

Abstract
The phosphoinositide-3 kinase (PI3K) pathway plays a critical role in cancer cell growth and survival. PI3K is activated in human cancers by elevated receptor tyrosine kinase activity, RAS mutation, as well as by mutation, amplification, and deletion of genes encoding components of the pathway. Additionally, PI3K pathway activation plays an important role in acquired resistance to both chemotherapy and targeted agents. The essential role of PI3K in human cancer has led to the development of PI3K pathway inhibitors that have shown promise in preclinical models and have entered phase 1 clinical trials. This article reviews preclinical and clinical data on members of this novel drug class, as well as data justifying the combination of PI3K inhibitors with other anticancer agents.
Keywords: Phosphoinositide-3 kinase, Targeted therapy, Drug development
Introduction
Phosphoinositide-3 kinase (PI3K) is a unique kinase because it catalyzes the production of the lipid second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PIP2), acting as a central signal transduction molecule that regulates several cellular processes. For example, PI3K plays a key role in the insulin receptor pathway and contributes to the regulation of glucose homeostasis [1, 2]. In drosophila, insulin receptor–mediated activation of PI3K also regulates cell growth. Genetic manipulation of drosophila wings resulting in higher levels of PI3K activity causes them to grow larger in size; conversely, drosophila wings with low level of PI3K activity are smaller in size [2].
In mammals, PI3K function has expanded to additional pathways beyond the insulin receptor pathway. PI3K becomes activated in response to the activation of multiple tyrosine kinases including epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER-2), HER-3, c-KIT, Abl, FMS-like tyrosine kinase 3, platelet-derived growth factor receptor α (PDGFRα), PDGFRβ, insulin-like growth factor 1 receptor (IGF-1R), MET, and vascular endothelial growth factor 2 (VEGFR2) [3•, 4•]. In addition to these tyrosine kinases, RAS is also able to activate PI3K [5]. The PI3K pathway also functions as an important signaling molecule in the immune system [6]. PI3K (particularly p110δ and p110γ) is activated in response to antigen activation of B and T cell receptors and is also activated by co-stimulatory molecules.
There are three classes of PI3Ks: type I, II, and III [1, 4•]. The role of type I PI3K is best established in cancer; the four type I PI3Ks are p110α, p110β, p110δ, and p110γ. Type 1A PI3Ks (p110α, p110β, and p110δ) are heterodimers consisting of a p85 regulatory subunit and the p110 catalytic domain [1, 4•]. The regulatory and catalytic units are encoded by different genes [1, 4•]. The p85α and p85β subunits are encoded by PIK3R1, PIK3R2, and PIK3R3, whereas the p110α, p110β, p110δ, p110γ subunits are encoded by PIK3CA, PIK3CB, PIK3CD, and PIK3CG, respectively.
The p85 regulatory subunit inhibits the activity of the p110 catalytic subunit and ensures that the catalytic domain only becomes activated at the appropriate time [1, 4•]. The binding to p85 of activated tyrosine kinases, adapter proteins, or in some cases activated G proteins, is the molecular switch that releases its inhibition of the catalytic activity of p110 (Fig. 1). Additionally, the binding of the p85 subunit to specific phosphotyrosine residues on activated tyrosine kinases in the plasma membrane localizes the PI3K heterodimer to the plasma membrane. This allows the p110 catalytic subunit to phosphorylate membrane-bound PIP2 to produce PIP3 [1].
Fig. 1.
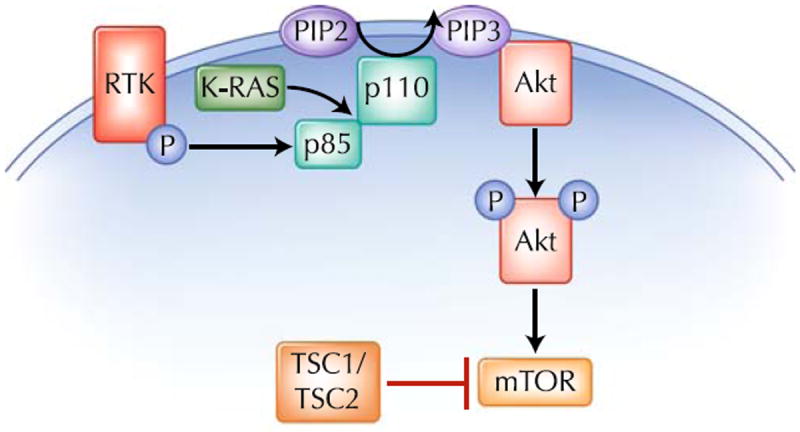
Phosphoinositide-3 kinase (PI3K) is a heterodimer consisting of p85 and p110 subunits. PI3K becomes activated when phosphorylated receptor tyrosine kinases (RTK), or adapter proteins (P), bind to p85. K-RAS is also able to activate PI3K. Once PI3K is activated, the p110 catalytic subunit converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3). Akt binds to PIP3 in the plasma membrane and becomes activated. Akt is then able to activate mammalian target of rapamycin (mTOR) by blocking the ability of TSC2 to inhibit mTOR
A major PIP3 binding protein is the serine/threonine kinase Akt (also called protein kinase B). The binding of Akt to PIP3 localizes Akt to the plasma membrane and allows it to be phosphorylated by PDK1 and mammalian target of rapamycin (mTOR) complex-2 [7]. Once phosphorylated, Akt moves to the cytoplasm and activates an array of proteins critical to cell growth and survival. For example, Akt inhibits proapoptotic proteins, including BAD, caspase 9, and the forkhead (FOX) family of transcription factors [8]. In addition, Akt also phosphorylates the tuberous sclerosis gene TSC2. Under basal conditions, TSC2 negatively regulates mTOR, so that Akt-mediated TSC2 phosphorylation activates mTOR, which in turns activates p70S6K and 4E-BP1, promoting translation of mRNAs important to cancer cell growth [7, 9]. An important negative regulator of PI3K is the phosphatase and tensin homolog (PTEN) phosphatase that converts PIP3 to PIP2 [10]. The ability of PTEN to counteract PI3K by reducing PIP3 levels is important in preventing overactivation of this pathway.
Genetic PI3K Pathway Aberrations in Cancer
Genetic aberrations leading to the activation of the PI3K pathway are common in human malignancies [1, 3•, 11]. One of the best examples of this is PIK3CA-activating mutations and amplifications. PIK3CA-activating mutations cluster in “hot-spot” regions within p110α and two of the most common PIK3CA mutations are H1047R and E545K [3•, 12]. The H1047R mutation may increase p110α binding to the plasma membrane, whereas the E545K mutation releases p110α from inhibition by p85α [13]. Cancers that harbor frequent PIK3CA mutation include breast cancer, colorectal cancer, glioblastoma, hepatocellular cancer, and ovarian cancer [1, 3•]. Notably, mutations in the other three p110 isoforms (p110β, p110γ, and p110δ) have not been reported. However, overexpression of p110δ and p110γ has been described in both acute myeloid leukemia (AML) and chronic myeloid leukemia [14, 15].
Recently, PIK3R1 mutations have also been described. Interestingly, p85α mutants are able to bind, but not inhibit, p110α [13]. Additionally, these mutants increase downstream Akt activation and result in leukemogenesis when cells expressing the p85 mutant are injected into mice [16]. Furthermore, p110β and p110δ also can become activated when they are bound to mutated p85α [13].
An Akt1-activating mutation, E17K, has been described in breast, colorectal, and ovarian cancers. The E17K mutation alters the lipid-binding properties of Akt1 and allows it to bind nonspecifically to the plasma membrane [12, 17]. As a result of the E17K mutation, Akt1 inappropriately localizes to the plasma membrane and becomes activated [17]. The oncogenic potential of the E17K mutation was demonstrated by the observation that mice injected with cells expressing this mutation developed leukemia [17].
Just as activating mutations of PIK3CA, PIK3R1 and Akt1 promote cancer, inactivating mutation and deletion of the tumor suppressor PTEN are also frequently found in human malignancy. PTEN is a negative regulator of PI3K and its deletion leads to inappropriately high levels of PI3K activation [10]. Cancers with high frequencies of genetic aberrations in PTEN include glioblastoma, prostate cancer, breast cancer, melanoma, endometrial cancer, colorectal cancer, and gastric cancer [1, 3•]. Two genetic syndromes that convey an increased risk of cancer, Cowden’s Disease and Bannayan–Riley–Ruvalcaba syndrome, are caused by germline PTEN mutations [10]. Interestingly, unlike most tumor suppressors, the loss of just one copy (ie, haploinsufficiency) of PTEN is often sufficient to cause cancer [10].
The activation of the PI3K pathway has also been associated with acquired resistance to molecularly targeted therapies. For example, in EGFR-mutated lung cancer, erlotinib blocks EGFR activation and prevents it from activating PI3K and the mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK)–extracellular signal-regulated protein kinase (ERK) pathways [18••]. Resistance to erlotinib can develop when molecular alterations, such as MET amplification or acquisition of the secondary T790M EGFR mutation, restore PI3K activation [19•, 20]. Similarly, in vitro models have demonstrated that PI3KCA mutations or PTEN deletions can lead to acquired resistance to cetuximab and trastuzumab [21, 22].
“First Generation” PI3K Inhibitors: Wortmannin and LY294002
The first PI3K inhibitor, wortmannin, was isolated from the fungus Penicillium wortmannin in 1957 [4•]. Wortmannin has been a widely used reagent in basic science laboratories but has not been developed clinically because of several pharmacologic shortcomings. Wortmannin is not a specific PI3K inhibitor and has activity against proteins that are structurally related to PI3K, including DNA-PK, ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), and mTOR [4]. Wortmannin is extremely reactive, has a short half-life, and causes liver dysfunction, lymphocytopenia, and hyperglycemia in animal models [23].
In 1994, Eli Lilly (Indianapolis, IN) synthesized the reversible PI3K inhibitor LY294002, developed as a structural analog of quercetin, a bioflavonoid produced by plants that can inhibit several protein kinases including PI3K [24]. LY294002 is more stable but less potent than wortmannin [4•]. Similar to wortmannin, it is not a specific PI3K inhibitor. Poor aqueous solubility impeded its clinical development [25].
“Second Generation” PI3K Inhibitors
The current generation of PI3K inhibitors was designed to improve upon the pharmacologic characteristics of wortmannin and LY294002. PX-866 (Oncothyreon) is a structural analog of wortmannin that was selected for development because it is more stable and less toxic than wortmannin [23]. Like wortmannin, it is a potent irreversible pan-PI3K inhibitor (Table 1) [26]. It has demonstrated significant antitumor activity in murine xenograft models of lung, ovarian, and colon cancer [23]. Phase 1 clinical trials on PX-866 are presently underway and preliminary results have shown several patients with stable disease, with abdominal pain and mild diarrhea described as possible associated side effects [27]. Reduced phosphorylation of mTOR and p70S6K have been demonstrated in peripheral blood mononuclear cells [27].
Table 1.
Summary of phosphoinositide-3 kinase inhibitors
| Study | Drug |
IC50 (nM)
|
|||||
|---|---|---|---|---|---|---|---|
| p110α | p110β | p110γ | P110δ | mTOR | Phase 1 trials | ||
| Ihle et al. [26]; Toral-Barza et al. [54] | Wortmannin | 2 | 0.6 | 10 | 3.9 | 200 | No |
| Garlich et al. [25], Toral-Barza et al. [54] | LY294002 | 720 | 306 | 1,600 | 1,330 | 1,500 | No |
| Ihle et al. [26] | PX-866 | 0.1 | > 300 | 1.0 | 2.9 | NA | Yes |
| Chiorean et al. [28] | PWT-458 | NAa | NAa | NAa | NAa | NAa | No |
| Garlich et al. [25]; Chiorean et al. [29] | SF1126 | NAb | NAb | NAb | NAb | NAb | Yes |
| Folkes et al. [30];Wagner et al. [31]; Sarker et al. [32] | GDC-0941 | 3 | 33 | 75 | 3 | 580 | Yes |
| Foster et al. [33]; Shapiro et al. [34]; Faulkner et al. [35]; Wheler et al. [36] | XL147 | 39 | 383 | 23 | 36 | 15,000 | Yes |
| Maira et al. [38] | NVP-BEZ235 | 4 | 75 | 5 | 7 | 21 | Yes |
| GDC-0980 | NA | NA | NA | NA | NA | Yes | |
| LoRusso et al. [37] | XL765 | 39 | 113 | 9 | 43 | 157 | Yes |
| Flinn et al. [43••] | CAL-101 | NA | NA | NA | 2.5 | NA | Yes |
PWT-458 is a pegylated form of wortmannin
SF1126 is a close derivative of LY294002
mTOR mammalian target of rapamycin, NA not available
Another approach to bring wortmannin to the clinic was to increase its stability by pegylation. PWT-458 (Pfizer; New York, NY), pegylated-17-hydroxywortmannin, is the product of this strategy [28]. Although PWT-458 has demonstrated antitumor activity in xenograft models, it has yet to enter into human clinical trials [28].
Efforts have also been directed at modifying the pharmaceutical properties of LY294002. SF1126 (Semafore Pharmaceuticals; Westfield, IN) was developed by attaching the RGD targeting peptide onto LY294002 [25]. The RGD peptide binds readily to integrins, and its addition onto LY294002 makes the drug water-soluble, correcting a major limitation. The integrin-targeting RGD peptide is expected to cause the drug to preferentially accumulate in endothelial cells and tumor cells. Preclinically, SF1126 has shown activity against murine models of breast cancer and glioblastoma [25]. In addition, it has demonstrated antiangiogenic activity in xenografted LN229 glioma cells by substantially decreasing microvessel formation [25]. SF1126 is currently in phase 1 clinical trials, and is one of the few PI3K inhibitors administered intravenously. The best response to date is stable disease of up to 20 weeks in patients with gastrointestinal stromal tumors (GIST), endometrial cancer, and prostate cancer [29]. The most serious side effect reported has been grade 3 diarrhea [29].
Other second-generation PI3K inhibitors are chemically distinct from wortmannin and LY294002. For example, PI-103 is a potent inhibitor of PI3K; as with wortmannin and LY294002, this compound has undesirable pharmacologic properties because of its poor solubility and rapid metabolism. However, a series of structural analogs has been studied, with GDC-0941 (Genentech; South San Francisco, CA) emerging with favorable pharmacokinetic properties and retention of potent PI3K inhibition [30]. Unlike wortmannin, GDC-0941 is selective for PI3K and is a relatively weak inhibitor of mTOR (Table 1). GDC-0941 has been well-tolerated in phase 1 clinical trials of once and twice daily dosing, 3 weeks of every 4, with mild nausea, fatigue, diarrhea, peripheral edema, and liver function test abnormalities observed [31, 32]. Scans from a HER-2–positive breast cancer patient demonstrated diminished fluorine-18 2-fluoro-2-deoxy-D-glucose (FDG) uptake and improvement of target lesions. Patients with sarcoma, endometrial cancer, and ovarian cancer have also achieved prolonged stable disease, the latter with improvement of CA125 [31, 32].
Like GDC-0941, XL147 (under co-development by Exelixis [South San Francisco, CA] and Sanofi-Aventis [Bridgewater, NJ]) selectivity inhibits PI3K without inhibiting mTOR (Table 1) [33]. XL147 is also currently in phase 1 clinical trials evaluating schedules 3 weeks of every 4 and continuous once-daily dosing [34, 35]. The dose-limiting toxicity is drug-induced rash; elevated liver function tests and fatigue have also occurred. XL147 likely augments food-induced changes in plasma insulin, but blood glucose is only minimally affected with mild hyperglycemia noted in four of 39 patients. Extensive pharmacodynamic studies utilizing pre- and post-treatment hair follicles, skin biopsies, and tumor biopsies demonstrated substantial inhibition of phosphorylated PI3K pathway components, with the PI3K pathway extinguished by ≥70% in some tumors, without compensatory upregulation of MEK/ERK phosphorylation [34]. Six patients, including several with EGFR wild-type non–small cell lung cancer (NSCLC), continued on treatment more than 6 months, with one partial response noted in an NSCLC patient. Reduced FDG–positron emission tomography (PET) activity was demonstrated in a patient with GIST, and a patient with hormone-refractory prostate cancer had his prostate-specific antigen (PSA) level normalize for more than 5 months. Notably, phase 1 clinical trials combining XL147 with chemotherapy (carboplatin and paclitaxel) or erlotinib are also underway [35, 36]. Preliminary results indicate that these combinations are feasible, reasonably well tolerated, and have produced responses [35, 36].
Dual PI3K and mTOR Inhibitors
The catalytic subunit of mTOR is structurally similar to PI3K, so that many PI3K inhibitors under development also potently inhibit mTOR. At the present time, it is unclear whether dual inhibition of mTOR and PI3K is an advantage over PI3K inhibition alone. However, both PI3K and dual PI3K–mTOR inhibitors may have advantages over agents that solely inhibit mTOR, largely because mTOR functions in a negative feedback loop, so that its activation limits upstream PI3K activity [9]. Therefore, a consequence of mTOR inhibition is activation of both PI3K and Akt, a problem circumvented by concomitant PI3K inhibition [12].
Several dual PI3K and mTOR inhibitors have entered clinical trials, including NVP-BEZ235 (Novartis; Basel, Switzerland), GDC-0980 (Genentech), and XL765 (Exelixis/Sanofi-Aventis) [37]. NVP-BEZ235 was derived from the imidazo[4,5-c]quinoline chemical scaffold that was predicted to inhibit the ATP binding site of PI3K based on its crystal structure [38]. This agent competitively inhibits all of the PI3K isoforms, and has been shown to be effective in preclinical models of glioblastoma, melanoma, pancreatic cancer, Waldenström’s macroglobulinemia, and multiple myeloma [38]. In breast cancer models, NVP-BEZ235 has been shown to selectively induce apoptosis in cell lines harboring HER-2 amplification, PIK3CA mutation or both, and has also been shown to reverse lapatinib resistance [39]. All three agents are in phase 1 trials, with XL765 investigated on twice daily and once-daily regimens. Although overall well tolerated, dose-limiting toxicities have occurred, including elevated transaminases, nausea/vomiting, anorexia, and rash. Pharmacodynamic assessments in hair follicles, skin, and tumor demonstrated about 60% to 90% PI3K inhibition when phosphorylated PI3K pathway proteins were analyzed [37].
Isotype-Selective PI3K Inhibitors
p110 Isotype-specific inhibitors are also under development [3•]. This strategy may minimize potential side effects. For example, p110α, and to a lesser extent p110β, play an important role in transducing PI3K signals from the insulin receptor [40]. Hence, a PI3K inhibitor that does not have activity against p110α and p110β potentially would induce less severe hyperglycemia as a side effect. Similarly, because p110γ and p110δ play important roles in lymphocyte signal transduction, having an inhibitor that lacked activity against p110γ and p110δ could minimize potential immunological side effects [3•].
Because p110δ is abundantly expressed in lymphocytes and plays an important role in antiapoptotic signaling pathways in some lymphomas and leukemias, there has been great interest in the development of a selective PI3Kδ inhibitor [41, 42]. CAL-101 (Calistoga Pharmaceuticals; Seattle, WA) is the first PI3Kδ specific inhibitor to be tested clinically and thus far the results have been encouraging. Preliminary results reported from the phase 1 clinical trial have indicated that 56% of patients with non-Hodgkin’s lymphoma have had a partial response [43••]. Significant activity was also seen in chronic lymphocytic leukemia (CLL), with 35% of the patients achieving partial response and another 41% of patients demonstrating at least 50% decrease in lymphocytosis [43]. CAL-101 has been well tolerated, causing a reversible elevation in liver function tests in 11% of the patients in this study [43••].
Inhibitors of Akt
Several inhibitors of Akt are under development. Perifosine, recently extensively reviewed, inhibits the ability of Akt to localize to the plasma membrane [44]. Perifosine has reached phase 2 trials and has demonstrated activity in sarcoma and Waldenström’s macroglobulinemia patients [44].
MK-2206 (Merck; Whitehouse Station, NJ) is an allosteric inhibitor of Akt1, 2, and 3 [45]. A phase 1 trial is underway evaluating every-other-day dosing in 28-day cycles; dose-limiting toxicities have included rash and mucositis. At tolerable doses, MK-2206 treatment resulted in significant Akt inhibition throughout the dosing interval [45]. Pharmacodynamic studies showed diminished phospho-Akt at all dose levels, as well as mild hyperglycemia. Six patients achieved stable disease for at least two cycles of MK-2206 treatment [45].
Determinants of PI3K Addiction and Development of Rational Combinations
Although prolonged stable disease has been observed in several clinical trials utilizing PI3K inhibitors, robust response rates among patients with solid tumors have not been achieved. This is prompting efforts to define determinants of PI3K addiction, with the ultimate goal of identifying tumors most likely to respond when the pathway is extinguished. For example, the dual PI3K/mTOR inhibitor NVP-BEZ235 causes apoptosis in breast cancer cell lines harboring either HER2 amplification and/or PIK3CA mutation [46••]. These data have corroborated those of other reports, in which NVP-BEZ235 effectively inhibits the entire PI3K pathway in HER2-amplified cells, with no residual mTORC1 activity detected, as evidenced by ablated p70S6K phosphorylation [18]. Similar results have also been described with GDC-0941, more selectively inhibiting PI3K alone, in transgenic mice bearing lung tumors induced by expression of mutant HER-2 [47]. These data are also consistent with observations demonstrating that resistance to lapatinib and trastuzumab can be overcome in in vitro models of breast cancer by NVP-BEZ235, SF1126, and GDC-0941 [39, 48, 49].
Importantly, among breast cancer cell lines with inactivated PTEN, NVP-BEZ235 did not induce substantial apoptosis [46••]. These cells expressed higher baseline levels of activated ERK, which was capable of phosphorylation of the mTORC1 target p70S6K; continued phospho-p70S6K expression after NVP-BEZ235 exposure correlated with preserved cell viability in response to the drug [46••]. Therefore, in breast cancer, not all genetic aberrations of the PI3K pathway lead to strict PI3K addiction, and resistance to NVPBEZ235 in PTEN-deficient cell lines potentially could be overcome by addition of an MEK inhibitor [46••].
Whether the genetic pathway alterations in other tumor types (eg, PIK3CA mutation in colon cancer; PIK3CA amplification in NSCLC or ovarian cancer; PTEN deficiency in glioblastoma, prostate cancer, ovarian or endometrial cancer) will result in sufficient PI3K pathway addition to dictate objective response or prolonged disease stability to a PI3K or PI3K/mTOR inhibitor alone remains to be determined. This issue has not been explored in depth in the context of phase 1 clinical trials to date. Ultimately, the assessment of molecular subsets of patients in phase 2 studies in individual tumor types will be important for defining those patients most likely to benefit.
Similarly, it is unclear whether the activation of receptor tyrosine kinases other than HER2 will lead to addiction to the PI3K pathway alone. Recently, it has been reported that among a large panel of NSCLC cancer cell lines, those most susceptible to PI-103–mediated PI3K/mTOR inhibition were those with receptor tyrosine kinase activation, including cell lines harboring EGFR mutation or amplification, MET amplification, or HER2 mutation or amplification [47]. EGFR mutant NSCLC xenografts, including those harboring the secondary T790M mutation conferring erlotinib resistance, underwent substantial tumor growth inhibition with the PI-103 pharmalog GDC-0941 [47]. However, data in the transgenic murine model of lung adenocarcinoma induced by EGFR L858R/T790M, was not as promising for PI3K inhibition alone; only stable disease was documented, in contrast to the results in mice harboring lung cancers driven by mutant HER2, where pronounced tumor shrinkage was observed [47]. Additionally, EGFR mutant NSCLC cell lines did not undergo substantial apoptosis in response to NVP-BEZ235 alone, but rather required the addition of the MEK inhibitor ARRY-142886 (AZD6244) [18]. The combination was as effective as gefitinib alone in gefitinib-sensitive models, and also induced marked regression in the EGFR L858R/T790M murine transgenic model [18]. Taken together, these data suggest that the degree of addiction to the PI3K pathway among activated receptor tyrosine kinases is not equivalent. In contrast to HER2-driven tumors, EGFR driven tumors may have only partial dependency on PI3K, so that the combination of PI3K inhibition and MEK inhibition may be necessary to achieve tumor regression. Furthermore, differences among compounds may also exist, with the possibility of unchanged, increased, or even decreased degrees of ERK activation present after exposure to individual compounds [34, 37].
The preclinical data in PTEN-deficient breast cancer cell lines and EGFR mutant NSCLC models suggest that the combination of PI3K and MEK inhibition may overcome the lack of responses observed to single-agent PI3K and PI3K/mTOR inhibitors to date. The same is likely true in K-RAS–driven tumors. These represent a major unmet medical need because direct pharmacologic inhibition of K-RAS has thus far been elusive. Both breast and NSCLC cell lines with activated K-RAS did not respond to PI3K inhibition, but K-RAS mutant NSCLC cell lines did achieve some degree of growth arrest and/or apoptosis in response to MEK inhibition. In this case, residual PI3K activity likely mediated resistance, such that the addition of a PI3K inhibitor to an MEK inhibitor resulted in synergy. Importantly, these results corroborated those in the K-RAS driven model of murine lung adenocarcinoma [50••]. In this model, the PI3K/mTOR inhibitor NVP-BEZ235 had no antitumor activity, whereas the MEK inhibitor AZD6244 had only modest activity. However, the combination led to a substantial reduction in tumor size and FDG avidity [50••].
These results have generated optimism that a dual pathway inhibitory approach will be successful in a large variety of tumor types, including both receptor tyrosine kinase-and K-RAS–driven cancers. A number of clinical trials have been designed to develop such combinations that will ultimately test this hypothesis, including a trial utilizing the PI3K inhibitor GDC-0941 and the MEK inhibitor GDC-0973 (Genentech). Additionally, the AKT inhibitor MK-2206 will be combined with the MEK inhibitor AZD6244, and similarly, mTOR and MEK inhibitor combinations are under exploration.
On a cautionary note, it will first be important to establish the tolerability of each regimen in carefully designed phase 1 trials. For combinations of molecularly targeted agents, dose titrations permitting simultaneous exploration of more than one dose level in which one or the other drug is escalated may speed the achievement of optimal safe doses. In some cases, combination of maximum doses at the outset may be successful, with deescalation permitted, if appropriate.
In addition to combining PI3K inhibitors with other targeted therapy, data also suggest that PI3K inhibitors will potentiate the effects of cytotoxic chemotherapy. Because activated Akt can mediate resistance to chemotherapy, suppression of the PI3K-Akt axis may sensitize cells. For example, experiments in doxorubicin-resistant CML cell lines demonstrated high levels of PI3K and Akt activity; importantly, doxorubicin resistance could be overcome by decreasing PI3K and Akt activity [51]. Further experimental evidence was observed in two pancreatic cancer cell lines in which wortmannin and LY294002 treatment decreased levels of phosphorylated Akt and increased gemcitabine-induced apoptosis [52]. Similarly, PX-866 has shown synergistic antitumor activity with cisplatin in a murine xenograft model of lung cancer [23].
Conclusions
The PI3K pathway inhibitors represent a novel drug class addressing a pathway universally activated in human cancer. PI3K inhibitors are still early in their development and are being tested in phase 1 monotherapy and combination clinical trials (Table 1). To date, the most significant clinical success has been the effectiveness of CAL-101, the oral PI3Kδ inhibitor, in B cell malignancies [43••]. Encouragingly, preliminary data indicate that PI3K inhibitors are well tolerated. As expected, given the importance of PI3K in insulin-receptor signaling, patients have been observed to have high insulin levels; however, in most cases, concomitant hyperglycemia has been mild at most. Other commonly observed toxicities thus far include rash, nausea, vomiting, diarrhea, and elevation of liver function tests [27, 29, 31, 32, 34, 37], usually of mild or moderate severity and reversible. Importantly, for several compounds, robust pharmacodynamic data have been generated during the course of phase 1 work, demonstrating PI3K pathway inhibition by assessment of phosphorylated targets in tumor biopsies acquired pre- and post-treatment. It remains unclear whether 60% to 90% decreases in pathway activation, as measured by immunofluorescence or immunohistochemistry, are sufficient to achieve anti-tumor activity. Nonetheless, the work suggests that the PI3K pathway is substantially modulated by several agents, so that single-agent phase 2 trials in individual tumor types are appropriate. In the context of these trials, it will be important to examine multiple molecular subsets both in retrospective and ultimately prospective fashion, so that patients most likely to derive benefit will be identified.
The same is true for MK-2206 and other Akt inhibitors under development. Of note, the genomic determinants of response may differ from those for a PI3K inhibitor, because PI3K3CA-mutant cancers may signal through Akt-independent mechanisms in some cases, with a PI3K-PDK1-SGK3 axis recently described [53].
The demonstration of expected pharmacodynamic effects of these compounds will also allow the field to move forward with rationally designed combinations. Preclinical data suggest that the addition of an MEK inhibitor to a PI3K inhibitor (or an Akt inhibitor) represents a particularly promising strategy designed to block the two most dominant signaling pathways operating in most cancer cells. Additionally, PI3K and Akt inhibition may substantially improve chemotherapy-mediated cytotoxicity. Ultimately, for a large portion of solid tumors, these agents may most likely prove their efficacy as part of combination regimens.
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
References
Papers of particular interest, published recently, have been highlighted as:
-
•
Of importance
-
••
Of major importance
- 1.Engelman JA, Ji L, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Hietakangas V, Cohen SM. Regulation of tissue growth through nutrient sensing. Annu Rev Genet. 2009;43:389–410. doi: 10.1146/annurev-genet-102108-134815. [DOI] [PubMed] [Google Scholar]
- 3•.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. This article provides an extremely informative review on the role of PI3K in cancer. It also reviews therapeutic approaches of targeting the PI3K pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4•.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. This excellent review thoroughly details the development of PI3K inhibitors. [DOI] [PubMed] [Google Scholar]
- 5.Gupta S, Ramjaun AR, Haiko P, Wang Y, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 6.Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–272. doi: 10.1111/j.1600-065X.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 7.Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–6488. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- 8.Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest. 2005;115:2618–2624. doi: 10.1172/JCI26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 11.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004:304–554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 12.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 13.Martin-Berenjeno I, Vanhaesebroeck B. PI3K regulatory subunits lose control in cancer. Cancer Cell. 2009;16:449–450. doi: 10.1016/j.ccr.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 14.Sujobert P, Bardet V, Cornillet-Lefebvre P, et al. Essential role for the p110δ isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005;106:1063–1066. doi: 10.1182/blood-2004-08-3225. [DOI] [PubMed] [Google Scholar]
- 15.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486–5496. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaiswal BS, Janakiraman V, Kljavin NM, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16:463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 18••.Faber AC, Li D, Song Y, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009;106:19503–19508. doi: 10.1073/pnas.0905056106. This paper demonstrates that combination therapy with PI3K and MEK inhibitors can be effective in EGFR-addicted cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. This paper demonstrated that acquired resistance to EGFR inhibitors can arise through MET amplification. The MET overexpression results in PI3K activation. [DOI] [PubMed] [Google Scholar]
- 20.Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jhawer M, Goel S, Wilson AJ, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68:1953–1961. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 23.Ihle NT, Williams R, Chow S, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3:763–772. [PubMed] [Google Scholar]
- 24.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 25.Garlich JR, De P, Dey N, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;68:206–215. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 26.Ihle NT, Paine-Murrieta G, Berggren MI, et al. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther. 2005;4:1349–1357. doi: 10.1158/1535-7163.MCT-05-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jimeno A, Hong DS, Hecker S, et al. Phase I trial of PX-866, a novel phosphoinositide-3-kinase (PI-3K) inhibitor. J Clin Oncol. 2009;27:3542. Meeting Abstracts. [Google Scholar]
- 28.Yu K, Lucas J, Zhu T, et al. PWT-458, a novel pegylated-17-hydroxywortmannin, inhibits phosphatidylinositol 3-kinase signaling and suppresses growth of solid tumors. Cancer Biol Ther. 2005;4:538–545. doi: 10.4161/cbt.4.5.1660. [DOI] [PubMed] [Google Scholar]
- 29.Chiorean EG, Mahadevan D, Harris WB, et al. Phase I evaluation of SF1126, a vascular targeted PI3K inhibitor, administered twice weekly IV in patients with refractory solid tumors. J Clin Oncol. 2009;27:2558. Meeting Abstracts. [Google Scholar]
- 30.Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 31.Wagner AJ, Von Hoff DH, LoRusso PM, et al. A first-in-human phase I study to evaluate the pan-PI3K inhibitor GDC-0941 administered QD or BID in patients with advanced solid tumors. J Clin Oncol. 2009;27:3501. Meeting Abstracts. [Google Scholar]
- 32.Sarker D, Kristeleit R, Mazina KE, et al. A phase I study evaluating the pharmacokinetics (PK) and pharmacodynamics (PD) of the oral pan-phosphoinositide-3 kinase (PI3K) inhibitor GDC-0941. J Clin Oncol. 2009;27:3538. Meeting Abstracts. [Google Scholar]
- 33.Foster P. Potentiating the antitumor effects of chemotherapy with the selective PI3K inhibitor XL147. AACR Meeting Abstracts. 2007;2007:C199. [Google Scholar]
- 34.Shapiro G, Kwak E, Baselga J, et al. Phase I dose-escalation study of XL147, a PI3K inhibitor administered orally to patients with solid tumors. J Clin Oncol. 2009;27:3500. Meeting Abstracts. [Google Scholar]
- 35.Faulkner N, LoRusso PM, Guthrie T, et al. A phase 1 safety and pharmacokinetic (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination with erlotinib in patients with advanced solid tumors. Mol Cancer Ther. 2009;8:C197. [Google Scholar]
- 36.Wheler JJ, Traynor AM, Bailey HH, et al. A phase 1 safety and pharmacokinectic (PK) study of the PI3K inhibitor XL147 in combination with paclitaxel and carboplatin in patients with advanced solid tumors. Presented at the American Association for Cancer Research–National Cancer Institute–European Organization for Research and Treatment of Cancer International Conference on Molecular Targets and Cancer Therapeutics; Boston, MA. November 15–19; 2009. p. B247. [Google Scholar]
- 37.LoRusso P, Markman B, Tabernero J, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765, a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced solid tumors. J Clin Oncol. 2009;27:3502. Meeting Abstracts. [Google Scholar]
- 38.Maira S-M, Stauffer Fdr, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 39.Eichhorn PJ, Gili M, Scaltriti M, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68:9221–9230. doi: 10.1158/0008-5472.CAN-08-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaywitz AJ, Courtney KD, Patnaik A, Cantley LC. PI3K enters beta-testing. Cell Metabolism. 2008;8:179–181. doi: 10.1016/j.cmet.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Billottet C, Grandage VL, Gale RE, et al. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene. 2006;25:6648–6659. doi: 10.1038/sj.onc.1209670. [DOI] [PubMed] [Google Scholar]
- 42.Uddin S, Hussain AR, Siraj AK, et al. Role of phosphatidylinositol 3’-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood. 2006;108:4178–4186. doi: 10.1182/blood-2006-04-016907. [DOI] [PubMed] [Google Scholar]
- 43••.Flinn IW, Byrd JC, Furman RR, et al. Evidence of clinical activity in a phase 1 study of CAL-101, an oral P110δ isoform-selective inhibitor of phosphatidylinositol 3-kinase, in patients with relapsed or refractory B-Cell malignancies. Presented at the 51st American Society of Hematology Annual Meeting; New Orleans, LA. December 5–8, 2009; p. 922. This abstract describes preliminary results showing that single agent CAL-101 has produced several partial responses in B cell malignancies. [Google Scholar]
- 44.Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11:102–110. doi: 10.1007/s11912-009-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tolcher AW, Yap TA, Fearen I, et al. A phase I study of MK-2206, an oral potent allosteric Akt inhibitor (Akti), in patients (pts) with advanced solid tumor (ST) ASCO Meeting Abstracts. 2009;27:3503. [Google Scholar]
- 46••.Brachmann SM, Hofmann I, Schnell C, et al. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci U S A. 2009;106:22299–22304. doi: 10.1073/pnas.0905152106. This paper demonstrates that NVP-BEZ235 is effective in HER2 amplified and PIK3CA mutant breast cancer cells. Importantly, it also shows that breast cancer cells with loss of PTEN function are resistant to NVP-BEZ235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009;106:18351–6. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ozbay T, Durden DL, Liu T, et al. In vitro evaluation of pan-PI3-kinase inhibitor SF1126 in trastuzumab-sensitive and trastuzumab-resistant HER2-over-expressing breast cancer cells. Cancer Chemother Pharmacol. 2009 Jul 28; doi: 10.1007/s00280-009-1075-9. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15:429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 50••.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. This paper shows that combined PI3K and MEK inhibition is effective in a RAS mutated model of murine lung cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hui RC, Gomes AR, Constantinidou D, et al. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol Cell Biol. 2008;28:5886–5898. doi: 10.1128/MCB.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ng SSW, Tsao MS, Chow S, Hedley DW. Inhibition of phosphatidylinositide 3-kinase enhances gemcitabine-induced apoptosis in human pancreatic cancer cells. Cancer Res. 2000;60:5451–5455. [PubMed] [Google Scholar]
- 53.Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toral-Barza L, Zhang WG, Lamison C, et al. Characterization of the cloned full-length and a truncated human target of rapamycin: activity, specificity, and enzyme inhibition as studied by a high capacity assay. Biochem Biophys Res Commun. 2005;332:304–310. doi: 10.1016/j.bbrc.2005.04.117. [DOI] [PubMed] [Google Scholar]