:cytochrome P-450 reductase activity is expressed in nmol of cytochrome c reduced/min/mg of protein.
NADH:cytochrome b5 reductase activity is expressed in nmol of cytochrome c reduced/min/mg of protein.
Xanthine dehydrogenase/oxidase activity is expressed in nmol of uric acid formed/min/mg of protein. All XDH/XO activity was NAD+ dependent.
Discussion
Because MMC is considered to be the prototype bioreductive alkylating anticancer agent, an understanding of the mechanisms of its activation has important clinical implications (35). Considerable evidence exists that NQO1 is important for the reductive activation of MMC to toxic species (2, 11, 12, 17). Clearly, MMC is a substrate for NQO1 (2), and NQO1 can metabolize MMC to a species that can cross-link DNA (17). Activation of MMC by human NQO1 has been shown in cell-free systems at physiological pH (17), but there is still some question regarding the role of NQO1 in activation of MMC in cells. Numerous studies have shown relationships of MMC toxicity to differences in NQO1 activity levels (2, 11, 12). The underlying mechanism investigated was based solely on bioreductive capacity. This approach largely ignores other mechanisms, such as DNA repair capacity, that can also be related to the cytotoxicity of MMC (36) and potentially play a role in changes to MMC sensitivity. In our study, we attempted to isolate the bioreductive component of antitumor quinone toxicity by expressing human NQO1 in cells that have the same genetic background as the NQO1-deficient parental cell.
To investigate the importance of NQO1 levels in determining the sensitivity of cells to killing by quinone antitumor agents, we transfected human NQO1 into CHO cells. It has previously been reported that CHO cells have very low levels of NQO1 activity (~10 nmol of DCPIP reduced/min/mg of protein) (11, 16). The glyA− auxotroph of CHO-K1 (772–56) that we chose to use in this study has no detectable NQO1 activity or protein. We transfected cells with NQO1 and used these human NQO1-expressing CHO cell lines to assess the importance of NQO1 in the reductive activation of the antitumor quinones MMC, EO9, STPN, and MeDZQ. NQO1 has been shown to activate a number of antitumor quinones (1, 2) and to be elevated in tumor tissue compared with normal tissue (3–6). These results led to a great deal of interest in the potential use of NQO1 for selective activation of antitumor compounds in the target tissue (7, 8).
Considerable evidence implicating NQO1 as an important enzyme in MMC toxicity comes from studies using the NQO1 inhibitor dicumarol and correlative studies using cell lines with varying NQO1 activities. A major complicating factor with dicumarol is that it is not selective for just NQO1 (13) but rather interacts with other enzymes that are involved in reductive activation processes (14, 15). For example, dicumarol has been shown to inhibit NADH:cytochrome b5 reductase-mediated MMC metabolism (15) and potentiate xanthine dehydrogenase- and xanthine oxidase-mediated MMC metabolism (14). Our results show that dicumarol inhibits the toxicity of EO9 to CHO cells that are devoid of detectable NQO1 activity. Previous studies by Keyes et al. (9) have shown that dicumarol can potentiate the toxicity of MMC to L1210 cells, which are devoid of NQO1 activity, under both aerobic and hypoxic conditions. These data taken together strongly suggest that the inhibiting/potentiating effects of dicumarol to MMC toxicity are not necessarily mediated by NQO1 inhibition.
The expression of NQO1 in CHO cells sensitized these cells to killing by EO9, STPN, and MeDZQ. Studies on the kinetics of the metabolism of these compounds by recombinant human NQO1 have shown that they are metabolized at rates at least 50-fold greater than those of MMC. For example, the rates of metabolism for EO9, STPN, and MeDZQ are 7.7 ± 2.0, 51 ± 4, and 25 ± 4 μmol of NADH oxidized/min/mg of protein, respectively, compared with 0.15 ± 0.01 μmol of NADH oxidized/min/mg of protein for MMC (22). These kinetic data are consistent with the sensitivities of the human NQO1 expressing CHO cells to these compounds. The DTD1 clone, which expresses the lowest level of NQO1, was sensitized only to STPN, which is the best substrate for human NQO1 (22). No increase in the sensitivity was seen between the two highest NQO1-expressing clones. This suggests that a threshold effect is occurring relative to the levels of NQO1 and cytotoxicity of these compounds (32).
There is evidence from at least one study that the expression of rat liver NQO1 in CHO cells leads to an increase in the sensitivity to killing by MMC and porfiromycin (24). A second study using human NQO1 failed to confirm this conclusion with the use of MMC (37). It has also been shown that rat NQO1 metabolizes MMC at a 5-fold faster rate than does human NQO1 (22, 25). This 5-fold difference in the rate of MMC metabolism using rat versus human NQO1 may explain why rat NQO1 expression increases MMC toxicity, whereas human NQO1 expression does not. These data emphasize the importance of species-specific differences in metabolic systems. A recent study has shown that overexpression of human NQO1 in NllI3T3 cells does not increase the sensitivity of these cells to MMC or diaziquone (37), but the parental NIH3T3 cell line used in these studies showed appreciable NQO1 activity. Due to the differences in the kinetics of rodent and human NQO1, as well as the apparent threshold effect seen with cytotoxicity and NQO1 activity, conclusions regarding the role of human NQO1 overexpression in cells with background rodent NQO1 activity may not be valid.
An advantage of our system is that the cell lines used have large differences in NQO1 activity while having similar levels of other reductive enzyme systems. The activities of NADPH:cytochrome c reductase, NADH:cytochrome b5 reductase, and xanthine dehydrogenase in the parental CHO cell line and the NQO1 transfectants are similar to the activity levels of these enzymes in other cell lines. NADPH: cytochrome c reductase and NADH:cytochrome b5 reductase levels have been shown to be 0–68 nmol/min/mg of protein and 6–127 nmol/min/mg of protein, respectively, in human tumor cell lines used in the National Cancer Institute screening panel (38) as well as in CHO, V79, and EMT6 cells (16). Xanthine dehydrogenase/oxidase enzyme levels are also similar to what has been shown in mouse tumor and normal tissues (39) as well as in human and rabbit tissues (40). It remains a possibility that in the case of MMC, the basal levels of these other reductases are sufficient to activate MMC, and thus transfection of NQO1 makes little difference to overall cytotoxicity.
The role of NQO1 in the bioactivation of MMC has been controversial. It is clear that MMC can be bioactivated by NQO1 in cell-free systems (2), although metabolism is pH dependent due to pH-dependent mechanism-based inactivation of NQO1 at higher pH values (13). Despite the pH-dependent metabolism, MMC can be bioactivated by NQO1 in the physiological pH range of 7–7.4 to form DNA cross-links (17). In a cellular system, however, the role of NQO1 in MMC bioactivation is less clear because of the presence of other bioreductive activating systems and variation in the efficiency of cellular repair systems. Correlations have been observed between the toxicity of MMC in panels of cell lines and NQO1 content is some cases (22, 23) but not in others (21). Given the relatively poor efficiency of MMC as a substrate for NQO1 and the pH dependence of its metabolism, conflicting data perhaps are not surprising. In this study, we did not observe increased MMC toxicity after the transfection of human NQO1 into CHO cells, although increased toxicity has been observed after transfection of rat NQO1 cDNA into CHO cells (24). In view of the complexities of metabolism of MMC and its relatively poor efficiency as a substrate for NQO1, however, it is likely that the cytotoxic sequelae of NQO1 transfections will vary with the cellular system and the type of NQO1 transfected. In agreement with this interpretation, when compounds that were efficient substrates for NQO1 were examined, such as EO9, MeDZQ, and STPN, NQO1-transfected CHO lines showed markedly increased sensitivity to these agents. In summary, our results show that NQO1 levels do not necessarily determine the toxicity of MMC in cultured mammalian cells transfected with human NQO1 cDNA but do determine the sensitivity of EO9, STPN, and MeDZQ. These results are consistent with the relative ability of human NQO1 to use these compounds as substrates for reduction and suggest that efficient substrates for NQO1 may be useful as bioreductive antitumor agents.
Acknowledgments
This work was supported by United States Public Health Service Grants CA36447, CA66392, and CA51210. D.L.G. was supported by National Research Service Award Postdoctoral Fellowship CA64039.
Glossary
ABBREVIATIONS
- NQO1
NAD(P)H:quinone oxidoreductase
- EO9
3-hydroxymethyl-5-aziridinyl-1-methyl-2-(1H-indole-4,7-dione)-propenol
- CHO
Chinese hamster ovary
- MeOZQ
2,5-diaziridinyl-3,6-dimethyl-1,4-benzoquinone
- MMC
mitomycin C
- STPN
streptonigrin
- DCPIP
2,6-dichlorophenolindophenol
- ECL
enhanced chemiluminescence
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- TBST
Tris-buffered saline/Tween 20
- G418
geneticin
- LB
Luria-Bertani
References
- 1.Walton MI, Smith PJ, Workman P. The role of NAD(P)H:quinone reductase (EC 1.6.99.2, DT-diaphorase) in the reductive bioactivation of the novel indolquinone antitumor agent EO9. Cancer Commun. 1991;3:199–206. doi: 10.3727/095535491820873164. [DOI] [PubMed] [Google Scholar]
- 2.Siegel D, Gibeon NW, Preusch PC, Ross D. Metabolism of mitomycin C by DT-diaphorase: role in mitomycin C-induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990;50:7483–7489. [PubMed] [Google Scholar]
- 3.Schlager JJ, Powis G. NAD(P)H:quinone acceptor oxidoreductase in human normal and tumor tissue: effects of cigarette smoking and alcohol. Int. J. Cancer. 1990;45:403–409. doi: 10.1002/ijc.2910450304. [DOI] [PubMed] [Google Scholar]
- 4.Koudstaal J, Makkink B, Overdiep SH. Enzyme histochemical pattern in human tumours: II. Oxidoreductases in carcinoma of the colon and the breast. Eur. J. Cancer. 1975;11:111–115. doi: 10.1016/0014-2964(75)90188-7. [DOI] [PubMed] [Google Scholar]
- 5.Schor NA, Cornelisse CJ. Biochemical and quantitative histochemical study of reduced pyridine nucleotide dehydrogenation by human colon carcinomas. Cancer Res. 1983;43:4850–4855. [PubMed] [Google Scholar]
- 6.Cresteil T, Jaiswal AK. High levels of expression of the NAD(P)H:quinone oxidoreductase (NQO1) gene in tumor cells compared to normal cells of the same origin. Biochem. Pharmacol. 1991;42:1021–1027. doi: 10.1016/0006-2952(91)90284-c. [DOI] [PubMed] [Google Scholar]
- 7.Workman P, Walton MI, Powis G, Schlager JJ. DT-diaphorase: questionable role in mitomycin C resistance, but a target for novel bioreductive drugs. Br. J. Cancer. 1995;60:800–802. doi: 10.1038/bjc.1989.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riley RJ, Workman P. DT-diaphorase and cancer chemotherapy. Biochem. Pharmacol. 1992;43:1657–1669. doi: 10.1016/0006-2952(92)90694-e. [DOI] [PubMed] [Google Scholar]
- 9.Keyes SR, Rockwell S, Sartorelli AC. Modification of the metabolism and cytotoxicity of bioreductive alkylating agents by dicoumarol in aerobic and hypoxic murine tumor cells. Cancer Res. 1989;49:3310–3313. [PubMed] [Google Scholar]
- 10.Keyes SR, Rockwell S, Sartorelli AC. Enhancement of mitomycin C cytotoxicity to hypoxic tumor cells by dicoumarol in vivo and in vitro. Cancer Res. 1985;45:213–216. [PubMed] [Google Scholar]
- 11.Dulhanty AM, Whitmore GF. Chinese hamster ovary cell lines resistant to mitomycin C under aerobic but not hypoxic conditions are deficient in DT-diaphorase. Cancer Res. 1995;51:1860–1865. [PubMed] [Google Scholar]
- 12.Malkinson AM, Siegel D, Forrest GL, Gazdar AF, Oie HK, Chan DC, Bunn PA, Mabry M, Dykes DJ, Harrison SD, Jr., Ross D. Elevated DT-diaphorase activity and messenger RNA content in human non-small lung cell carcinoma: relationship to the response of lung tumor xenografts to mitomycin C. Cancer Res. 1992;52:4752–4757. [PubMed] [Google Scholar]
- 13.Ross D, Siegel D, Beall H, Prakash AS, Mulcahy RT, Gibson NW. DT-diaphorase in activation and detoxification of quinones: bioreductive activation of mitomycin C. Cancer Metastasis Rev. 1993;12:83–101. doi: 10.1007/BF00689803. [DOI] [PubMed] [Google Scholar]
- 14.Gustafson DL, Pritsos CA. Enhancement of xanthine dehydrogenase mediated mitomycin C metabolism by dicumarol. Cancer Res. 1992;52:6936–6939. [PubMed] [Google Scholar]
- 15.Hodnick WF, Sartorelli AC. Reductive activation of mitomycin C by NADH:cytochrome b5 reductase. Cancer Res. 1993;53:4907–4912. [PubMed] [Google Scholar]
- 16.Keyes SR, Fracasso PM, Heimbrook DC, Rockwell S, Sligar SG, Sartorelli AC. Role of NADPH:cytochrome c reductase and DT-diaphorase in the biotransformation of mitomycin C. Cancer Res. 1984;44:5638–5643. [PubMed] [Google Scholar]
- 17.Siegel D, Beall H, Senekowitsch C, Kasai M, Arai H, Gibson NW, Ross D. Bioreductive activation of mitomycin C by DT-diaphorase. Biochemistry. 1992;31:7879–7885. doi: 10.1021/bi00149a019. [DOI] [PubMed] [Google Scholar]
- 18.Pan SS, Andrews PA, Glover CJ, Bachur NR. Reductive activation of mitomycin C and mitomycin C metabolites catalyzed by NADPH-cytochrome P-450 reductase and xanthine oxidase. J. Biol. Chem. 1984;259:959–966. [PubMed] [Google Scholar]
- 19.Gustafson DL, Pritsos CA. Bioactivation of mitomycin C by xanthine dehydrogenase from EMT6 mouse mammary carcinoma tumors. J. Natl. Cancer Inst. 1992;84:1180–1185. doi: 10.1093/jnci/84.15.1180. [DOI] [PubMed] [Google Scholar]
- 20.Marshall RS, Paterson MC, Rauth AM. Deficient activation by a human cell strain leads to mitomycin resistance under aerobic but not hypoxic conditions. Br. J. Cancer. 1989;39:341–346. doi: 10.1038/bjc.1989.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson N, Stratford IJ, Houlbrook S, Carmichael J, Adams GE. The sensitivity of human tumour cells to quinone bioreductive drugs: what role for DT-diaphorase? Biochem. Pharmacol. 1992;44:409–412. doi: 10.1016/0006-2952(92)90429-m. [DOI] [PubMed] [Google Scholar]
- 22.Beall HD, Murphy AM, Siegel D, Hargreaves RHJ, Butler J, Ross D. NAD(P)H quinone oxidoreductase (DT-diaphorase) ae a target for bioreductive antitumor quinones: quinone cytotoxicity and selectivity in human lung and breast cancer cell lines. Mol. Pharmacol. 1995;48:499–504. [PubMed] [Google Scholar]
- 23.Paull K, Camalier R, Fitzsimmons SA, Lewis AD, Workman P, Grever M. Correlations of DT-diapborase expression with cell sensitivity data obtained from the NCI human tumor cell line panel. Proc. Am. Assoc. Cancer Res. 1994;35:369. [Google Scholar]
- 24.Hodnick HF, Belcourt MF, Kemple B, Rockwell S, Sartorelli AC. Potentiation of mitomycin C and porfiromycin toxicity to Chinese hamster ovary (CHO) cells by overexpression of DT-diaphorase (DTD) cDNA. Proc. Am. Assoc. Cancer Res. 1995;36:602. [Google Scholar]
- 25.Beall HD, Mulcahy RT, Siegel D, Traver RD, Gibson NW, Ross D. Metabolism of bioreductive antitumor compounds by purified rat and human DT-diaphorase. Cancer Res. 1994;54:3196–3201. [PubMed] [Google Scholar]
- 26.Boland MP, Knox RJ, Roberts JJ. The differences in kinetics of rat and human DT diaphorase result in a differential sensitivity of derived cell lines to CB 1954 (5-(aziridinyl-1-yl)-2,4-dinitrobenzamide) Biochem. Pharmacol. 1991;41:867–875. doi: 10.1016/0006-2952(91)90190-g. [DOI] [PubMed] [Google Scholar]
- 27.Ernster L. DT-diaphorase. Methods Enzymol. 1967;10:309–317. [Google Scholar]
- 28.Benson AM, Hunkler MJ, Talalay P. Increase of NAD(P)H: quinone reductase by dietary antioxidants: possible role in protection against carcinogenesis and toxicity. Proc. Natl. Acad. Sci. USA. 1980;77:5216–5220. doi: 10.1073/pnas.77.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strobel HW, Dignam JD. Purification and properties of NADPH-cytochrome P-450 reductase. Methods Enzymol. 1978;52:89–96. doi: 10.1016/s0076-6879(78)52009-0. [DOI] [PubMed] [Google Scholar]
- 30.Stirpe F, Della Corte E. The regulation of rat liver xanthine dehydrogenase: conversion in vitro of the enzyme activity from dehydrogenase (type D) to oxidase (type O) J. Biol. Chem. 1969;244:3855–3863. [PubMed] [Google Scholar]
- 31.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 32.Ross D, Beall HD, Siegel D, Traver RD, Gustafson DL. Enzymology of bioreductive drug activation. Br. J. Cancer. 1996;74(Suppl. 27):51–58. [PMC free article] [PubMed] [Google Scholar]
- 33.Martins EAL, Mori L, Birnboim HC, Meneghini R. Menadione-resistant Chinese hamster cell variants are cross-resistant to hydrogen peroxide and exhibit stable chromosomal and biochemical alterations. Mol. Cell. Biochem. 1992;118:181–189. doi: 10.1007/BF00299397. [DOI] [PubMed] [Google Scholar]
- 34.Thor H, Smith MT, Hartzell P, Bellomo G, Jewell SA, Orrenius S. The metabolism of menadione (2-methyl-1,4-napthoquinone) by isolated hepatocytes: a study of the implications of oxidative stress in intact cells. J. Biol. Chem. 1982;257:12419–12425. [PubMed] [Google Scholar]
- 35.Doroshow JH. Reductive activation of mitomycin C: a delicate balance. J. Natl. Cancer Inst. 1992;84:1138–1139. doi: 10.1093/jnci/84.15.1138. [DOI] [PubMed] [Google Scholar]
- 36.Jones NJ. Genetic analysis of mitomycin C-hypersensitive Chinese hamster cell mutants. Mutagenesis. 1994;9:477–482. doi: 10.1093/mutage/9.5.477. [DOI] [PubMed] [Google Scholar]
- 37.Powis G, Gasdaska PY, Gallegos A, Sherrill K, Goodman D. Over-expression of DT-diaphorase in transfected NIH3T3 cel1s does not lead to increased anticancer quinone drug sensitivity: a questionable role for the enzyme as a target for bioreductively activated anticancer drugs. Anticancer Res. 1995;15:1141–1146. [PubMed] [Google Scholar]
- 38.Fitzsimmons SA, Workman P, Grever M, Paull K, Lewis AD. Differential expression of DT-diaphorase, cytochrome P450 reductase and cytochrome b5 reductase in the NCI human tumour cell line panel. Proc. Am. Assoc. Cancer Res. 1994;35:369. [Google Scholar]
- 39.Anderson RF, Patel KB, Reghebi K, Hill SA. Conversion of xanthine dehydrogenase to xanthine oxidase as a possible marker for hypoxia in tumours and normal tissue. Br. J. Cancer. 1989;60:193–197. doi: 10.1038/bjc.1989.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wajner M, Harkness RA. Distribution of xanthine dehydrogenase and oxidase activities in human and rabbit tissues. Biochim. Biophys. Acta. 1989;991:79–84. doi: 10.1016/0304-4165(89)90031-7. [DOI] [PubMed] [Google Scholar]