

Abstract
Objective
Multidrug resistance is the major cause of failure of many chemotherapeutic agents. While resistance can arise from several factors, it is often dominated by drug efflux mediated by P-glycoprotein (P-gp), a membrane-bound polysubstrate export pump expressed at high levels in resistant cells. While co-administration of pump inhibitors and a drug could suppress efflux, this two-drug strategy has not yet advanced to therapy. We recently demonstrated that the reversible attachment of a guanidinium-rich molecular transporter, polyarginine, to a drug provides a conjugate that overcomes efflux-based resistance in cells and animals. This study is to determine whether this strategy for overcoming resistance is effective against human disease.
Methods
Tumor samples from ovarian cancer patients, both malignant ascites cells and dissociated solid tumor cells, were exposed to Taxol-oligoarginine conjugates designed to release free drug only after cell entry. Cell Viability was determined via propidium-iodide uptake by flow cytometry. To analyze bystander effect, toxicity of the drug conjugates was also tested on peripheral blood leucocytes.
Results
Human ovarian carcinoma specimens resistant to Taxol in vitro demonstrated increased sensitivity to killing by all Taxol-transporter conjugates tested. These studies also show that the drug conjugates were not significantly more toxic to normal human peripheral blood leukocytes than Taxol.
Conclusions
These studies with human tumor indicate that oligoarginine conjugates of known drugs can be used to overcome the efflux-based resistance to the drug, providing a strategy that could improve the treatment outcomes of patients with efflux-based drug-resistance.
Keywords: human, ovarian carcinoma, drug resistance, taxol, and oligoarginine
Introduction
Ovarian carcinoma (OVCA) is commonly diagnosed at a late stage, two thirds of patients eventually relapse and a subpopulation develops resistance to Taxol drugs [1,2,3,4,5]. A significant problem in the treatment of OVCA is multidrug resistance (MDR). Chemotherapeutic agents initially produce a cytoreductive response, but the development of MDR leads to failure of the drug and frequently cross-resistance to other therapeutics. While many factors contribute to MDR, including drug target mutation, drug metabolism and sequestration [6], a major contributor is the active export of drugs by transmembrane polysubstrate efflux pumps that prevent drugs from reaching their intracellular targets. The recently reported X-ray crystal structure of the mouse P-gp efflux pump, which has 87% sequence identity to human P-gp [7], along with earlier binding studies on the interactions of P-gp with its substrates [8,9], provides a rationale for how this protein recognizes and mediates the unidirectional efflux of numerous drugs. Hydrophobic drugs with high membrane solubility can enter the internal cavity of the pump through two portals positioned on the inner leaflet of the membrane after which an energy-dependent conformational change in the pump results in drug efflux into the extracellular milieu. Drug efflux by P-gp represents one of the best-studied mechanisms of resistance to hydrophobic anticancer drugs [10].
Much effort has been invested in the development of new drug candidates that are less susceptible to efflux, often requiring synthesis and evaluation of numerous analogs to identify a candidate that maintains target efficacy but is not a P-gp substrate. An alternative approach has focused on using a second agent, to suppress pump expression, accelerate the post-translational degradation of the pump, or inhibit pump efflux by the co-administered drug. However, because export pumps are ubiquitously expressed and required for normal function and because the co-administration of two agents can produce highly variable and often undesired pharmacokinetic responses, this approach has not advanced beyond clinical trials.
We have designed a method that involves the attachment of a drug to oligoarginine, a guanidinum-rich molecular transporter through a releasable linker [11]. The guanidinum-rich molecular transporters, which include oligoarginine and other cationic peptides, have been shown in recent years to facilitate the delivery of attached cargo into cells by rendering them water-soluble [12,13]. Significantly, because the drug-transporter conjugate is not a substrate for P-gp export and passes rapidly through the cell membrane, it evades efflux-mediated resistance. After cell entry the conjugate is cleaved and the free drug is released at a rate controlled by the linker design.
From a clinical perspective, this approach offers a number of advantages. Taxol, is water-insoluble and must be administered with solubilizing agents like Cremephor-EL that often elicit an acute hypersensitive reaction [14,15,16,17]. In contrast, Taxol conjugated to the transporter can be formulated in minimal volumes (as little as 1-2 ml) of saline. Furthermore, the conjugate is designed to be inactive until it enters cells, thereby minimizing off-target toxicity. Upon entering cells, the conjugate releases free drug over time at a pre-determined rate controlled by linker design, thus avoiding peak-trough effects often associated with a bolus administration. Additionally, the conjugates can be given intraperitoneally (IP), taking advantage of a mode of administration recommended in a National Cancer Institute announcement indicating that IP-administered paclitaxel, when used with intravenously (IV)-administered paclitaxel and cisplatin, provides a 16-month extension to median survival in ovarian cancer patients [18]. Previous biodistribution studies in transgenic mice where the bioluminescent probe luciferin is conjugated to an oligoarginine transporter provide evidence that transporter conjugates injected IP remain localized within the cavity [11]. IP administration of the Taxol-oligoarginine conjugates in larger volumes of saline will minimize localized toxicity associated with the transporter and maintain uniform distribution confined to the IP region. Thus, the balance between drug delivery/maintenance in cancer cells and the drug toxicity profile can be regulated in this drug-conjugate linker system to achieve maximum efficacy and manageable side effects.
To determine if the drug-conjugate linker system is applicable to the complexity and heterogeneity of human disease, we examined the effectiveness of our approach in an in-vitro study using human specimens' from ovarian cancer patients. By testing the cancer cells without selecting for growth advantages or expanding particular subpopulations, the assay emulates the heterogeneity of the clinical disease. The growth rate of primary tumor samples (ascites cells and dissociated solid tumor cells) in vitro replicates the physiological environment where cancer cells grow more slowly than established cell lines [19,20]. The diverse set of transporter-drug conjugates evaluated includes prodrugs that exhibit extended stabilities under assay conditions, allowing them to be administered and remain intact until cell entry, after which they are rapidly cleaved in the intracellular reducing environment, releasing the free drug at tunable rates ranging from minutes to hours and controlled by linker design.
Materials and Methods
Synthesis of Conjugates
Synthesis of compounds is described in the supporting information.
Determination of the Prodrug Half-Lives
To evaluate the half-lives of the synthesized conjugates under the assay conditions, each conjugate (0.3 mg) was dissolved in 200 μl HEPES buffered saline, pH 7.4 and incubated at 37°C containing 10 μl of a solution of 1-naphthalenemethanol in methanol (0.42 mg/ml), which served as an internal standard. At appropriate intervals 20 μl of the solutions were removed and analyzed by reverse-phase HPLC. The percent decomposition was calculated from the integrated peak areas of the conjugate, the internal standard, and the various decomposition products. To evaluate the half-lives under reducing conditions, the conjugates were incubated as described above with 10 mM dithiothreitol.
In-vitro Cytotoxicity Assay
All tissue samples were obtained with IRB certified informed patient consent, and the study approved by the Stanford Research Compliance Office. Ascites cells were centrifuged, filtered through sterile gauze, cryopreserved or tested immediately. For frozen ascites samples, cells were thawed, washed twice with media, assessed for >80% cell viability and resuspended and incubated at 37°C for 90 minutes before plating. Freshly harvested solid tumor specimens were processed overnight with Accutase (Sigma), washed twice before plating.
For cytotoxicity assays, 200 μl of a 2 × 105 cells/ml in Iscove's media with 10% FCS was distributed into each well of 48-well polypropylene plates. Conjugates and Taxol were added to the wells in duplicate at appropriate concentrations. Control wells were established in parallel and the plates were incubated at 37°C for 72 hours (unless otherwise noted). Cells were analyzed by flow cytometry. Due to limited quantities of the patient samples, results are given as the mean ± SEM of duplicate wells unless otherwise noted.
Human Peripheral Blood Leukocyte Assay
Neutrophils were obtained by a modified technique of Boyum [21]. In brief, a double gradient was prepared by layering an equal volume of HISTOPAQUE®-1077 over HISTOPAQUE®-1119 (Sigma). Freshly obtained heparinized blood sample (10 ml) diluted in 20 ml PBS was carefully layered on top of the double gradient. After centrifugation at 800g for 20 minutes, granulocytes were recovered at the 1077/1119 interphase and mononuclear cells at the plasma/1077 interphase. Recovered cells were washed twice with PBS and re-suspended to 1 × 106 cells/ml in medium with FCS. 200 μl of the solution was distributed into each well of 48-well plates. Conjugates and Taxol were added to the wells in duplicate at appropriate concentrations. Control wells were run in parallel and the plates were incubated at 37°C overnight and then analyzed by flow cytometry.
Flow Cytometry Analysis
200 μl of a propidium-iodide (PI) solution (10 μg/ml) was added to each well, mixed and transferred to tubes with the cells. Analysis was performed on a FACScan or LSR flow cytometer with data management using Cell Quest (Becton-Dickinson) and FlowJo (TreeStar Inc., Oregon). In some experiments cells were stained with CD45-FITC (Caltag) according to the manufacturer's protocol to exclude hematopoietic cells. However, electronic gates on forward and side scatter were sufficient to exclude hematopoietic cells for most samples. Live cells were determined by PI exclusion. EC50 values were calculated from semi-logarithmic dose response curves (GraphPad Prism, CA).
Results
Design, Synthesis and Properties of Releasable Taxol-Octaarginine Conjugates
An octaarginine (r8) transporter was attached to either the C2′ or C7 position of Taxol using different biocleavable disulfide linkers. At the outset, it was expected that C2′ derivatives would be inactive until free drug was released, because C2′-modification of Taxol is known to attenuate or eliminate activity [22]. C7 conjugates were also prepared because this position in Taxol can often be modified without loss of activity [22]. Eight conjugates were prepared and evaluated for in-vitro activity (Figure 1, C2′-conjugates: 2a-d, C7-conjugates: 3a-d). Conjugates 2a and 3a contain unhindered ester linkages attached directly to Taxol at the C2′ or C7 position, respectively. Conjugates 2b and 3c incorporate linkers with two or one methyl groups, respectively, attached at the alpha carbon of the ester linkage. Conjugate 2c extends the linker by an additional carbon resulting in additional flexibility. Finally, conjugates 2d, 3b, and 3d connect the transporter through carbonate linkers of varying length and functionality. The linkers are designed to provide shelf-stable conjugates that would release drug only after cell entry and at tunable rates. For example, installation of methyl groups adjacent to an ester linkage (compounds: 2b, 3c) increases the stability of the prodrugs (due to increased steric hindrance at a potential hydrolysis site) under assay conditions while retaining rapid kinetics of induced release of free Taxol. Replacement of the ester linkage with a carbonate moiety (compounds: 2d, 3b, 3d) also improves the intrinsic stability of the prodrug to hydrolytic degradation.
Figure 1.
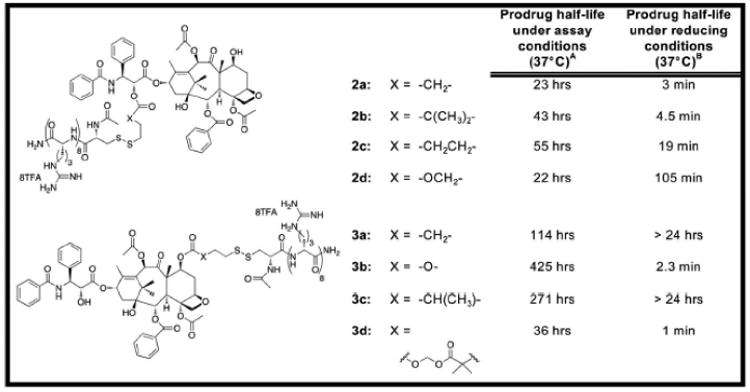
Structures of Taxol-transporter conjugates and their half-lives under assay or reducing conditions: (A) Prodrug half-life under assay conditions (HBS, pH=7.4, 37°C). (B) Prodrug half-life under reducing conditions (10mM DTT, HBS, pH=7.4, 37°C).
In-vitro Human Ascites and Primary Tumor Assay
Tumor spread via survival and proliferation in body cavity fluids, including the peritoneal fluid, is an important route of cancer metastasis [19]. As a prelude to clinical evaluation, the efficacy of the Taxololigoarginine conjugates against eight different OVCA ascites (patients-A-H) and one solid tumor sample (patient-I) was examined (Table 1). All but two patient samples were derived from serous papillary tumor, the most common subtype of OVCA. Patient-G presented with clear cell carcinoma, a rare subtype that is often refractory to standard therapies. This 7-to-1 ratio of serous papillary to clear cell approximates the actual percentage of patients who normally present with each subtype of OVCA. Patient-H presented with an unusual carcinosarcoma (formerly called malignant-mixed mesodermal tumor).
Table 1.
| Patient* | Age (yrs)† | Histology | Stage‡/grade | Chemotherapy Treatment | Specimen Type | Patient Status§ |
|---|---|---|---|---|---|---|
| A | 60 | Serous/high grade OVCA | IIIC/3 | Post surgery: 8 cycles taxane/carboplatin and etoposide | Ascites at surgery, frozen | DOD, 19 months |
| B | 65 | Serous/papillary OVCA | NIC/3 | Pre specimen collection: 3 cycles taxane/cisplatin, recurrence. Post specimen: 4 cycles taxane/cisplatin | Ascites from paracentesis, frozen | DOD, 11 months |
| C | 65 | Serous/high grade OVCA | IIIC/3 | Neoadjuvant pre specimen: 3 cycles taxane/carboplatin. Post surgery: 6 cycles paclitaxel/carboplatin IP | Ascites at surgery, frozen | DOD, 10 months |
| D | 51 | Serous/papillary OVCA | IIIC/3 | Post surgery: 6 cycles taxane/cisplatin | Ascites at surgery, frozen | DOD, 13 months |
| E | 54 | Serous/high grade OVCA | IIIC/3 | Post surgery: 6 cycles taxane IV, cisplatin IP | Ascites at surgery, frozen | AWD, 11 months |
| F | 74 | Serous/papillary OVCA | IIIC/3 | Post surgery: 6 cycles taxane/cisplatin | Ascites at surgery, frozen | AWD, 11 months |
| G | 69 | Clear cell OVCA | IIIC/3 | None | Ascites at surgery, fresh | DOD, 2 months |
| H | 86 | Carcinosarcoma [Malignant Mixed Mesodermal] | IIB/3 | None | Ascites at surgery, fresh | AWD, 6 months |
| I | 46 | Serous/high grade OVCA | IIIC/3 | Post surgery: 8 cycles taxane/carboplatin | Metastasis to omentum, fresh | NED, 10 months |
All patients were chemo naive at the time of surgery, except Patients B and C, who received standard taxane/platinum combination chemotherapy.
At the time of diagnosis.
International Federation of Obstetrics and Gynecology (FIGO) System.
Post-Diagnosis; DOD = died of disease; AWD = alive with disease; NED = alive with no evidence of disease.
Taxol was inactive in vitro against eight of the nine tissue samples tested. Five of nine patients succumbed to disease in less than 19 months post sample collection. The only sample in which Taxol displayed modest activity was the fresh solid-tumor (patient-I). In contrast, each of the Taxol-oligoarginine conjugates displayed good cytotoxic activity against both ascites cells and solid tumor samples. The dose-response curves in Figure 2 demonstrate the efficacy of the drug-conjugates against both clear cell carcinoma (Figure 2A) and serous papillary carcinoma (Figure 2B) ascites cells. Demonstrating efficacy against clear cell carincoma is significant as the prognosis is extremely poor for patients with this highly resistant subtype [23]. EC50 values for each of the conjugates and Taxol are provided in Table 2. The activity of the conjugates is independent of histology or treatment prior to sample collection. Furthermore, the doses at which the conjugates are effective against the primary OVCA samples (1-20 μM) are in the pharmacokinetic range for IP-administered Taxol (60 mg/m2) [24]. The fact that micromolar concentrations are required for cytotoxicity is expected in patient ascites and cell-lines grown as multicellular clumps or spheroids [25,26,27], unlike adherent OVCA cell-lines where EC50 is achieved by nanomolar concentrations of Taxol [28].
Figure 2.
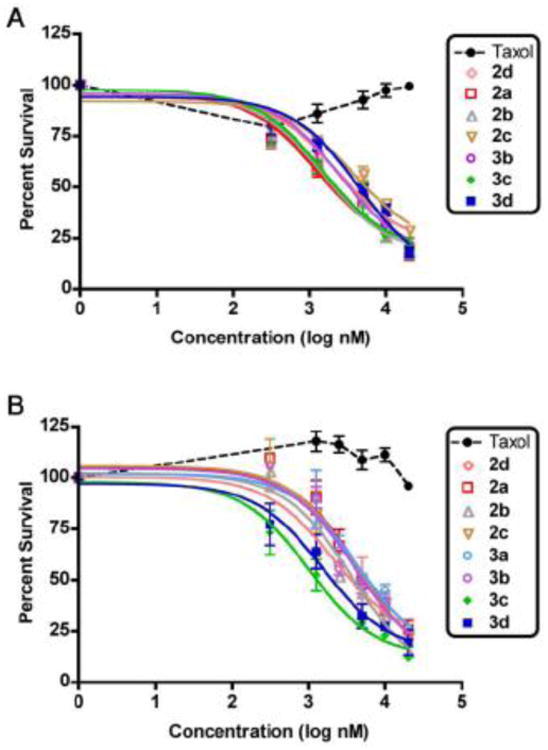
Dose-response curves of Taxol and Taxol-r8 conjugates against two subtypes of ovarian carcinoma (mean ± SEM of two independent experiments each performed in duplicate), (A) clear cell carcinoma (patient G) and (B) serous papillary carcinoma (patient F).
Table-2. EC50 values of Taxol.
| A: First generation Taxol-r8 conjugates | |||
|---|---|---|---|
|
| |||
| EC50 (μM) | |||
| Patient | Taxol | 2a | 3a |
| A ascites* | ⋙20 | 1.4 | 1.3 |
| B ascites | ⋙20 | 7.7 | 4.6 |
| C ascites | ⋙20 | 15 | 11 |
| D ascites‡ | ⋙20 | 17 | 24 |
| E ascites†,‡ | ⋙20 | 14 | 17 |
| F ascites* | ⋙20 | 4.2 | 4.4 |
| G ascites*,§ | ⋙20 | 1.2 | --- |
| H ascites†,¶ | ⋙20 | 21 | --- |
| I solid tumor† | 16.2 | 3.9 | 3.1 |
| B: Second generation Taxol-r8 conjugates | ||||||
|
| ||||||
| EC50 (μM) | ||||||
| Patient | 2b | 2c | 2d | 3b | 3c | 3d |
|
| ||||||
| A ascites* | 1.0 | --- | --- | 2.2 | --- | 1.1 |
| B ascites | 8.0 | --- | --- | 1.2 | --- | --- |
| F ascites* | 3.4 | 4.2 | 2.3 | 3.7 | 1.5 | 1.0 |
| G ascites*,§ | 1.5 | 3.7 | 2.3 | 2.5 | 4.3 | 1.5 |
| H ascites†,¶ | 23.0 | --- | --- | --- | 20.0 | --- |
Refer to Figure 1 for chemical structures of Taxol-r8 conjugates (2a, 3a) and (2b, 2c, 2d, 3b, 3c, and 3d)
Values calculated from 5 different compound concentrations ranging from 0.625 μM to 20 μM
Results are the mean of two independent experiments
48-hr incubation.
Clear cell carcinoma.
Carcinosarcoma
Toxicity Profile Control Assays
To confirm that ascites cells-killing activity was the result of intracellular delivery and release of free Taxol rather than transporter-associated toxicity, the transporter by itself was also tested for toxicity. Octaarginine alone showed very little toxicity against all three different OVCA subtypes at concentrations up to 2.5 μM (Figure-3A). At concentrations higher than 2.5 μM, octaarginine transporter was toxic but the conjugates consistently yielded 2-4X more cell death than the transporter by itself. Previous studies have shown that changes in the length or stereochemistry of a transporter can be used to modulate its toxicity and activity as well as its lifetime through protease degradation [29].
Figure 3.
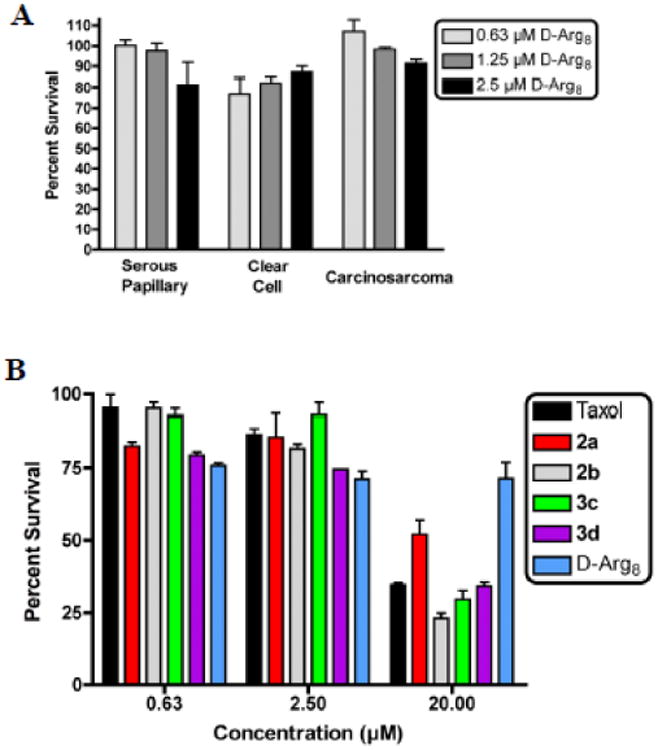
A. Toxicity of D-Arg8 towards OVCA tumor specimens. B. Toxicity of Taxol, Taxol-r8 conjugates and D-Arg8 towards normal human neutrophils; Taxol (black), conjugates 2a (red), 2b (gray), 3c (green), 3d (purple), and D-Arg8 alone (blue).
As part of the preclinical development of Taxol-oligoarginine conjugates as therapeutic agents, the toxicity of the conjugates against normal human neutrophils was assessed (Figure 3B). Cells were incubated for 48 hours with Taxol, octaarginine alone, or Taxol-r8 conjugate. Taxol dilutions were made from a DMSO stock whereas octaarginine and Taxol-r8 conjugate were from PBS stock. It is encouraging that the octaarginine transporter alone (r8, blue bars) showed very little toxicity towards neutrophils, even at the highest administered dose. Notably, none of the Taxol- r8 conjugates (2a-red bars, 2b-gray bars, 3c-green bars, and 3d-purple bars) were significantly more toxic to neutrophils compared to Taxol (black bars). Toxicity to mononuclear cells was also assessed and again the Taxol-r8 conjugates demonstrated similar toxicity profiles to Taxol (data not shown).
Discussion
Efflux-based resistance is a significant contributor to the failure of drugs. It has been demonstrated in cancer cell lines and animal models that the attachment of clinically tested molecular transporter to a known drug (Taxol) affords a conjugate that can overcome efflux-based multidrug resistance in vitro. Here we demonstrate that this strategy is applicable to human disease in an assay that evaluates efficacy against tumor samples from ovarian cancer patients in-vitro. The readily prepared conjugates utilize known drugs with validated pathways, are highly water-soluble thereby improving formulation and administration, and are taken up rapidly by cells.
Considering the heterogeneous and complex environment surrounding tumors and our intent to identify preferred compounds for preclinical advancement, we designed and synthesized several Taxol-octaarginine conjugates differing by site of transporter attachment to Taxol and by linker composition. Overall, the conjugates exhibit a wide range of stabilities (with half-lives ranging from hours to weeks) far exceeding the time required for administration and cell entry but they release free drug in the presence of a reducing environment in minutes to hours. Shelf life of the conjugates as solids at room temperature extends for months. The rates of both intrinsic linker degradation as well as designed instability in a reducing environment can be tuned and appropriately selected based on the application or mode of administration.
While various bioactivatable linker-cleavage strategies (e.g., protease, esterase, or phosphatase) are compatible with this approach, all conjugates in this initial in-vitro study incorporated a cleavable disulfide linker. Cleavage of such linkers would occur only after entry of the conjugate into cells where a high glutathione concentration is encountered (generally 1-10 mM intracellular compared to 10 μM extracellular) [30]. Glutathione is reported to be an important stimulus for cancer cell proliferation and thus disulfide linkers were selected in part to exploit the elevated levels of glutathione in cancer cells [31] and even higher levels in resistant disease [32,33]. The specific structure of the linker was varied to determine whether the stability of the conjugate under assay conditions or the rate of release of free drug would influence activity. Although all eight conjugates tested in this in-vitro study gave identical results, further studies will determine which conjugate has the necessary stability and release rates essential for in-vivo efficacy.
In conclusion, the Taxol-oligoarginine conjugates contain bioreleasable disulfide linkers and exhibit a number of beneficial physical properties, including prolonged shelf stability, prodrug stability during administration, and tunable release rates of the free drug in the intracellular reducing environment. Significantly, in all the human tumor samples, the conjugates outperform Taxol, which was inactive in vitro against the ascites cells. Furthermore, when evaluated against normal human leukocytes, neutrophils and monocuclear cells, the conjugates were comparable in toxicity to the parent drug alone. The ability to improve both the administration and efficacy of a therapeutic drug, particularly against resistant disease, by attaching a molecular transporter could significantly improve the prognosis of OVCA patients
Supplementary Material
Research Highlights.
‘Taxol-Oligoarginine Conjugates Overcome Drug Resistance in-vitro in Human Ovarian Carcinoma’ by Paul A. Wender, Wesley C. Galliher, Neelima M. Bhat, Thomas H. Pillow, Marcia M. Bieber, Nelson N. H. Teng
Overcome efflux-based resistance of Taxol in tumor samples from OVCA patients
Reversible attachment of polyarginine to Taxol allows for easy drug delivery
Tunable release rates of active drug in intracellular reducing environment
Acknowledgments
Grant Support: This work was supported by National Institutes of Health Grants CA31841 and CA31845 (to PAW), the Pacific Ovarian Cancer Research Consortium (to NNHT), and a Stanford Cancer Center Developmental Cancer Research Award (to PAW and NNHT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: A Cancer J for Clinicians. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Greenlee RT, Hill-Harmon MB, Murray T, Thun M. Cancer statistics, 2001. CA: A Cancer J for Clinicians. 2001;51:5–36. doi: 10.3322/canjclin.51.1.15. [DOI] [PubMed] [Google Scholar]
- 3.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154–9. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen-Gunther J, Mannel RS. Ascites as a predictor of ovarian malignancy. Gynecol Oncol. 2002;87:77–83. doi: 10.1006/gyno.2002.6800. [DOI] [PubMed] [Google Scholar]
- 5.Burges A, Wimberger P, Kümper C, Gorbounova V, Sommer H, Schmalfeldt B, et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM × anti-CD3 antibody: a phase I/II study. Clin Cancer Res. 2007;13:3899–905. doi: 10.1158/1078-0432.CCR-06-2769. [DOI] [PubMed] [Google Scholar]
- 6.Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 7.Aller S, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science. 2009;323:1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gatlik-Landwojtowicz E, Aanismaa P, Seelig A. Quantification and characterization of P-glycoprotein-substrate interactions. Biochemistry. 2006;45:3020–32. doi: 10.1021/bi051380+. [DOI] [PubMed] [Google Scholar]
- 9.Seelig A. A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem. 1998;251:252–61. doi: 10.1046/j.1432-1327.1998.2510252.x. [DOI] [PubMed] [Google Scholar]
- 10.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nature reviews Drug discovery. 2006;5:219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 11.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci U S A. 2008;105:12128–33. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv Drug Deliv Rev. 2008;60:452–72. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fonseca SB, Pereira MP, Kelley SO. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev. 2009;61:953–64. doi: 10.1016/j.addr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–7. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 15.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93:2325–7. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 16.Straubinger RM. Biopharmaceutics of Paclitaxel [Taxol]: Formulation, Activity, and Pharmacokinetics. In: Suffness M, editor. Taxol Science and Applications. Boca Raton, FL: CRC Press; 1995. pp. 237–58. [Google Scholar]
- 17.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–8. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 18.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal Cisplatin and Paclitaxel in Ovarian Cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 19.Liu B, Wang T, Qian X, Liu G, Yu L, Ding Y. Anticancer effect of tetrandrine on primary cancer cells isolated from ascites and pleural fluids. Cancer Letters. 2008;268:166–75. doi: 10.1016/j.canlet.2008.03.059. [DOI] [PubMed] [Google Scholar]
- 20.Richard C, Matthews D, Duivenvoorden W, Yau J, Wright PS, Th'ng JPH. Flavopiridol sensitivity of cancer cells isolated from ascites and pleural fluids. Clin Cancer Res. 2005;11:3523–9. doi: 10.1158/1078-0432.CCR-04-2507. [DOI] [PubMed] [Google Scholar]
- 21.Boyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl. 1968;97:77–89. [PubMed] [Google Scholar]
- 22.Kingston DG. Recent advances in the chemistry of taxol. J Nat Prod. 2000;63:726–34. doi: 10.1021/np000064n. [DOI] [PubMed] [Google Scholar]
- 23.Ho CM, Huang YJ, Chen TC, Huang SH, Liu FS, Chang Chien CC, et al. Pure-type clear cell carcinoma of the ovary as a distinct histological type and improved survival in patients treated with paclitaxel-platinum-based chemotherapy in pure-type advanced disease. Gynecol Oncol. 2004;94:197–203. doi: 10.1016/j.ygyno.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Francis P, Rowinsky E, Schneider J, Hakes T, Hoskins W, Markman M. Phase I feasibility and pharmacologic study of weekly intraperitoneal paclitaxel: a Gynecologic Oncology Group pilot Study. J Clin Oncol. 1995;13:2961–7. doi: 10.1200/JCO.1995.13.12.2961. [DOI] [PubMed] [Google Scholar]
- 25.Frankel A, Buckman R, Kerbel RS. Abrogation of taxol-induced G2-M arrest and apoptosis in human ovarian cancer cells grown as multicellular tumor spheroids. Cancer Res. 1997;57:2388–93. [PubMed] [Google Scholar]
- 26.Nicholson KM, Bibby MC, Phillips RM. Influence of drug exposure parameters on the activity of paclitaxel in multicellular spheroids. Eur J Cancer. 1997;33:1291–8. doi: 10.1016/s0959-8049(97)00114-7. [DOI] [PubMed] [Google Scholar]
- 27.Konecny G, Crohns C, Pegram M, Felber M, Lude S, Kurbacher C, et al. Correlation of drug response with the ATP tumorchemosensitivity assay in primary FIGO stage III ovarian cancer. Gynecol Oncol. 2000;77:258–63. doi: 10.1006/gyno.2000.5728. [DOI] [PubMed] [Google Scholar]
- 28.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 29.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97:13003–8. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito G, Swanson JA, Lee KD. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv Drug Deliv Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 31.Obrador E, Navarro J, Mompo J, Asensi M, Pellicer JA, Estrela JM. Regulation of tumour cell sensitivity to TNF-induced oxidative stress and cytotoxicity: role of glutathione. Biofactors. 1998;8:23–6. doi: 10.1002/biof.5520080105. [DOI] [PubMed] [Google Scholar]
- 32.Godwin AK, Meister A, O'Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci U S A. 1992;89:3070–4. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kigawa J, Minagawa Y, Kanamori Y, Itamochi H, Cheng X, Okada M, et al. Glutathione concentration may be a useful predictor of response to second-line chemotherapy in patients with ovarian cancer. Cancer. 1998;82:697–702. doi: 10.1002/(sici)1097-0142(19980215)82:4<697::aid-cncr12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.