

Abstract
Anti-MHC class I alloantibodies have been implicated in the process of acute and chronic rejection because these Abs can bind to endothelial cells and transduce signals leading to the activation of cell survival and proliferation pathways. To characterize the role of the MHC class I-signaling pathway in the pathogenesis of Ab-mediated rejection, we developed a mouse vascularized heterotopic cardiac allograft model in which B6.RAG1 KO hosts (H-2Kb/Db) received a fully MHC-incompatible BALB/c (H-2Kd/Dd) heart transplant and were passively transfused with anti-donor MHC class I Ab. We demonstrate that cardiac allografts of mice treated with anti-MHC class I Abs show characteristic features of Ab-mediated rejection including microvascular changes accompanied by C4d deposition. Phosphoproteomic analysis of signaling molecules involved in the MHC class I cell proliferation and survival pathways were elevated in anti-class I-treated mice compared with the isotype control-treated group. Pairwise correlations, hierarchical clustering, and multidimensional scaling algorithms were used to dissect the class I-signaling pathway in vivo. Treatment with anti-H-2Kd Ab was highly correlated with the activation of Akt and p70S6Kinase (S6K). When measuring distance as a marker of interrelatedness, multidimensional scaling analysis revealed a close association between members of the mammalian target of rapamycin pathway including mammalian target of rapamycin, S6K, and S6 ribosomal protein. These results provide the first analysis of the interrelationships between these signaling molecules in vivo that reflects our knowledge of the signaling pathway derived from in vitro experiments.
Antibody-mediated (AMR)3 rejection remains a major obstacle to solid organ transplantation. In cardiac transplantation, AMR has been shown to be associated with acute hemodynamic compromise, accelerated coronary allograft vasculopathy (CAV), and decreased graft survival (1, 2). The histologic hallmarks of AMR include microvascular changes, consisting of endothelial cell injury and increased intravascular macrophages, interstitial edema and/or hemorrhage, and neutrophilic infiltration. Immunohistochemistry demonstrates capillary Ig and complement deposition, intravascular CD68-positive macrophages, and fibrin staining in vessels of grafts with AMR (1, 2).
The development of posttransplant Abs to MHC class I Ags are generally regarded as a risk factor for AMR and chronic rejection (2, 3). However, under certain conditions, anti-MHC class I Abs have been implicated in facilitating graft accommodation (4–7). Accommodation is the absence of Ab-mediated injury and continuing functioning of the graft, despite the presence of circulating anti-donor MHC Abs (4, 8). Accommodation is thought to reflect an acquired resistance of the graft to Ab-mediated injury and is associated with increased expression of the survival proteins Bcl-2, Bcl-xL, A20, and HO-1 (5, 6) and resistance to complement (8). The potential detrimental vs beneficial effects of anti-HLA Ab on the state of the graft remain to be elucidated.
Previous studies have demonstrated that Ab ligation and cross-linking of MHC class I molecules in cultured human endothelial cells (EC) transduces signals that both stimulate EC proliferation and activate cell survival pathways that may be involved in promoting rejection and accommodation, respectively (4, 9–13). Ligation of MHC class I molecules on cultured EC induces tyrosine phosphorylation of Src family protein tyrosine kinases, c-Src, Fyn, and the focal adhesion proteins focal adhesion kinase (FAK) and paxillin (14). Class I-mediated activation of FAK triggers a pro-survival signaling cascade, resulting in the activation of the PI3K/Akt-signaling pathway and up-regulation of the antiapoptotic proteins Bcl-2 and Bcl-xL (11, 13, 15, 16). Class I-mediated up-regulation of antiapoptotic proteins renders endothelial cells refractory to activation and resistant to complement-mediated lysis (11). Class I-mediated activation of FAK can also elicit cell proliferation through phosphorylation of ERK and S6 ribosomal protein (S6RP) (14, 17). Analysis of human cardiac transplant biopsies with evidence of AMR exhibited increased Bcl-2 expression and phosphorylation of S6RP at site Ser235/236 on the vascular endothelium, suggesting that class I-mediated activation of survival and proliferation pathways is both tightly linked and operational during AMR (15, 17).
Only a limited number of in vivo models have been described to study the mechanisms underlying AMR. Arguably, the most convincing models have capitalized on the use of animals with a genetic defect in B cell function where the specific effects of Abs could be assessed in the absence of alloreactive T and B lymphocytes (18–22). The aim of our study was to develop an experimental transplant system that would permit us to characterize the specific effects of anti-MHC Ab on signal transduction in endothelial cells in the absence of alloreactive T and B cells. Because intravascular macrophages and complement deposition play an important role in AMR (23), we selected the B6.RAG1 KO animal as a host with its intact innate immune system comprised of macrophages and complement. The mouse is devoid of the adaptive immune system allowing manipulations of the humoral immune response via passive transfer of anti-donor MHC Ab.
We report that passive transfer of anti-donor MHC class I Ab in B6.RAG1 KO recipients of fully MHC-incompatible BALB/c (H-2Kd/Dd) heart transplant mimics characteristics of human AMR as evidenced by microvascular changes with complement deposition in capillaries. In addition, treatment of mice with anti-MHC class I Ab stimulates prominent phosphorylation of Akt Ser473 by day 15 posttransplant and is accompanied by increased expression of the prosurvival protein Bcl-2. Treatment with anti-MHC class I Ab-induced phosphorylation of ERK and the mammalian target of rapamycin (mTOR), S6K, S6RP pathway by day 30. Thus, the pattern of protein phosphorylation suggests that these markers could provide meaningful signatures that can be used to understand the mechanisms underlying AMR.
Materials and Methods
Mice
Male C57BL/6(H-2b) RAG1 knockout (KO) and male BALB/c (H-2d) inbred mice were purchased from The Jackson Laboratory. The mice were housed in the Animal Facilities at University of California (Los Angeles, CA; UCLA) under pathogen-free conditions and used at 8–12 wk of age. The mouse studies described within have been reviewed and approved by the UCLA Animal Research Committee.
Heterotopic heart transplantation
BALB/c (H-2d) hearts were heterotopically transplanted into B6.RAG1 KO (H-2b) recipients as previously described (24). Briefly, under methoxyflurane inhalational anesthesia, the donor aorta and pulmonary artery were anatomized to the recipient abdominal aorta and inferior vena cava, respectively. On day 3 posttransplant, 750 μg of purified anti-H-2Kd Ab, 250 μg of anti-H-2Kd F(ab′)2 Ab or an equivalent dose of isotype control murine IgG (mIgG) were injected i.v. A subset of mice received a second injection of mAb or isotype control IgG 24 h before organ harvest. No statistically significant differences were found by histology or Western blot between animals receiving one or two doses of Ab. Therefore, for the purposes of data analysis and presentation, these two animal groups have been combined. Graft function was monitored by abdominal palpation daily until recipient mice were sacrificed on either day 15 or 30. After removing the allograft, it was analyzed using routine histology, immunohistochemistry, and Western blot techniques. Blood samples were collected after animals were sacrificed to measure circulating levels of anti-H-2Kd Ab on days 15 and 30.
Monoclonal Abs
The hybridoma (HB-159) anti-H-2Kd Ab was purchased from the American Type Culture Collection and the IgG was purified by protein A-agarose affinity chromatography (Sigma-Aldrich). The isotype control mouse IgG2a mAb was purchased from Jackson ImmunoResearch Laboratories. The F(ab′)2 of the anti-H-2Kd mAb or isotype control IgG was prepared as previously described (25).
Protein isolation and Western blot
Mouse tissue was homogenized in tissue extraction II lysis buffer (Invitrogen Life Technologies). Total protein concentration was measured by the BCA protein assay (Pierce). Proteins in the cell lysate were fractionated by SDS-PAGE. Proteins were transferred to polyvinylidene difluoride membrane. The membrane was blocked using 5% milk in TBST and then probed with primary rabbit anti-mouse Abs against signaling molecules involved in the MHC class I-signaling pathway, Akt Ser473, S6RP Ser235/236, S6K Thr421/Ser424, ERK Thr202/Tyr204, and mTOR Ser2448 were purchased from Cell Signaling Technology. Bcl-2 was purchased from Santa Cruz Biotechnology. Equal loading was performed using an Ab against the housekeeping protein vinculin (Sigma-Aldrich). The phosphorylated bands were scanned with the GS-710 Calibrated Imaging Densitometer and quantified by the Quantity One software program from Bio-Rad.
Circulating anti-H-2Kd Ab testing
Sera were collected from B6.RAG1 KO transplant recipients treated with 750 μg of anti-H-2Kd or mIgG Ab on day 15 or 30 and tested for reactivity with BALB/c splenocytes by flow cytometry cross-match. Briefly, 0.5 × 106 BALB/c donor splenocytes were incubated with various dilutions of recipient sera for 30 min at 4°C. The cells were washed twice with PBS containing 1% BSA and 0.01% azide and were incubated with FITC-conjugated goat anti-mouse Ig Ab (Jackson ImmunoResearch Laboratories) and PE-conjugated hamster anti-mouse CD3 Ab (eBioscience) for 30 min at 4°C. Cells were washed three times and anti-H-2Kd bound to T cells was detected on a FACSCalibur flow cytometer and analyzed using CellQuest software (BD Biosciences).
Histology and immunohistochemistry
The cardiac allografts were preserved for both paraffin-embedded and frozen sections. Immediately after the recipients were sacrificed, the samples for paraffin sections were preserved by fixation in 10% formalin. The samples for frozen section were preserved in 2-ME on dry ice and frozen in OCT before cryostat sectioning. The frozen sections (10 μm) were fixed in acetone and incubated with 0.5% H2O2 to block endogenous peroxidase activity. To analyze morphological features, sections were stained with H&E as well as immunohistochemistry stains. For immunohistochemistry, first, normal heat-inactivated goat serum was used for blocking. Appropriate primary rabbit mAb against mouse S6RP Ser235/236, S6K Thr421/Ser424, S6RP, S6K (Cell Signaling Technology) and Bcl-2 (Santa Cruz Biotechnology) were used. Macrophage staining was performed using rat anti-mouse Mac-2 Ab from Acris. Bound primary Ab was detected using biotinylated goat anti-mouse IgG and streptavidin peroxidase-conjugated complexes obtained from Vector Laboratories. The control sections were performed by replacing the primary antisera with normal rabbit serum. The peroxidase reaction was developed with 3,3′ diaminobenzidine tetrahydrochloride (Sigma-Aldrich). EC were double stained using rabbit mAb against mouse S6RP Ser235/236, S6K Thr421/Ser424 (Cell Signaling Technology), and Bcl-2 (Santa Cruz Biotechnology). EC were stained using rat anti-mouse CD34 (Santa Cruz Biotechnology). Double immunostaining of Bcl-2 and CD34 was performed using a HRP avidin tag for CD34 and developed using a DAB kit with nickel (Vector Laboratories). Bcl-2 was visualized using a VECTASTAIN ABC-AP kit (Vector Laboratories). Additionally, double staining of S6K Thr421/Ser424 or S6RP Ser235/236 with CD34 was performed with a HRP avidin tag for S6K Thr421/Ser424 or S6RP Ser235/236 (Vector Laboratories) and developed using a DAB kit (Vector Laboratories). CD34 was visualized using a VECTASTAIN ABC-AP kit (Vector Laboratories).
To evaluate the histologic features of the allografts, all slides were interpreted and scored by the same pathologist who was blinded to the experimental group. Microvascular changes, consisting of increased prominence and number of intravascular cells, and expansion of capillaries were graded 0–3 (0, negative; 1, focal; 2, multifocal; 3, diffuse) as previously described (26). It has been shown that these microvascular changes consist of endothelial cell injury as well as an increased number of intravascular macrophages, with edema. A score of 2 or higher was considered positive for microvascular changes. The presence of MAC2-positive intravascular macrophages was scored on a scale of 0–3 (0, no staining; 1, focal staining; 2, multifocal staining; 3, diffuse staining). A score of 1 or higher was considered positive. Positive EC staining in capillaries and intramyocardial arteries and veins for S6K Thr421/Ser424, S6RP Ser235/236, and Bcl-2 was scored on a scale of 0–3 (0, no staining; 1, focal staining; 2, multifocal staining; 3, diffuse staining). A score of 1 or higher was considered positive. Positive leukocyte staining for S6K Thr421/Ser424, S6RP Ser235/236, and Bcl-2 was scored on a scale of 0–3 (0, no staining; 1, focal staining; 2, multifocal staining; 3, diffuse staining). A score of 1 or higher was considered positive.
C4d staining
C4d was detected using affinity-purified polyclonal rabbit Abs to a 14-mer peptide that is specific to the C4d fragment of mouse C4 (27). The mouse C4d peptide is analogous to the peptides used to produce polyclonal rabbit Abs to rat and human C4d (28, 29). These Abs bind to both C4b and C4d. C4b is the first split product of C4 that binds to cell membranes and factor I cleaves C4b to the smaller fragment C4d. The cleavage of C4b to produce C4d is a regulation step, and, unless factor I is blocked, all C4b is rapidly converted to C4d. Staining with this affinity-purified polyclonal Ab to C4d has been shown to correlate absolutely with activation of complement by Abs (27). Capillary C4d staining was scored on a scale of 0–2 (0, no staining; ±, rare staining; 1, focal staining; 2, diffuse staining). A score of 1 or higher was considered positive.
Statistical analysis
Unless otherwise noted, values in text are shown as mean ± SE or counts (percents). Differences in histological features of AMR and protein signal transduction between H-2Kd Ab-treated and control mice were tested using Fisher’s exact test, and differences in phosphorylation levels of individual proteins (measured by their relative percents of maximum values) were tested using the Student t test (regarding the latter, visual examination of box plots supported normality assumptions, and log-transforming values did not alter results (data not shown)). To assess multiple phosphorylation differences simultaneously, Hoteling’s T2 generalized means test was used and, to gauge covariate effects, multivariate ANOVA was used. Pearson correlations, multidimensional scaling (via dissimilarities), and hierarchical cluster tree (complete linkage) analyses were performed to examine relationships, reduce dimensions, and visualize patterns among the protein expression levels. All statistical analyses were preformed using STATA software (Stata Statistical Software, Release 9; StataCorp). All p values were two-sided and, when comparing groups, a significance level of 0.05 was used.
Results
Effects of donor-specific anti-H-2Kd Ab on cardiac allograft rejection
To determine whether treatment with anti-donor MHC class I Ab induces histologic changes in the allograft consistent with AMR, B6.RAG1 KO recipients were transplanted with H-2Kd-incompatible BALB/c hearts, treated with 750 μg of anti-donor anti-H-2Kd Ab (n = 19), anti-mIgG isotype control (n = 13) or no Ab (n = 6) on posttransplant day 3 and grafts were analyzed on day 15 or 30 following transplant. Circulating levels of anti-donor H-2Kd Ab were measured by two-color flow cytometric cross-match using donor splenocytes as target cells. High titers (>1:64) of anti-H-2Kd Ab were found in the circulation of recipient mice throughout the 30-day period of treatment, whereas, treatment with isotype control IgG displayed no Ab binding to target cells (Fig. 1).
FIGURE 1.

Time course and titer of donor-specific anti-H-2Kd Ab levels detected by flow cytometry cross-matching. Circulating anti-H-2Kd mAb was measured by flow cytometry in B6.RAG1 KO recipients on (A) day 15 (n = 3) or (B) day 30 (n = 3) following BALB/c cardiac transplantation. No alloantibody responses were detected in mice receiving isotype control IgG Ab. Each data point represents the average median channel of fluorescence ±SE.
Passive transfer of anti-H-2Kd IgG2a mAb did not cause B6.RAG1 KO recipients to reject their hearts within 30 days (100% survival). However, characteristic features of human AMR, including microvascular changes with intravascular macrophage accumulation (2, 26), were observed in the majority of cardiac allografts harvested on day 30. Histology showed a significant increase in microvascular changes (score ≥2) in 8 of 11 BALB/c cardiac allografts from mice treated with anti-H-2Kd Ab (Fig. 2A) in comparison to 3 of 10 mIgG isotype controls that displayed normal capillary morphology (Fig. 2B). Immunohistochemistry confirmed that there was a significant accumulation of intravascular Mac2-positive macrophages in the myocardial capillaries from animals treated with anti-H-2Kd Ab compared with isotype control mIgG Ab. Six of 12 recipients treated with donor-specific anti-MHC Ab showed Mac2-positive infiltrates (Fig. 2C) compared with only 1 of 10 isotype control-treated recipients (Fig. 2D). Because the specific isotype (IgG2a) of the anti-H-2Kd Ab used in these experiments activates complement, we tested for the presence of C4d deposition. Strong C4d staining was observed in the capillary endothelium in 17 of 19 anti-H-2Kd recipients (Fig. 2E). In contrast, no C4d deposition was observed in cardiac allografts from mIgG isotype control Ab-treated mice (Fig. 2F).
FIGURE 2.

Treatment with anti-donor H-2Kd Ab induces histological features of AMR. BALB/c cardiac allografts were transplanted into B6.RAG1 KO recipients and passively transfused with anti-H-2Kd Ab or mIgG Ab and grafts were examined on days 15 and 30. Microvascular abnormalities were observed by H&E staining in recipients treated with anti-H-2Kd Ab. Arrows indicate prominent nuclei of cells in distended capillaries (A) not seen in isotype control mIgG Ab where arrows indicate flat and thin nuclei in collapsed capillaries (B) on day 30. Intravascular macrophages were assessed by MAC2 immunoperoxidase staining in recipients treated with anti-H-2Kd Ab (C) or isotype control mIgG Ab (D) on day 30. Complement activation was measured by C4d deposition in recipients treated with anti-H-2Kd Ab (E), isotype control mIgG Ab (F) or anti-H-2Kd F(ab′)2 Ab (G). Grafts represent C4d staining on day 30.
We next explored the duration of exposure to anti-donor class I Ab in relation to pathologic patterns of AMR. BALB/c heart allografts transplanted into B6.RAG1 KO recipients received passive transfer of 750 μg of anti-H-2Kd Ab on posttransplant day 3 and grafts were examined on day 15. Grafts from animals treated with anti-donor H-2Kd mAb displayed microvascular changes (n = 6/7, p = N/S) with numerous intravascular macrophages confirmed by immunohistochemistry staining (n = 6/7, p = N/S). Grafts examined for C4d deposition on day 15 all showed strong capillary staining (n = 7/7, p < 0.001).
To test whether the capacity of anti-donor class I Ab to induce histopathologic changes in the cardiac allograft requires complement, we designed studies to compare the effect of the intact IgG Ab to the F(ab′)2. No C4d was present on the endothelium in anti-H-2Kd F(ab′)2-treated mice (n = 4) (Fig. 2G). Furthermore, grafts from animals treated with the F(ab′)2 showed microvascular changes (four of four) with positive staining for Mac2-positive cells (four of four). Taken together, these results indicated that passive transfer of anti-donor MHC Abs, in the absence of alloreactive T and B cells and complement, was sufficient to induce pathologic changes in the allograft consistent with AMR.
Passively transferred anti-H-2Kd Ab stimulates phosphorylation of intracellular proteins involved in cell survival and cell proliferation
Ligation of MHC class I molecules on the surface of cultured EC by anti-HLA class I Ab stimulates an intracellular signaling cascade resulting in cell proliferation and survival, and has been implicated in the process of acute and chronic AMR (11, 13, 15, 17).
To determine the effect of Ab to MHC class I on cells of the allograft, B6.RAG1 KO recipients of BALB/c cardiac allografts were passively transfused with anti-H2-Kd IgG and studied by Western blot for expression of phosphorylated ERK Thr202/Tyr204, S6K Thr421/Ser424, Akt Ser473, S6RP Ser235/236, and mTOR Ser2448. Generally, treatment with anti-donor H-2Kd IgG stimulated increases in protein phosphorylation in the anti-H-2Kd Ab-treated group (Fig. 3, p = 0.002, Hoteling’s T2 test) compared with isotype control-treated mice. Analysis of the phosphorylation status of each individual protein showed significant increases in phosphorylation of ERK Thr202/Tyr204 (Fig. 3A, p = 0.004), Akt Ser473 (Fig. 3C, p = 0.004), and mTOR Ser2448 (Fig. 3E, p = 0.05) indicating that treatment with anti-MHC class I Ab stimulates both survival and proliferation pathways.
FIGURE 3.

Analysis of phospho-proteins in the MHC class I-signaling pathway in B6.RAG1 KO recipients of BALB/c cardiac allografts following passive transfer of anti-H-2Kd Ab. B6.RAG1 KO mice (n = 32) received fully MHC mismatched cardiac transplants from BALB/c donors. Three days after transplantation, mice were injected with either the anti-H-2Kd IgG Ab (n = 19) or an isotype control IgG Ab (n = 13). Mice were sacrificed on day 15 (n = 13) or 30 (n = 19) and the heart allografts were studied by Western blot for expression of phosphorylated ERK Thr202/Tyr204 (A), S6K Thr421/Ser424 (B), Akt Ser473 (C), S6RP Ser235/236 (D), and mTOR Ser2448 (E). Densitometry results are expressed as the percent maximal increase in phosphorylation divided by total protein (mean ± SE).
We next analyzed the relationship between the duration of treatment with anti-class I Ab and protein phosphorylation status in the allograft. Grafts harvested from anti-H-2Kd Ab-treated mice on day 30 exhibited a significant increase in protein phosphorylation (p = 0.04). Although grafts examined on day 15 showed increased protein phosphorylation, this finding did not reach statistical significance (p = 0.09). Examination of the phosphorylation status of each individual protein showed a prominent increase in the phosphorylation of Akt Ser473 in grafts harvested on days 15 and 30. Animals treated with anti-MHC class I Ab and examined on day 30 also showed a significant increase in the phosphorylation of ERK Thr202/Tyr204. Interestingly, phosphorylation of S6K Thr421/Ser424 and S6RP Ser235/236 were lower in cardiac allografts from B6.RAG1 KO animals harvested at day 15 compared with grafts harvested on day 30. These preliminary results suggest that class I-mediated activation of the Akt pathway occurred within 2 wk of administration of anti-donor MHC Ab in this setting, and remained at high levels through day 30. In contrast, activation of S6K Thr421/Ser424 and S6RP Ser235/236, involved in cell proliferation, appear to require a longer duration of exposure to anti-class I Ab (30 days).
Anti-H-2Kd Ab-induced survival and proliferation signal transduction does not require complement
To determine whether the capacity of anti-H-2Kd Ab to stimulate protein phosphorylation required complement, we compared the effect of the intact IgG Ab to a F(ab′)2. BALB/c cardiac allografts were transplanted into B6.RAG1 KO recipients and passively transfused with either the whole IgG (n = 3) or the F(ab′)2 portion (n = 4) of the anti-H-2Kd class I Ab. After 30 days, BALB/c cardiac allograft tissues were analyzed by Western blot for the phosphorylation of Akt Ser473, ERK Thr202/Tyr204, S6K Thr421/Ser424, S6RP Ser235/236, and mTOR Ser2448. Similar to the intact IgG molecule, treatment with the F(ab′)2 of the anti-donor class I Ab stimulated concomitant phosphorylation of proteins involved in class I-mediated cell survival and proliferation (Fig. 4, A–E, p = 0.0003). There was no significant difference in the level of phosphorylation of proteins in the MHC class I-signaling pathway between mice injected with the intact anti-H-2Kd IgG or anti-H-2Kd F(ab′)2.
FIGURE 4.

Anti-MHC class I-induced protein phosphorylation and Bcl-2 expression in the absence of complement. Lysates from of BALB/c cardiac allografts treated with anti-H-2Kd F(ab′)2 or intact anti-H-2Kd IgG were analyzed for phosphorylation of ERK Thr202/Tyr204 (A), S6K Thr421/Ser424 (B), Akt Ser473 (C), S6RP Ser235/236 (D), and mTOR Ser2448 (E) and Bcl-2 protein expression (F).
PI3K/Akt kinase activity promotes cell survival by regulating levels of the antiapoptotic family members Bcl-2 and Bcl-xL (11, 13, 15). Because we observed a positive association between treatment with anti-donor H-2Kd Ab and increased Akt Ser473 phosphorylation, we examined the possibility that exposure of graft endothelium to anti-donor class I Ab stimulates Bcl-2 expression. The results presented in Fig. 4F confirm the involvement of Bcl-2 as a downstream target of the Akt pathway and show that exposure to either the intact IgG or F(ab′)2 of the anti-class I Ab induces Bcl-2 up-regulation in the graft. These studies are consistent with the effect of anti-class I Ab on Akt Ser473 phosphorylation in EC. Furthermore, these experiments rule out the possibility that phosphorylation was due to signaling by FcRs or complement.
Passively transferred anti-H-2Kd Ab stimulates signal transduction in the endothelium of BALB/c donor cardiac allografts
The results of the Western blot studies indicated that passive transfer of anti-MHC class I Ab stimulates activation of cell survival and proliferation pathways in BALB/c cardiac allografts; however, this finding does not pinpoint the graft cells involved in the signaling process. We therefore examined protein expression by immunohistochemistry to assess the activation of the class I-mediated cell survival pathway in capillary and intramyocardial artery and vein endothelium of the graft. Fig. 5 shows the staining pattern of representative cardiac transplant samples. To confirm EC localization of proteins involved in the MHC class I-signaling pathway, cardiac allografts were double stained with an endothelial cell marker CD34. Ab treatment was significantly correlated with the expression of Bcl-2 on days 15 and 30 (Fig. 5A). Capillary EC stained positive for the expression of Bcl-2 in 18 of 19 (95%) mice treated with anti-H-2Kd Ab (Fig. 5B, p < 0.001). Likewise, intramyocardial artery and vein endothelium stained positive for the expression of Bcl-2 in 18 of 19 (95%) mice treated with anti-H-2Kd Ab. In contrast, 2 of 18 cardiac allografts from either IgG isotype control or no Ab-treated mice showed evidence (11%) of Bcl-2 staining in capillary EC (Fig. 5C) and only 1 of 18 control allografts was positive for Bcl-2 in intramyocardial artery and vein endothelium (6%). Furthermore, 3 of 4 (75%) mice treated with F(ab′)2 of the anti-H-2Kd Ab displayed positive Bcl-2 staining whereas none of the F(ab′)2 isotype control-treated mice displayed increased Bcl-2 staining. MHC class I-induced Bcl-2 staining was limited to EC of the graft and not observed in other cell types. These results are consistent with the Western blot studies and support the role of anti-H-2Kd Ab-activating antiapoptotic cell survival machinery in the EC of the graft.
FIGURE 5.
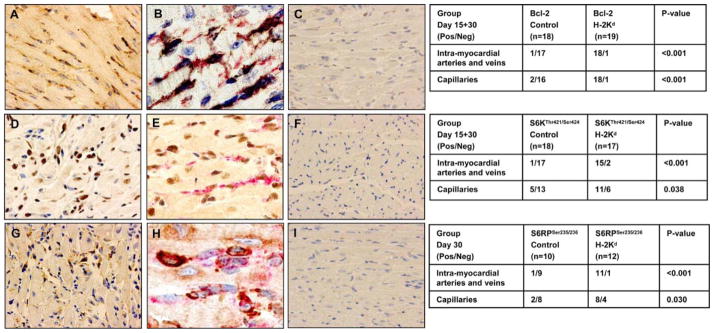
Representative histology of phosphorylated cell survival and proliferation proteins in cardiac allografts following passive transfer of anti-H-2Kd Ab. Immunohistochemical analysis of proteins involved in MHC class I-induced cell survival or proliferation pathway. Bcl-2 expression in the vascular endothelium of BALB/c cardiac allografts treated with anti-H-2Kd Ab (A) vs control mIgG Ab (C). To localize staining of Bcl-2 in capillary endothelium of cardiac allografts treated with H-2Kd, sections were double stained for Bcl-2 (red) and anti-CD34 (black) (B). Phosphoproteomic analysis was determined by immunoperoxidase single staining for S6K Thr421/Ser424 (D) and S6RP Ser235/236 (G) in allografts treated with anti-H-2Kd Ab or mIgG isotype controls (F and I). Sections of cardiac allografts treated with H-2Kd were double stained for S6K Thr421/Ser424 (brown) and anti-CD34 (red) (E) or S6RP Ser235/236 (brown) and anti-CD34 (red) (H).
The next experiment analyzed whether treatment with anti-H-2Kd Ab stimulated phosphorylation of S6K Thr421/Ser424 and S6RP Ser235/236 in capillary and intramyocardial artery and vein endothelium of cardiac allografts. In both mice treated with anti-H-2Kd and controls, we observed positive staining of S6K and S6RP total protein in all cell types including EC, leukocytes, and myocytes in the allograft. Ab treatment was significantly correlated with the phosphorylation of S6K at Thr421/Ser424 on days 15 and 30 (Fig. 5D). Capillary EC stained positive for the phosphorylation of S6K Thr421/Ser424 in 11 of 17 (65%) mice treated with anti-H-2Kd Ab (Fig. 5E, p = 0.038). Positive S6K Thr421/Ser424 phosphorylation was also observed in 15 of 17 (88%) intramyocardial artery and vein endothelium of anti-H-2Kd Ab-treated mice (p < 0.001). In contrast, only 3 of 17 (18%) cardiac allografts treated with anti-H-2Kd Ab stained positive for S6K Thr421/Ser424 in leukocytes. Ab treatment was also significantly associated with staining for phosphorylated S6RP at Ser235/236 on day 30 (Fig. 5G). Positive capillary EC staining for the phosphorylation of S6RP Ser235/236 was observed in 8 of 12 (67%) cardiac allografts treated with anti-H-2Kd Ab (Fig. 5H, p = 0.03). Intramyocardial artery and vein endothelium stained positive for S6RP Ser235/236 in 11 of 12 (92%) anti-H-2Kd Ab-treated mice (p < 0.001). S6RP Ser235/236 also stained positive in 3 of 17 (18%) cardiac allografts for leukocytes. No significant association was observed between treatment with control isotype mIgG or no Ab and activation of the S6K Thr421/Ser424 or S6RP Ser235/236 (Fig. 5, F and I). These results indicate that treatment with anti-donor H-2Kd Ab activates both survival and cell proliferation pathways in the endothelium of the cardiac allograft. The lack of reliable Abs with acceptable performance characteristics in fixed and frozen tissues prevented us from studying protein phosphorylation of Akt Ser473, mTOR Ser2448, and ERK Thr202/Tyr204 in the endothelium of the graft by immunohistochemistry.
Pairwise correlations between proteins in the MHC class I-signaling pathway
We performed pairwise correlations between phosphorylation levels of ERK, S6K, Akt, S6RP, and mTOR to identify coordinately phosphorylated proteins in anti-H-2Kd-treated mice. Two protein cophosphorylation patterns were detected in anti-H-2Kd-treated animals. The strongest pairwise correlation was observed between S6K Thr421/Ser424 and Akt Ser473 (p = 0.004) (Table I). Treatment with anti-H-2Kd Ab was also highly correlated with cophosphorylation among proteins involved in the mTOR-signaling pathway including mTOR Ser2448, S6K Thr421/Ser424, and S6RP Ser235/236. No correlation between phosphorylation status of Akt Ser473 and ERK Thr202/Tyr204 was observed. These data identify strong interrelationships between the S6K/Akt and mTOR/S6K/S6RP pathway and suggest that these proteins may be coordinately phosphorylated during AMR.
Table I.
Pairwise correlations between phosphorylated proteins involved in cell proliferation and survivala
| p-ERK | p-S6K | p-Akt | p-S6RP | p-mTOR | |
|---|---|---|---|---|---|
| p-ERK | |||||
| p-S6K | 0.1263 0.6538 |
||||
| p-Akt | −0.0145 0.9590 |
0.7016 0.0036 |
|||
| p-S6RP | −0.1628 0.5621 |
0.4612 0.0836 |
0.1698 0.5452 |
||
| p-mTOR | 0.2380 0.3930 |
0.0315 0.9114 |
−0.2131 0.4458 |
0.5849 0.0220 |
|
| (n = 15) | |||||
Pairwise correlations of protein cophosphorylation patterns in B6.RAG1 KO mice treated with anti-H-2Kd. The upper number represents the correlation coefficient and the lower number represents the p value; bold values, p < 0.05.
Multidimensional scaling (MDS) analysis of protein interactions following MHC class I stimulation
To investigate higher order relationships among the five phosphorylated proteins, we performed MDS analysis. Using this approach, we plotted the phosphorylation results for members of the MHC class I-signaling pathway (ERK Thr202/Tyr204, S6K Thr421/Ser424, Akt Ser473, S6RP Ser235/236, and mTOR Ser2448) in two dimensions and the distance between each protein represents a measure of their interrelatedness (30). As shown in Fig. 6, phosphorylated Akt Ser473 and S6K Thr421/Ser424 were closely related, as detected by the short distance between them. Furthermore, mTOR Ser2448 and S6RP Ser235/236 appear to be closely related. In contrast, phosphorylation of ERK Thr202/Tyr204 is distant from Akt Ser473, S6K Thr421/Ser424, S6RP Ser235/236, and mTOR Ser2448 implying the activation of this protein occurs independently from the others.
FIGURE 6.
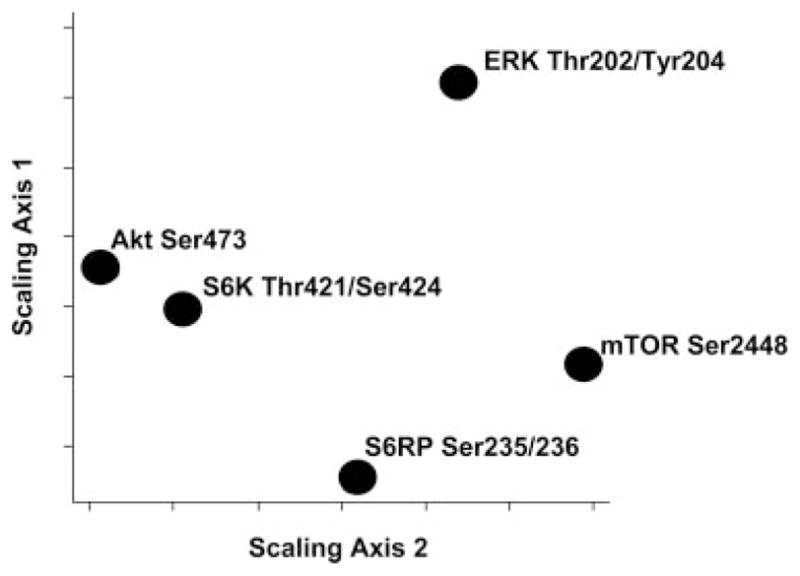
Demonstration of interrelationships between proteins involved in the MHC class I-signaling pathway in vivo. MDS analysis of protein phosphorylation data obtained by Western blot shows that S6K and Akt are closely linked and members of the mTOR pathway: mTOR, S6K, S6RP, are linked to Akt and related to each other. The great distance separating ERK from members of the mTOR pathway suggests multiple signaling inputs are required for ERK activation.
Heterogeneity in the class I signal transduction pathway revealed by hierarchical clustering
To visualize patterns among the proteins that become phosphorylated following class I ligation, we performed hierarchical clustering of the cardiac allografts based on the phosphorylation levels (scale of 0.2–1.0) of ERK Thr202/Tyr204, S6K Thr421/Ser424, Akt Ser473, S6RP Ser235/236, and mTOR Ser2448. Two distinct subsets of mice were identified based on anti-H-2Kd Ab treatment vs treatment with isotype control IgG (Fig. 7, main branches of the dendrogram) (p < 0.001). Furthermore, mice treated with anti-H-2Kd Ab and examined at day 15 clustered together and this hierarchical group was defined by high ERK Thr202/Tyr204 and Akt Ser473 phosphorylation (Fig. 7, top panel, red/orange squares; bottom panel, red squares) and low protein phosphorylation of S6K Thr421/Ser424 and S6RP Ser235/236 (Fig. 7, top panel, green/blue squares; lower panel, black squares). A third hierarchical cluster group was principally comprised of day 30 mice and the phosphorylated proteins that defined this group were primarily involved in cell proliferation including ERK Thr202/Tyr204, mTOR Ser2448, S6K Thr421/Ser424, and S6RP Ser235/236 (Fig. 7, top panel, red/orange squares; bottom panel, red squares). To determine whether the patterns of protein expression correlated with the histologic features of AMR, we compared the phosphorylation levels of the five proteins to the grade of microvascular changes. Interestingly, most of the anti-H-2Kd-treated grafts displaying microvascular changes with positive staining for macrophages showed concomitant activation and phosphorylation of S6K Thr421/Ser424 and S6RP Ser235/236.
FIGURE 7.

Hierarchical clustering of BALB/c cardiac allografts based on phosphoproteomic patterns. Two main branches emerge from this analysis based on treatment with anti-H-2Kd Ab or control mIgG Ab. Top panel, The degree of phosphorylated protein on a scale of low to high (0–1 or blue-red). Bottom panel uses the mean cutoff values of each phosphorylated protein presented in Fig. 3 to determine the pattern of phosphorylated survival and cell proliferation proteins. Red squares indicate phosphorylated protein levels above the mean cutoff value. Black squares indicate phosphorylated protein levels below the mean cutoff.
Discussion
We developed a murine heart allograft model to study the effect of anti-MHC class I Ab-mediated signal transduction on the development of AMR. Using B6.RAG1 KO mice lacking T and B cells, we demonstrated that passive transfer of anti-donor HLA class I Ab can induce pathological changes in the graft consistent with the AMR phenotype. Furthermore, we showed that treatment of mice with anti-class I Abs stimulates phosphorylation of key signaling proteins involved in cell survival and proliferation in the capillary endothelial cells of the graft. These data indicate that the signaling relationships we previously observed in vitro when primary cultured EC were stimulated with anti-class I Ab (11, 13, 15, 17) are also operational in vivo after passive administration of anti-donor MHC class I Ab.
Passive transfer of anti-class I mAb-mediated microvascular changes of AMR. Although characteristic features of human AMR were present, none of the grafts were lost to acute rejection. These results differ from previous reports in which passive transfer of complement binding alloantibodies into B cell-deficient, T cell competent mice resulted in irreversible acute rejection of cardiac allografts (19, 31) and suggest that acute Ab-mediated rejection requires a concomitant T cell immune response. Alternatively, the relative weakness of the mouse complement system may also limit the functional effect of the Ab (32). We also did not observe development of CAV in mice treated with anti-donor H-2Kd class I Ab. This observation maybe explained by the short duration of Ab stimulation (i.e., 30 days). Extending the period of transplant following anti-MHC class I treatment to 60–90 days may highlight more definitive signs of CAV. Also, we did not harvest the most proximal coronary arteries that might be more likely to develop allograft vasculopathy. As shown in a recent report by Uehara et al. (33), passive transfer of anti-MHC class I Ab H-2Kk at biweekly intervals for a period of 14–28 days lead to progressive chronic transplant arteriopathy in the proximal coronary arteries in RAG1KO recipients of B10.BR hearts. In addition, these studies differed from our experiments in that the mice were exposed to frequent i.p. injections of 30 μg of anti-MHC class I Ab twice weekly throughout the duration of the experiment, whereas our treatment regimen consisted of a single high dose i.v. injection. We intentionally used a single high dose of mAb (750 μg) because in preliminary studies, this protocol showed persistent circulating Ab titers >1:64 at day 30. It is possible, however, that chronic stimulation of EC by anti-class I Ab is required to promote development of CAV. The capacity to elicit signal transduction events is dependent upon the fine specificity of the Ab and the degree of MHC cross-linking (16, 34). Therefore, a second explanation for the lack of evidence of CAV following treatment with anti-H-2Kd mAb may be due to differences in the capacity of anti-H-2Kd vs -H-2Kk Abs to aggregate corresponding MHC molecules and transduce survival or proliferation signals. Further studies are required to determine the effect of Ab specificity, dose, and threshold of Ab exposure on development and location of graft injury in CAV.
Of interest, the passively transferred anti-H-2Kd IgG2a mAb showed an unusually long half-life, and appeared at similar levels on days 15 and 30 posttransplant. Recent studies show that expression of the neonatal FcR (FcRn) functions to protect IgG from degradation, increasing the half-life of this class of Ab in the serum (35–37). Vascular EC regulate the levels of IgG by salvaging and exocytosis of the Abs that penetrate the cell surface from intra- and extravascular compartments (38). B6.RAG1 KO mouse pups express a higher level of FcRn when fed by wild-type mothers compared with B6.RAG1 KO mothers at day 15 during development (39). This raises the possibility that in our model, passive transfer of anti-H-2Kd Ab in the B6.RAG1 KO mouse increases FcRn expression and extends the half-life of the circulating Ab. Anti-class I Ab treatment led to coordinately phosphorylated proteins engaged in cell proliferation including mTOR Ser2448, S6K Thr421/Ser424, and S6RP Ser235/236.
mTOR is a central regulator of cell growth and nutrient signaling (40). mTOR controls cell proliferation through formation of mTOR complex 1 (mTORC1) requiring two associated proteins, regulatory associated protein of mTOR (Raptor) and GβL. Raptor acts as a mTOR scaffold protein binding to the TOS motif of mTOR substrates; whereas, GβL is required to create a functional signaling complex (41). mTORC1 regulates the phosphorylation of S6K and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) (42). S6K then activates its downstream target S6RP (43). mTORC1 activation to downstream targets leads to increased ribosomal biosynthesis and translation of critical mRNAs of proteins required for G (1) to S phase transition (44). In murine studies, inhibition of the mTOR kinase domain leads to impaired cell proliferation (45). mTOR knockout mice display an aberrant developmental phenotype including decreased cell size and arrested cell proliferation (46). Similar to mTORnull mice, Raptornull blastocysts exhibit defective inner cell mass and trophoblast cell outgrowth (47). Furthermore, cardiac xenografts undergoing acute vascular rejection showed increased p70S6K activity (48). These data highlight the importance of mTOR in regulating cell proliferation and suggest that drugs such as rapamycin that block mTORC1 may prevent endothelial cell proliferation and neoangiogenesis by inhibiting mTOR and its downstream targets S6K and S6RP. Indeed, clinical trials in transplantation have shown that rapamycin can protect cardiac transplant recipients from transplant vasculopathy (49, 50).
Bcl-2 is an antiapoptotic protein and a marker of accommodation in allografts and xenografts (5, 6, 10, 13, 15). The present study confirms and extends the evidence that anti-MHC class I Abs can stimulate phosphorylation of Akt at Ser473 and expression of Bcl-2 that have the potential to promote accommodation. However, additional studies are required to assess whether activation of Akt and up-regulated Bcl-2 can decrease susceptibility to allograft damage in our model. We propose a model whereby Ab-mediated clustering of MHC class I molecules stimulates the phosphorylation of Src and complex formation between Src and FAK and FAK phosphorylation. FAK activation facilitates the phosphorylation of many associated proteins including PI3K, Akt, and paxillin (51, 52). PI3K and Akt kinase activity promote cell survival by regulating levels of the antiapoptotic proteins Bcl-2 and Bcl-xL (15). In accordance with these findings, ligation of HLA class I molecules has been shown to trigger a prosurvival signaling cascade resulting in phosphorylation of PI3K and Akt and up-regulation of the antiapoptotic proteins Bcl-2 and Bcl-xL in EC (11, 15). Biopsies from heart allograft recipients with evidence of Ab-mediated rejection also displayed increased Bcl-2 expression on the vascular endothelium of the graft (15, 17). These results are consistent with previous studies showing class I-induced activation of Akt and up-regulation of Bcl-2 confer resistance to Ab-complement-mediated cell death (4, 11, 13). Anti-class I Ab treatment led to cophosphorylation of Akt Ser473 and S6K Thr421/Ser424 and this close relationship was confirmed by MDS analysis. S6K has been identified as an mTOR-phosphorylating kinase at site Ser2448 (53). Formation of mTOR complex 2 containing adaptor proteins rapamycin-insensitive companion of mTOR, GβL, and stress-activated protein kinase-interacting protein 1 phosphorylates Akt at site Ser473 (54, 55). In vascular EC, mTOR regulates the activation and phosphorylation of Akt at Ser473 providing further evidence linking S6K and Akt (56).
Hierarchical clustering revealed two major subsets within the anti-H-2Kd treatment group based on patterns of phosphorylated proteins. Grafts treated with anti-H-2Kd Ab and harvested on day 15 comprised one cluster and a second cluster comprised the day 30 mice. The day 15 subset of mice were defined primarily by high ERK Thr202/Tyr204 and Akt Ser473 phosphorylation and lacked evidence of phosphorylation of proteins involved in the proliferation pathway such as S6K Thr421/Ser424 and S6RP Ser235/236. In contrast, the second subset comprised day 30 mice that demonstrated high ERK Thr202/Tyr204, mTOR Ser2448, S6K Thr421/Ser424, and S6RP Ser235/236 phosphorylation. These data suggest that short-term exposure to anti-MHC class I mAb may promote phosphorylation and activation of proteins involved in accommodation whereas longer-term exposure might promote phosphorylation and activation of proteins associated with cell proliferation. Microvascular abnormalities with increased numbers of macrophages were often found in allografts that were dually positive for S6K Thr421/Ser424 and S6RP Ser235/236 phosphorylation in the capillary endothelium. This data suggests that the differences in the phosphorylation patterns may influence pathological changes such as macrophages infiltration in the graft. We speculate that increased staining of S6RP Ser235/236 phosphorylation in endothelial cells may be linked to the synthesis of proteins involved in inflammatory macrophage recruitment. This hypothesis is supported by recent studies showing that alloantibodies stimulate mouse endothelial cells to produce MCP-1 (57). Alternatively, previous studies have reported increased expression of ribosomes in necrotic tissues (58). This could explain why we observed staining of phosphorylated S6RP and S6K in day 30 cardiac allografts from recipients treated with anti-H-2Kd mAb.
We do not yet understand the basis for the differences in the phosphorylation patterns in grafts harvested on day 15 compared with day 30 day even though the Ab titers appear to remain the same. One explanation is that the duration of exposure to Ab influences class I-mediated signal transduction in the endothelium of the graft resulting in differences in the phosphorylation patterns seen in day 15 and 30 allografts. Recent studies show that a persistent threshold of Ab exposure is required to cause cardiac allograft vasculopathy in a similar experimental model (33). Passive transfer of anti-donor mAb into B6.RAG1 KO heart allograft recipients over 14–28 days led to progressive chronic transplant arteriopathy. If treatment was stopped on day 14, lesions showed no progression. This data suggests that the duration of Ab exposure is an important factor in the development of CAV. An analogous situation may also occur in patients diagnosed with AMR. Increased phosphorylation of S6RP Ser235/236 in endomyocardial biopsies was significantly associated with circulating Abs and diagnosis of AMR (17). Furthermore, a study of the temporal relationship between S6RP and AMR showed sustained phosphorylation (mean = 133 days) of S6RP Ser235/236 in patients with two or more Ab-mediated rejection episodes. Using Kaplan-Meier estimates of CAV-free survival rates, 58% of patients with Ab-mediated rejection and persistent phosphorylated S6RP, were estimated to develop CAV within 3 years compared with only 29% of patients without sustained phosphorylated S6RP (17). These data support the hypothesis that the duration of exposure to anti-MHC Ab contributes to differences in phosphorylation patterns in the endothelial cells of the allografts and may possibly lead to differences in transplant outcome.
A hallmark of AMR is complement deposition within the allograft. Numerous studies have reported a positive association between circulating anti-MHC Ab, C4d staining, and diagnosis of AMR (59 – 62), using a newly developed Ab detecting the complement split product C4d. Although the class I-mediated histologic changes and signaling events were associated with C4d deposition, these same pathological changes and signaling events occurred in the absence of C4d deposition suggesting that anti-MHC Ab can activate EC independent of complement.
In summary, we have developed a mouse model system to study the effect of anti-MHC class I Ab in vivo. We have shown that mice treated with anti-class I Ab exhibit morphologic features of human AMR and that passive transfer of anti-MHC class I Ab is significantly correlated with activation of proteins regulating cell survival and proliferation. This experimental model should allow us to explore the mechanisms underlying accommodation and CAV and permit us to identify key biomarkers relevant to AMR. It is also possible to use this model to characterize the effect of antirejection drugs such as rapamycin on the MHC class I-signaling pathway in vivo.
Footnotes
This work was supported by National Institutes of Health Grant RO1AI42819 and American Heart Association Grant 0555081Y (to E.F.R.) and National Institutes of Health Grants RO1AI23847 and RO1AI42223 (to J.K.-W.).
Abbreviations used in this paper: AMR, Ab-mediated rejection; CAV, coronary allograft vasculopathy; EC, endothelial cell; FAK, focal adhesion kinase; S6RP, S6 ribosomal protein; mTOR, mammalian target of rapamycin; KO, knockout; MDS, multidimensional scaling; FcRn, neonatal FcR; mTORC, mTOR complex; Raptor, regulatory associated protein of mTOR.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Michaels PJ, Espejo ML, Kobashigawa J, Alejos JC, Burch C, Takemoto S, Reed EF, Fishbein MC. Humoral rejection in cardiac transplantation: risk factors, hemodynamic consequences and relationship to transplant coronary artery disease. J Heart Lung Transplant. 2003;22:58–69. doi: 10.1016/s1053-2498(02)00472-2. [DOI] [PubMed] [Google Scholar]
- 2.Reed EF, Demetris AJ, Hammond E, Itescu S, Kobashigawa JA, Reinsmoen NL, Rodriguez ER, Rose M, Stewart S, Suciu-Foca N, et al. Acute antibody-mediated rejection of cardiac transplants. J Heart Lung Transplant. 2006;25:153–159. doi: 10.1016/j.healun.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez ER, Skojec DV, Tan CD, Zachary AA, Kasper EK, Conte JV, Baldwin WM., 3rd Antibody-mediated rejection in human cardiac allografts: evaluation of immunoglobulins and complement activation products C4d and C3d as markers. Am J Transplant. 2005;5:2778–2785. doi: 10.1111/j.1600-6143.2005.01074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salama AD, Delikouras A, Pusey CD, Cook HT, Bhangal G, Lechler RI, Dorling A. Transplant accommodation in highly sensitized patients: a potential role for Bcl-xL and alloantibody. Am J Transplant. 2001;1:260–269. doi: 10.1034/j.1600-6143.2001.001003260.x. [DOI] [PubMed] [Google Scholar]
- 5.Lin Y, Soares MP, Sato K, Takigami K, Csizmadia E, Smith N, Bach FH. Accommodated xenografts survive in the presence of anti-donor antibodies and complement that precipitate rejection of naive xenografts. J Immunol. 1999;163:2850–2857. [PubMed] [Google Scholar]
- 6.Bach FH, Ferran C, Hechenleitner P, Mark W, Koyamada N, Miyatake T, Winkler H, Badrichani A, Candinas D, Hancock WW. Accommodation of vascularized xenografts: expression of “protective genes” by donor endothelial cells in a host Th2 cytokine environment. Nat Med. 1997;3:196–204. doi: 10.1038/nm0297-196. [DOI] [PubMed] [Google Scholar]
- 7.Platt JL. A perspective on xenograft rejection and accommodation. Immunol Rev. 1994;141:127–149. doi: 10.1111/j.1600-065x.1994.tb00875.x. [DOI] [PubMed] [Google Scholar]
- 8.Williams JM, Holzknecht ZE, Plummer TB, Lin SS, Brunn GJ, Platt JL. Acute vascular rejection and accommodation: divergent outcomes of the humoral response to organ transplantation. Transplantation. 2004;78:1471–1478. doi: 10.1097/01.tp.0000140770.81537.64. [DOI] [PubMed] [Google Scholar]
- 9.Bian H, Reed EF. Anti-HLA class I antibodies transduce signals in endothelial cells resulting in FGF receptor translocation, down-regulation of ICAM-1 and cell proliferation. Transplant Proc. 2001;33:311. doi: 10.1016/s0041-1345(00)02022-4. [DOI] [PubMed] [Google Scholar]
- 10.Jin YP, Jindra PT, Gong KW, Lepin EJ, Reed EF. Anti-HLA class I antibodies activate endothelial cells and promote chronic rejection. Transplantation. 2005;79:S19–S21. doi: 10.1097/01.tp.0000153293.39132.44. [DOI] [PubMed] [Google Scholar]
- 11.Narayanan K, Jaramillo A, Phelan DL, Mohanakumar T. Pre-exposure to sub-saturating concentrations of HLA class I antibodies confers resistance to endothelial cells against antibody complement-mediated lysis by regulating Bad through the phosphatidylinositol 3-kinase/Akt pathway. Eur J Immunol. 2004;34:2303–2312. doi: 10.1002/eji.200324843. [DOI] [PubMed] [Google Scholar]
- 12.Lepin EJ, Jin YP, Barwe SP, Rozengurt E, Reed EF. HLA class I signal transduction is dependent on Rho GTPase and ROK. Biochem Biophys Res Commun. 2004;323:213–217. doi: 10.1016/j.bbrc.2004.08.082. [DOI] [PubMed] [Google Scholar]
- 13.Narayanan K, Jendrisak MD, Phelan DL, Mohanakumar T. HLA class I antibody mediated accommodation of endothelial cells via the activation of PI3K/cAMP dependent PKA pathway. Transpl Immunol. 2006;15:187–197. doi: 10.1016/j.trim.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, Reed EF. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin, and focal adhesion kinase in an actin-dependent manner. J Immunol. 2002;168:5415–5423. doi: 10.4049/jimmunol.168.11.5415. [DOI] [PubMed] [Google Scholar]
- 15.Jin YP, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, Reed EF. Anti-HLA class I antibody-mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum Immunol. 2004;65:291–302. doi: 10.1016/j.humimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Jindra PT, Zhang X, Mulder A, Claas F, Veale J, Jin YP, Reed EF. Anti-HLA antibodies can induce endothelial cell survival or proliferation depending on their concentration. Transplantation. 2006;82:S33–S35. doi: 10.1097/01.tp.0000231447.34240.3c. [DOI] [PubMed] [Google Scholar]
- 17.Lepin EJ, Zhang Q, Zhang X, Jindra PT, Hong LS, Ayele P, Peralta MV, Gjertson DW, Kobashigawa JA, Wallace WD, et al. Phosphorylated S6 ribosomal protein: a novel biomarker of antibody-mediated rejection in heart allografts. Am J Transplant. 2006;6:1560–1571. doi: 10.1111/j.1600-6143.2006.01355.x. [DOI] [PubMed] [Google Scholar]
- 18.Russell PS, Chase CM, Colvin RB. Alloantibody- and T cell-mediated immunity in the pathogenesis of transplant arteriosclerosis: lack of progression to sclerotic lesions in B cell-deficient mice. Transplantation. 1997;64:1531–1536. doi: 10.1097/00007890-199712150-00005. [DOI] [PubMed] [Google Scholar]
- 19.Wasowska BA, Qian Z, Cangello DL, Behrens E, Van Tran K, Layton J, Sanfilippo F, Baldwin WM., 3rd Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation. 2001;71:727–736. doi: 10.1097/00007890-200103270-00007. [DOI] [PubMed] [Google Scholar]
- 20.Brandle D, Joergensen J, Zenke G, Burki K, Hof RP. Contribution of donor-specific antibodies to acute allograft rejection: evidence from B cell-deficient mice. Transplantation. 1998;65:1489–1493. doi: 10.1097/00007890-199806150-00014. [DOI] [PubMed] [Google Scholar]
- 21.Shi C, Lee WS, He Q, Zhang D, Fletcher DL, Jr, Newell JB, Haber E. Immunologic basis of transplant-associated arteriosclerosis. Proc Natl Acad Sci USA. 1996;93:4051–4056. doi: 10.1073/pnas.93.9.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi C, Russell ME, Bianchi C, Newell JB, Haber E. Murine model of accelerated transplant arteriosclerosis. Circ Res. 1994;75:199–207. doi: 10.1161/01.res.75.2.199. [DOI] [PubMed] [Google Scholar]
- 23.Stewart S, Winters GL, Fishbein MC, Tazelaar HD, Kobashigawa J, Abrams J, Andersen CB, Angelini A, Berry GJ, Burke MM, et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J Heart Lung Transplant. 2005;24:1710–1720. doi: 10.1016/j.healun.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 24.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice: the role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Bian H, Harris PE, Reed EF. Ligation of HLA class I molecules on smooth muscle cells with anti-HLA antibodies induces tyrosine phosphorylation, fibroblast growth factor receptor expression and cell proliferation. Int Immunol. 1998;10:1315–1323. doi: 10.1093/intimm/10.9.1315. [DOI] [PubMed] [Google Scholar]
- 26.Lones MA, Czer LS, Trento A, Harasty D, Miller JM, Fishbein MC. Clinical-pathologic features of humoral rejection in cardiac allografts: a study in 81 consecutive patients. J Heart Lung Transplant. 1995;14:151–162. [PubMed] [Google Scholar]
- 27.Murata K, Fox-Talbot K, Qian Z, Takahashi K, Stahl GL, Baldwin WM, 3rd, Wasowska BA. Synergistic deposition of C4d by complement-activating and non-activating antibodies in cardiac transplants. Am J Transplant. 2007;7:2605–2614. doi: 10.1111/j.1600-6143.2007.01971.x. [DOI] [PubMed] [Google Scholar]
- 28.Minami K, Murata K, Lee CY, Fox-Talbot K, Wasowska BA, Pescovitz MD, Baldwin WM., 3rd C4d deposition and clearance in cardiac transplants correlates with alloantibody levels and rejection in rats. Am J Transplant. 2006;6:923–932. doi: 10.1111/j.1600-6143.2006.01281.x. [DOI] [PubMed] [Google Scholar]
- 29.Regele H, Exner M, Watschinger B, Wenter C, Wahrmann M, Osterreicher C, Saemann MD, Mersich N, Horl WH, Zlabinger GJ, Bohmig GA. Endothelial C4d deposition is associated with inferior kidney allograft outcome independently of cellular rejection. Nephrol Dial Transplant. 2001;16:2058–2066. doi: 10.1093/ndt/16.10.2058. [DOI] [PubMed] [Google Scholar]
- 30.Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–2746. [PubMed] [Google Scholar]
- 31.Wasowska BA, Qian Z, Cangello DL, Van Tran K, Layton JL, Sanfilippo F, Baldwin WM., 3rd Alloantibodies restore cardiac allograft rejection to IgKO mice. Transplant Proc. 2001;33:317. doi: 10.1016/s0041-1345(00)02025-x. [DOI] [PubMed] [Google Scholar]
- 32.Fuller TC, Winn HJ. Immunochemical and biologic characterization of alloantibody active in immunologic enhancement. Transplant Proc. 1973;5:585–587. [PubMed] [Google Scholar]
- 33.Uehara S, Chase CM, Cornell LD, Madsen JC, Russell PS, Colvin RB. Chronic cardiac transplant arteriopathy in mice: relationship of alloantibody, C4d deposition and neointimal fibrosis. Am J Transplant. 2007;7:57–65. doi: 10.1111/j.1600-6143.2006.01599.x. [DOI] [PubMed] [Google Scholar]
- 34.Reed EF. Signal transduction via MHC class I molecules in endothelial and smooth muscle cells. Crit Rev Immunol. 2003;23:109–128. doi: 10.1615/critrevimmunol.v23.i12.60. [DOI] [PubMed] [Google Scholar]
- 35.Kacskovics I, Kis Z, Mayer B, West AP, Jr, Tiangco NE, Tilahun M, Cervenak L, Bjorkman PJ, Goldsby RA, Szenci O, Hammarstrom L. FcRn mediates elongated serum half-life of human IgG in cattle. Int Immunol. 2006;18:525–536. doi: 10.1093/intimm/dxh393. [DOI] [PubMed] [Google Scholar]
- 36.Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179:4580–4588. doi: 10.4049/jimmunol.179.7.4580. [DOI] [PubMed] [Google Scholar]
- 37.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 38.Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, Ghetie V, Ward ES. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol. 1998;10:1289–1298. doi: 10.1093/intimm/10.9.1289. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins SL, Wang J, Vazir M, Vela J, Sahagun O, Gabbay P, Hoang L, Diaz RL, Aranda R, Martin MG. Role of passive and adaptive immunity in influencing enterocyte-specific gene expression. Am J Physiol. 2003;285:G714–G725. doi: 10.1152/ajpgi.00130.2003. [DOI] [PubMed] [Google Scholar]
- 40.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 41.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 42.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 43.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 44.Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000;19:6680–6686. doi: 10.1038/sj.onc.1204091. [DOI] [PubMed] [Google Scholar]
- 45.Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24:6710–6718. doi: 10.1128/MCB.24.15.6710-6718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G, Kozma SC. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. 2004;24:9508–9516. doi: 10.1128/MCB.24.21.9508-9516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 48.Holzknecht ZE, Kuypers KL, Plummer TB, Williams J, Bustos M, Gores GJ, Brunn GJ, Platt JL. Apoptosis and cellular activation in the pathogenesis of acute vascular rejection. Circ Res. 2002;91:1135–1141. doi: 10.1161/01.res.0000046236.20251.fa. [DOI] [PubMed] [Google Scholar]
- 49.Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10) Proc Natl Acad Sci USA. 2000;97:1695–1700. doi: 10.1073/pnas.97.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinney SP, Mancini D. Cardiac allograft vasculopathy: advances in understanding its pathophysiology, prevention, and treatment. Curr Opin Cardiol. 2004;19:170–176. doi: 10.1097/00001573-200403000-00019. [DOI] [PubMed] [Google Scholar]
- 51.Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001;20:6459–6472. doi: 10.1038/sj.onc.1204786. [DOI] [PubMed] [Google Scholar]
- 52.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 53.Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005;280:26089–26093. doi: 10.1074/jbc.M504045200. [DOI] [PubMed] [Google Scholar]
- 54.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 55.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dormond O, Madsen JC, Briscoe DM. The effects of mTOR-AKT interactions on anti-apoptotic signaling in vascular endothelial cells. J Biol Chem. 2007;282:23679–23686. doi: 10.1074/jbc.M700563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rahimi S, Qian Z, Layton J, Fox-Talbot K, Baldwin WM, 3rd, Wasowska BA. Non-complement- and complement-activating antibodies synergize to cause rejection of cardiac allografts. Am J Transplant. 2004;4:326–334. doi: 10.1111/j.1600-6143.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- 58.Larsen TH, Hesketh JE, Rotevatn S, Greve G, Saetersdal T. Ribosome distribution in normal and infarcted rat hearts. Histochem J. 1994;26:79–89. [PubMed] [Google Scholar]
- 59.Haas M, Rahman MH, Racusen LC, Kraus ES, Bagnasco SM, Segev DL, Simpkins CE, Warren DS, King KE, Zachary AA, Montgomery RA. C4d and C3d staining in biopsies of ABO- and HLA-incompatible renal allografts: correlation with histologic findings. Am J Transplant. 2006;6:1829–1840. doi: 10.1111/j.1600-6143.2006.01356.x. [DOI] [PubMed] [Google Scholar]
- 60.Ionescu DN, Girnita AL, Zeevi A, Duquesnoy R, Pilewski J, Johnson B, Studer S, McCurry KR, Yousem SA. C4d deposition in lung allografts is associated with circulating anti-HLA alloantibody. Transpl Immunol. 2005;15:63–68. doi: 10.1016/j.trim.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 61.Collins AB, Schneeberger EE, Pascual MA, Saidman SL, Williams WW, Tolkoff-Rubin N, Cosimi AB, Colvin RB. Complement activation in acute humoral renal allograft rejection: diagnostic significance of C4d deposits in peritubular capillaries. J Am Soc Nephrol. 1999;10:2208–2214. doi: 10.1681/ASN.V10102208. [DOI] [PubMed] [Google Scholar]
- 62.Feucht HE. Complement C4d in graft capillaries–the missing link in the recognition of humoral alloreactivity. Am J Transplant. 2003;3:646–652. doi: 10.1034/j.1600-6143.2003.00171.x. [DOI] [PubMed] [Google Scholar]