

Abstract
Chemically interfacing the inert basal plane of graphene with other materials has limited the development of graphene-based catalysts, composite materials, and devices. Here, we overcome this limitation by chemically activating epitaxial graphene on SiC(0001) using atomic oxygen. Atomic oxygen produces epoxide groups on graphene, which act as reactive nucleation sites for zinc oxide nanoparticle growth using the atomic layer deposition precursor diethyl zinc. In particular, exposure of epoxidized graphene to diethyl zinc abstracts oxygen, creating mobile species which diffuse on the surface to form metal oxide clusters. This mechanism is corroborated with a combination of scanning probe microscopy, Raman spectroscopy, and density functional theory, and can likely be generalized to a wide variety of related surface reactions on graphene.
Introduction
Because of its remarkable electronic, thermal, and mechanical properties, graphene is a leading candidate for a variety of applications including transistors,1,2 batteries,3 photocatalysts,4-6 solar cells,7 and supercapacitors.8 However, despite the widespread technological interest in graphene, the chemical inertness of this material hinders its integration with the other materials that are present in fully fabricated devices and systems. The most common solution to this problem has been to oxidize graphene or graphite using an aggressive solution-based treatment known as the Hummers Method.9 This approach creates a plethora of oxygen containing functional groups on both the edges and basal plane of graphene,10,11 which can be used as chemically active anchors to graphene oxide (GO).12-16 Subsequent reduction via thermal or chemical methods17 results in reduced graphene oxide (rGO), which partially restores the electrical conductivity of the original graphene.
Despite the chemical success of these methods, GO and rGO are fundamentally different from pristine graphene. GO and rGO possess a high concentration of defects including polyfunctionalization, holes, and edge states that act as scattering centers and thus compromise charge conduction that underlies performance in applications.18,19 For example, Liang et al. have shown that in composite films of TiO2 and graphitic nanomaterials, the defects inherent to rGO and GO lower their catalytic performance compared to pristine graphene.20 However, it should be noted that previously demonstrated solution-based methods for producing nanocomposites between pristine graphene and metal oxide nanoparticles require the presence of binding agents or surfactants,21-23 which occlude internal interfaces and likely compromise ultimate catalytic performance.
Here, we report an alternative method for chemically activating graphene via gas-phase atomic oxygen that avoids the irreversible defect formation characteristic of the Hummers Method. Functionalization of graphene by atomic radicals has become a proven method for imparting new properties to graphene.24 Following atomic oxygen exposure, graphene is functionalized with epoxide groups, which can then be used to nucleate the growth of metal oxide nanoparticles via organometallic precursors. In particular, epitaxial graphene (EG) on SiC(0001) is exposed to alternating cycles of atomic oxygen (AO) and diethyl zinc (DEZ) under ultrahigh vacuum (UHV) conditions. Atomic force microscopy (AFM) shows that this process creates regularly sized nanoparticles on the surface of EG, while X-ray photoelectron spectroscopy (XPS) confirms the chemical identity of these nanoparticles as ZnO. Density functional theory (DFT) calculations provide molecular-level insight into the underlying chemical mechanisms that underpin this process, which is then validated with Raman spectroscopy. While demonstrated here for metal oxide nanoparticle growth, epoxidation with atomic oxygen can serve as a general method for chemically activating graphene with minimal collateral defects.
Experimental Section
The reaction scheme and proposed reactive species are outlined in Figure 1, and a more detailed description is given in the Supporting Information. EG was produced by thermally evaporating silicon from n-doped SiC(0001) in a UHV chamber with a base pressure of 6 × 10-11 Torr using previously reported methods.25 EG was then exposed to cycles of AO and DEZ. For the AO half cycles, the EG was exposed to 10-6 Torr of molecular oxygen in the presence of a 1500 °C tungsten filament, which thermally cracks molecular oxygen into AO. Previous work has shown that this procedure decorates the surface of graphene with epoxide functional groups, producing graphene epoxide (GE).26 Exposures to AO were limited to the low density regime (< 3 % of carbons converted) to limit the potential for formation of WOx. Furthermore, high AO exposure has been shown to irreversibly damage the physical and electronic structure of graphene on metallic surfaces.27 For the DEZ half cycles, samples were transferred into an adjacent high vacuum chamber and exposed to the vapor pressure of liquid DEZ. To remove any physisorbed DEZ or other species following this step, the sample was heated to 100-200 °C between each cycle.
Figure 1.
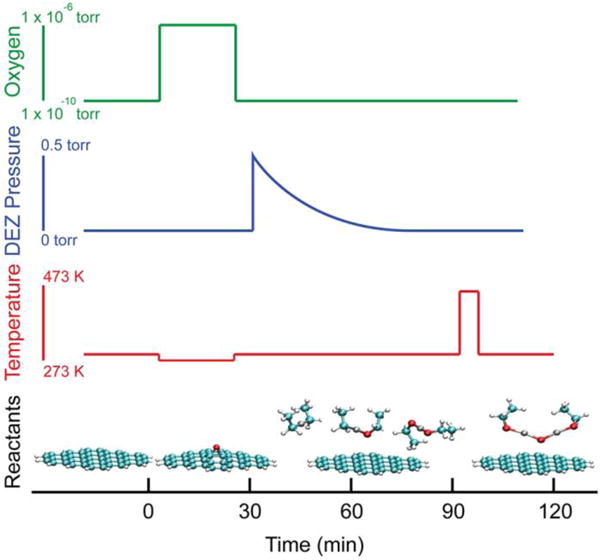
Synthetic steps for the creation of ZnO nanoparticles on the surface of graphene. The top three panels show the oxygen, diethyl zinc (DEZ), and thermal profiles during the reaction. The bottom panel shows the reactive species assumed to be present at each step.
Density functional calculations were performed using the ADF program to characterize the proposed reaction and potential energy surface.8,28 A supramolecular approach was used to calculate the interaction energies between the organometallic species and surface species. 29,30 Additional details concerning the calculations can be found in the supplemental information.
Results and Discussion
Ex situ ambient AFM is used to characterize the surface morphology following the growth of ZnO on graphene. Figure 2a shows a non-contact mode AFM image typical of a clean EG surface. Figure 2b shows the surface after exposure to 4 cycles of DEZ and H2O at room temperature. Although there is some streakiness that suggests physisorbed species on the surface, the image is nearly identical to the pristine EG sample. Evidently, the chemical inertness of graphene prevents any significant chemical interactions with either H2O or DEZ. The difficulty in seeding metal oxide growth on pristine graphene has been discussed by several groups within the context of atomic layer deposition (ALD) dielectrics.31-33
Figure 2.
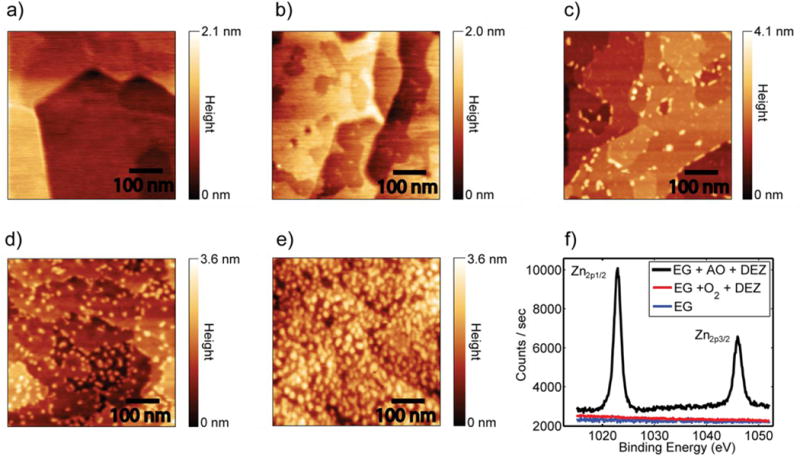
AFM images demonstrating the growth of ZnO nanoparticles on EG. a) Clean EG. b) EG after exposure to 4 cycles of DEZ and H2O showing no growth. Images c), d), and e) showing increasing coverage of nanoparticles with increasing exposure to atomic oxygen. Exposure levels were 1 AO and DEZ cycle with 360 L oxygen, 2 AO and DEZ cycles with 1000 L oxygen, and 2 AO and DEZ cycles with 3600 L oxygen. All images are 500 nm × 500 nm. f) XPS spectra of EG after exposure to 2 O2 and DEZ cycles with 1000 L oxygen, and 2 AO and DEZ cycles with 1000 L oxygen.
In contrast, Figure 2c shows EG after a single cycle of AO (360 L oxygen exposure) followed by saturating DEZ. In this case, the surface morphology is significantly changed, with nanoparticle features appearing on the surface. Although nanoparticles are predominantly clustered at the step edges, nanoparticles are also observed directly on the basal plane. Direct growth on the pristine basal plane distinguishes this method from previously presented methods of growing ZnO on graphene. From line profiles of the AFM images, the nanoparticle height is uniformly 5-6 Å. This value is significantly higher than the 1.7 Å/cycle observed for the growth of ZnO on silica.34
By increasing the exposure of EG to AO, the nanoparticle density correspondingly increases. Figures 2d and 2e show EG after 2 cycles of AO and DEZ with oxygen exposures of 1000 L and 3600 L AO, respectively. Increasing exposure to AO increases the density of epoxide moieties on the surface,26 which in turn leads to interactions with a larger number of DEZ molecules. Finally, XPS is utilized to confirm that the nanoparticles are only being formed in the presence of epoxide functional groups (see Figure 2f). When EG is exposed to 2 cycles of O2 and DEZ, there is no evidence of zinc deposition on the surface. However, when EG is dosed with molecular oxygen in the presence of the hot filament to create AO under otherwise identical conditions, zinc is detected in XPS. Furthermore, the energetic position of the Zn2p3/2 peak at 1022.6 eV is consistent with the formation of ZnO nanoparticles as opposed to the aggregation of metallic zinc.34 Additional support for zinc oxidation resulting from AO was obtained from in situ UHV STM (Figure SI-1).
The ZnO nanoparticles are highly stable on the graphene surface. In particular, the ZnO/EG samples were heated to 500 °C in UHV, similar to the stability of gold nanoparticles on suspended graphene membranes by Kim et al.35 The particles were also subjected to a mild bath sonication in acetone, isopropyl alcohol, and deionized water. Following this treatment, the nanoparticles survived on the surface with no evidence of reduction in number or further aggregation. Despite this stability, the nanoparticles are not strongly bound to the surface as indicated by contact mode AFM and Raman spectroscopy. In Figure 3a, contact mode AFM (with a constant force of 10 nN and estimated pressure of ∼ 1GPa) was used to scan a small square area of the sample. Afterwards, the sample was reimaged using non-contact AFM. Many of the nanoparticles were pushed aside by the AFM tip during contact mode imaging, as evidenced by the aggregation of nanoparticles at the boundary of the scan window. The ability to manipulate the nanoparticles at relatively small contact force illustrates the relatively weak bonding to the graphene surface.
Figure 3.
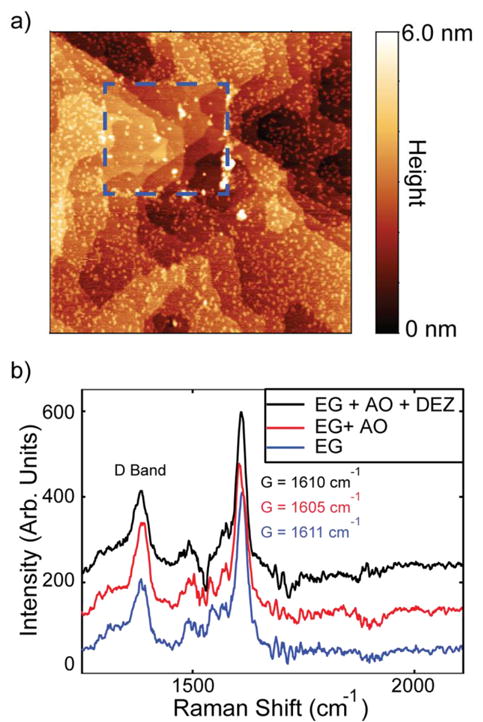
Evidence for weakly interacting nanoparticles on graphene. a) Non-contact AFM image (2 μm × 2 μm) of EG + ZnO following contact mode AFM with a constant force of 10 pN in the rectangular area marked in blue. The color-scale range is limited to 6 nm to improve contrast. b) Raman spectrum of EG, EG + 3600 L AO, and EG + 3600 L AO + DEZ. After exposure to DEZ, the Raman spectrum largely recovers the spectral features of pristine EG. Peak positions assignments are instrumentally limited to ±1 cm-1.
To further confirm the weak chemical interactions between the nanoparticles and graphene, Raman spectra were taken of EG after exposure to AO and to AO and DEZ as shown in Figure 3b. The ratio and positions of the D, G, and 2D peaks can be used to measure the density of defects, covalent functionalization, and charge doping.36 After dosing EG with AO, the ratio of the D/G peaks increases from 0.29 to 0.54, confirming that the oxygen is covalently bound to the surface.26 In addition, the G band shifts from 1612 ± 1 cm-1 to 1605 ± 1 cm-1, which is consistent with AO having a p-type doping effect. However, after dosing with DEZ, the D/G ratio partially recovers to 0.42, and the G band returns to 1611 cm-`1, providing evidence that the oxygen is no longer covalently attached to the surface.
In an effort to elucidate the underlying chemical mechanisms, the thermodynamic driving forces and bond energies associated with early stages of ZnO particle growth were evaluated using DFT calculations.28 The most important results are illustrated graphically in Figure 4, and additional informational can be found in Table SI-1. The calculations indicate that exposure of graphene epoxide (GE) to DEZ leads to two subsequent oxygen abstraction reactions on the surface to form the thermodynamically more stable ethyl-ethoxy zinc (EEoZ) and diethoxy zinc (DEoZ) molecules, respectively. The bond energy analysis (i.e., comparison of bond energies associated with reactants and products) suggests that the above reactions are primarily driven by the conversion of the weakest Zn-C bonds into strong Zn-O and C-O bonds in EEoZ and DEoZ. Further abstraction of oxygen atoms by DEoZ does not occur because the Zn coordination is saturated with two oxygen atoms in DEoZ, and the formation of peroxy-functionality (i.e., ethylperoxy-ethoxy zinc) is thermodynamically unfavorable.
Figure 4.
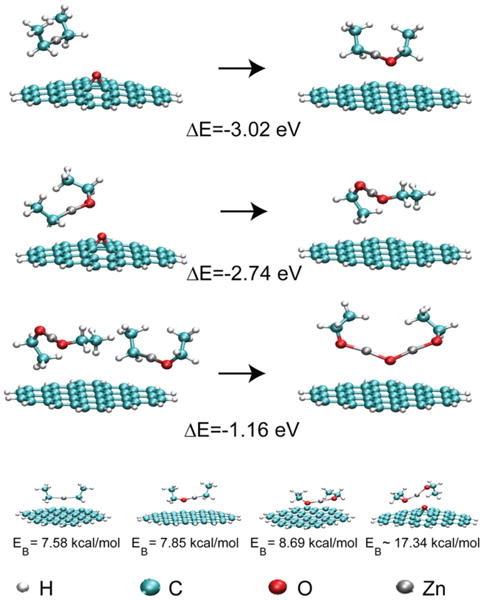
Key reactions and their energetics involved in the initial formation of ZnO on graphene through the epoxide intermediate. The top two reactions illustrate the abstraction of oxygen from GE. The third reaction is the most energetically favorable cluster formation pathway. The final row shows the geometry and binding energy of the organometallic precursors with graphene and GE.
To further elucidate the role of the GE surface in catalyzing ZnO growth, we have examined the potential energy surfaces governing interaction between the precursor/intermediate and the surface. Two key surface sites were identified, namely the pristine basal plane of graphene and the epoxy functionalities. The interactions of DEZ, EEoZ, and DEoZ molecules with the surface sites were calculated with the supramolecular approach (see Supporting Information for details).29,30 The molecules show weak interactions with the basal plane of graphene that increase with the number of oxygen atoms (7.58 kcal/mol for DEZ, 7.75 kcal/mol for EEoZ and 8.69 kcal/mol for DEoZ). On the other hand, at the epoxy site on the GE surface, DEoZ molecules form stronger van der Waals complexes (17.34 kcal/mol) (note that the DEZ and EEoZ molecules undergo oxygen abstraction at the epoxy site). Although subsequent growth of ZnO particles from EEOZ, DEoZ, and other Zn-alkoxides can be inferred from previous studies that utilized ozone or N2O gas as a source of oxygen atoms, the surface catalyzed oxygen abstraction by DEZ (being the differentiating step) requires further attention.37,38 Accordingly, we have additionally characterized the potential energy surface of this reaction (see Supporting Information and Figure SI-3 for more details).
The nonzero activation energy (∼23 kcal/mol) suggests a thermally activated process. However, the lower binding energy of the zinc alkoxides will lead to competition between desorption and oxygen abstraction. As a result, we anticipate that temperature optimization should yield a maximum in the growth rate of ZnO on graphene epoxide substrates.
The above theoretical analysis, in combination with the aforementioned experimental observations, leads to the following proposed mechanism for ZnO nanoparticle growth. First, the exposure of graphene to AO forms graphene epoxide (GE). Then, exposure to DEZ leads to two subsequent oxygen atom abstraction reactions to form DEoZ that docks to epoxy sites on the GE surface. In further AO and DEZ cycles, the epoxy functionalization of graphene and oxygen atom abstraction by DEZ continues, in addition to the growth of ZnO clusters. Additional information on the formation energies of ZnO clusters containing several Zn atoms from different reactants are provided in the Supporting Information.
Chemically, this process resembles recent reports of metal oxides grown via ALD using physisorbed ozone as the oxygen source,32,39,40 where weakly interacting oxygen can be abstracted from the surface by the organometallic precursor. The method presented here, however, has the advantage that the density of the oxygen source can be precisely controlled, allowing for control over the size and density of nanoparticles on the surface.
Conclusions
AO has been shown to be an effective strategy for chemically activating epitaxial graphene, thus enabling the growth of ZnO nanoparticles. Moreover, by using reversible epoxide functionalization, nanoparticle growth is achieved without introducing permanent defects or relying on the use of surfactants or binders. Finally, because this reaction sequence utilizes the oxygen scavenging nature of the transition metal, it should be transferrable to other metal oxide nanoparticles, thereby enabling diverse chemical functionalization of graphene.
Supplementary Material
Acknowledgments
This work was supported by the Department of Energy (Award Number DE-FG02-09ER16109), the Office of Naval Research (Award Number N00014-11-1-0463), and a W. M. Keck Foundation Science and Engineering Grant. J.E.J acknowledges an IIN Postdoctoral Fellowship and the Northwestern University International Institute for Nanotechnology, as well as the National Institutes of Health and National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH/NIAMS T32 AR007611). Public facilities were utilized in the Keck-II Facility of the NUANCE Center at Northwestern University. NUANCE is supported by the NSFNSEC, NSF-MRSEC, Keck Foundation, State of Illinois, and Northwestern University. Work by SP and GCS was supported by the ANSER center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DESC0001059.
Footnotes
Supporting Information Available: Additional experimental details concerning sample preparation, DFT calculations, and UHV STM images can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Avouris P, Chen Z, Perebeinos V. Nat Nano. 2007;2:605. doi: 10.1038/nnano.2007.300. [DOI] [PubMed] [Google Scholar]
- 2.Bunch JS, van der Zande AM, Verbridge SS, Frank IW, Tanenbaum DM, Parpia JM, Craighead HG, McEuen PL. Science. 2007;315:490. doi: 10.1126/science.1136836. [DOI] [PubMed] [Google Scholar]
- 3.Wang D, Choi D, Li J, Yang Z, Nie Z, Kou R, Hu D, Wang C, Saraf LV, Zhang J, Aksay IA, Liu J. ACS Nano. 2009;3:907. doi: 10.1021/nn900150y. [DOI] [PubMed] [Google Scholar]
- 4.Liang Y, Wang H, Casalongue HS, Chen Z, Dai H. Nano Research. 2010;3:701. [Google Scholar]
- 5.Zhang H, Lv X, Li Y, Wang Y, Li J. ACS Nano. 2010;4:380. doi: 10.1021/nn901221k. [DOI] [PubMed] [Google Scholar]
- 6.Liang YT, Vijayan BK, Lyandres O, Gray KA, Hersam MC. J Phys Chem Lett. 2012;3:1760. doi: 10.1021/jz300491s. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Zhi L, Muellen K. Nano Lett. 2008;8:323. doi: 10.1021/nl072838r. [DOI] [PubMed] [Google Scholar]
- 8.Sun X, Xie M, Wang GK, Sun HT, Cavanagh AS, Travis JJ, George SM, Lian J. J Electrochem Soc. 2012;159:A364. [Google Scholar]
- 9.Hummers WS, Offeman RE. J Am Chem Soc. 1958;80:1339. [Google Scholar]
- 10.Dreyer DR, Park S, Bielawski CW, Ruoff RS. Chem Soc Rev. 2010;39:228. doi: 10.1039/b917103g. [DOI] [PubMed] [Google Scholar]
- 11.Gao W, Alemany LB, Ci LJ, Ajayan PM. Nat Chem. 2009;1:403. doi: 10.1038/nchem.281. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi H, Lightcap IV, Tsujimoto M, Takano M, Umeyama T, Kamat PV, Imahori H. J Am Chem Soc. 2011;133:7684. doi: 10.1021/ja201813n. [DOI] [PubMed] [Google Scholar]
- 13.Kamat PV. J Phys Chem Lett. 2009;1:520. [Google Scholar]
- 14.Li B, Cao H. J Mater Chem. 2011;21:3346. [Google Scholar]
- 15.Singh G, Choudhary A, Haranath D, Joshi AG, Singh N, Singh S, Pasricha R. Carbon. 2012;50:385. [Google Scholar]
- 16.Yoo H, Kim Y, Lee J, Lee H, Yoon Y, Kim G, Lee H. Chem Eur J. 2012;18:4923. doi: 10.1002/chem.201103967. [DOI] [PubMed] [Google Scholar]
- 17.Sarkar SB, Durga Chem Phys Lett. 2013;561 [Google Scholar]
- 18.Gómez -Navarro C, Meyer JC, Sundaram RS, Chuvilin A, Kurasch S, Burghard M, Kern K, Kaiser U. Nano Lett. 2010;10:1144. doi: 10.1021/nl9031617. [DOI] [PubMed] [Google Scholar]
- 19.Mkhoyan KA, Contryman AW, Silcox J, Stewart DA, Eda G, Mattevi C, Miller S, Chhowalla M. Nano Lett. 2009;9:1058. doi: 10.1021/nl8034256. [DOI] [PubMed] [Google Scholar]
- 20.Liang YT, Vijayan BK, Gray KA, Hersam MC. Nano Lett. 2011;11:2865. doi: 10.1021/nl2012906. [DOI] [PubMed] [Google Scholar]
- 21.Lotya M, Hernandez Y, King PJ, Smith RJ, Nicolosi V, Karlsson LS, Blighe FM, De S, Wang Z, McGovern IT, Duesberg GS, Coleman JN. J Am Chem Soc. 2009;131:3611. doi: 10.1021/ja807449u. [DOI] [PubMed] [Google Scholar]
- 22.Liang Y, Wu D, Feng X, Müllen K. Adv Mater. 2009;21:1679. [Google Scholar]
- 23.Green AA, Hersam MC. J Phys Chem Lett. 2010;1:544. doi: 10.1021/jz900235f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johns JE, Hersam MC. Acc Chem Res. 2013;46:77. doi: 10.1021/ar300143e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang QH, Hersam MC. Nat Chem. 2009;1:206. doi: 10.1038/nchem.212. [DOI] [PubMed] [Google Scholar]
- 26.Hossain MZ, Johns JE, Bevan KH, Karmel HJ, Liang YT, Yoshimoto S, Mukai K, Koitaya T, Yoshinobu J, Kawai M, Lear AM, Kesmodel LL, Tait SL, Hersam MC. Nat Chem. 2012;4:305. doi: 10.1038/nchem.1269. [DOI] [PubMed] [Google Scholar]
- 27.Vinogradov NA, Schulte K, Ng ML, Mikkelsen A, Lundgren E, Mårtensson N, Preobrajenski AB. J Phys Chem C. 2011;115:9568. [Google Scholar]
- 28.te Velde G, Bickelhaupt FM, Baerends EJ, Guerra CF, Van Gisbergen SJA, Snijders JG, Ziegler T. J Comput Chem. 2001;22:931. [Google Scholar]
- 29.Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215. [Google Scholar]
- 30.Zhao Y, Truhlar DG. J Chem Theory Comput. 2006;2:1009. doi: 10.1021/ct060044j. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Tabakman SM, Dai H. J Am Chem Soc. 2008;130:8152. doi: 10.1021/ja8023059. [DOI] [PubMed] [Google Scholar]
- 32.Jandhyala S, Mordi G, Lee B, Lee G, Floresca C, Cha PR, Ahn J, Wallace RM, Chabal YJ, Kim MJ, Colombo L, Cho K, Kim J. ACS Nano. 2012;6:2722. doi: 10.1021/nn300167t. [DOI] [PubMed] [Google Scholar]
- 33.Alaboson JMP, Wang QH, Emery JD, Lipson AL, Bedzyk MJ, Elam JW, Pellin MJ, Hersam MC. ACS Nano. 2011;5:5223. doi: 10.1021/nn201414d. [DOI] [PubMed] [Google Scholar]
- 34.Lim SJ, Kwon S, Kim H. Thin Solid Films. 2008;516:1523. [Google Scholar]
- 35.Kim K, Regan W, Geng B, Alemán B, Kessler BM, Wang F, Crommie MF, Zettl A. Phys Status Solidi RRL. 2010;4:302. [Google Scholar]
- 36.Ferrari AC, Basko DM. Nat Nano. 2013;8:235. doi: 10.1038/nnano.2013.46. [DOI] [PubMed] [Google Scholar]
- 37.Warner EJ, Cramer CJ, Gladfelter WL. J Vac Sci Technol A. 2013:041504. [Google Scholar]
- 38.Maejima K, Fujita S. In: Phys Status Solidi C. Stutzmann M, editor. Vol. 3. 2006. p. 1022. [Google Scholar]
- 39.Lee B, Mordi G, Kim MJ, Chabal YJ, Vogel EM, Wallace RM, Cho KJ, Colombo L, Kim J. Appl Phys Lett. 2010;97:043107. [Google Scholar]
- 40.Garces NY, Wheeler VD, Gaskill DK. J Vac Sci Technol B. 2012;30 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.