

Abstract
The accumulation of genetic and epigenetic alterations mediates colorectal cancer (CRC) formation by deregulating key signaling pathways in cancer cells. In CRC, one of the most commonly inactivated signaling pathways is the transforming growth factor-beta (TGF-β) signaling pathway, which is often inactivated by mutations of TGF-β type II receptor (TGFBR2). Another commonly deregulated pathway in CRC is the phosphoinositide-3-kinase (PI3K)-AKT pathway. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is an important negative regulator of PI3K-AKT signaling and is silenced in approximately 30% of CRC. The combination of TGFBR2 inactivation and loss of PTEN is particularly common in microsatellite unstable CRCs. Consequently, we determined in vivo if deregulation of these two pathways cooperate to affect CRC formation by analyzing tumors arising in mice that lack Tgfbr2 and/or Pten specifically in the intestinal epithelium. We found that lack of Tgfbr2 (Tgfbr2IEKO) alone is not sufficient for intestinal tumor formation and lack of Pten (PtenIEKO) alone had a weak effect on intestinal tumor induction. However, the combination of Tgfbr2 inactivation with Pten loss (PtenIEKO;Tgfbr2IEKO) led to malignant tumors in both the small intestine and colon in 86.0% of the mice and to metastases in 8.1% of the tumor-bearing mice. Moreover, these tumors arose via a β-catenin independent mechanism. Inactivation of Tgf-β signaling and loss of Pten led to increased cell proliferation, decreased apoptosis, and decreased expression of cyclin-dependent kinase inhibitors. Thus, inactivation of TGF-β signaling and loss of PTEN cooperate to drive intestinal cancer formation and progression by suppressing cell cycle inhibitors.
Keywords: colorectal cancer, transforming growth factor beta, PTEN, p21Cip1, colon cancer
Introduction
Colorectal cancer (CRC) is one of the most commonly diagnosed malignancies in industrialized countries, and it remains the second leading cause of cancer-related deaths in the USA, accounting for 51,690 deaths during 2012.(1) Obtaining a better understanding of the functionally significant genetic and epigenetic alterations that arise during the molecular pathogenesis of CRC will help lead to the development of more effective therapies for treating CRC.
The classic paradigm of CRC formation follows the adenoma→cancer sequence, in which CRC begins as a benign neoplasm, called an adenoma.(2) In this classic model, the adenomas are initiated by mutations that activate the Wnt signaling pathway (e.g. mutations in adenomatous polyposis coli (APC) or beta catenin (CTNNB1)).(3–5) Adenomas can then progress to malignancy if they acquire additional oncogenic mutations that lead to the deregulation of other pathways, such as the RAS-RAF-MAPK and/or the phosphoinositide-3-kinase (PI3K)-AKT signaling pathways.(6–8) It has also become clear that CRCs can be classified into three classes based on the form of genomic instability that they display: chromosomal instability (CIN), microsatellite instability (MSI), and CpG Island Methylator Phenotype (CIMP).(9) The Microsatellite Instability (MSI) class of CRC accounts for approximately 15% of CRC and results from inactivation of the DNA mismatch repair system. The MSI cancers often arise through a serrated polyp→cancer sequence.(10) They have a pronounced susceptibility to PI3K inhibitors compared to other CRCs, suggesting they are particularly dependent on the PI3K-AKT pathway.(11)
The PI3K-AKT signaling pathway is aberrantly activated in 40% of CRC.(7) Activation of PI3K signaling promotes cancer formation through a variety of mechanisms.(12–14) Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is an important negative regulator of PI3K-AKT signaling,(15) and its inactivation is a common cause of increased PI3K activity in cancers.(16) Loss of PTEN expression occurs in 20–40% of CRCs.(17, 18) and the MSI subtype has an especially high frequency of PTEN gene mutations compared to CIN CRC’s.(19, 20) Notably, PTEN loss may also affect tumorigenesis through mechanisms other than PI3K pathway activation,(21) including the induction of genomic stability,(22) and deregulated cellular senescence.(23, 24)
Like the PI3K pathway, the TGF-β signaling pathway is commonly deregulated in human cancers.(25, 26) The importance of TGF-β signaling inactivation in CRC is highlighted by the high frequency of resistance to TGF-β, a multifunctional cytokine that acts as a tumor suppressor in the colon.(27) TGF-β mediates its effects on cells through a heteromeric cell surface receptor that consists of two obligate serine-threonine kinase components, TGF-β receptor type I (TGFBR1) and type II (TGFBR2). In colon cancer, the mutational inactivation of TGFBR2 is a common mechanism for inactivating the TGF-β signaling pathway, especially in MSI CRCs.(28) Inactivation of TGFBR2 can result in the deregulation of a multitude of cellular processes that may affect tumorigenesis.(29, 30) The myriad of cellular processes that can be affected by TGF-β signaling inactivation raises the question of which ones are functionally relevant for CRC formation. Furthermore, the effects of TGF-β signaling deregulation in CRC appear to depend on the concurrent signaling pathways that are altered in the cancer cells.(31, 32) Indeed, it is increasingly appreciated that the TGF-β signaling pathway interacts intimately with a variety of pathways including the Wnt-APC-β-catenin, RAS-RAF-MAPK, and PI3K-AKT pathways, and that the interaction of these pathways may be a major factor that determines the biological consequences of TGF-β signaling inactivation in CRC cells.(33, 34)
In light of the common co-occurrence of mutations in TGFBR2 and PTEN in CRC, especially in MSI CRC, we generated an in vivo model to directly assess whether these genetic alterations cooperate in intestinal cancer formation. We found that inactivation of Tgfbr2 and loss of Pten converge to suppress cyclin dependent kinase inhibitors, which may promote intestinal cancer formation and progression in vivo.
Results
Inactivation of Tgfbr2 alone or loss of Pten alone has little to no effect on tumor formation in the intestinal epithelium
In order to assess the in vivo effects of TGF-β signaling inactivation and PI3K-AKT signaling deregulation on intestinal tumorigenesis, Villin-Cre transgenic mice were bred with mice carrying Ptenflx/flx and Tgfbr2flx/flx alleles to create three cohorts of mice: Villin-Cre;Tgfbr2flx/flx (no Tgfbr2 in the intestinal epithelium, designated Tgfbr2IEKO); Villin-Cre;Ptenflx/flx (no Pten in the intestinal epithelium, designated PtenIKEO) and Villin-Cre;Ptenflx/flx;Tgfbr2flx/flx (no Pten and no Tgfbr2 in the intestinal epithelium, designated PtenIKEO;Tgfbr2IEKO). No intestinal tumors were observed in the control mice lacking Villin-Cre (Control). Consistent with previous studies, mice that lacked Tgfbr2 in the intestinal epithelium did not develop tumors by 54 weeks of age. This observation supports our previous studies, suggesting that inactivation of TGF-β signaling alone is not sufficient for intestinal tumor formation.(31, 33)
Deletion of Pten in the intestinal epithelium of PtenIKEO mice was confirmed by immunohistochemistry (IHC) using an antibody specific for PTEN (Supplemental Figure S4). PtenIKEO mice (N=39) were necropsied and examined as noted in the Methods section. The majority of Pten-deficient mice did not have gross or microscopic abnormalities in their intestinal epithelium, which is similar to the findings of Marsh et al and Byun et al. (35, 36) However, unlike in the published studies, we did find gross lesions in 3 PtenIKEO mice (Table 1). These lesions varied from mild hyperplasia (small intestine and cecum) to a single adenocarcinoma (small intestine), which was characterized by dysplastic glands invading into the muscularis propria. There were no large mucinous adenocarcinomas found in the PtenIKEO mice.
Table 1.
Tumor incidence in Control, Tgfbr2IEKO, PtenIEKO and PtenIEKO;Tgfbr2IEKO mice
| Genotype | Number of mice | Number of mice with hyperplastic lesions | Number of mice with adenocarcinomas | Number of hyperplastic lesions | Number of adenocarcinomas |
|---|---|---|---|---|---|
| Control# | 20 | 0 | 0 | 0 | 0 |
| Tgfbr2IEKO | 20 | 0 | 0 | 0 | 0 |
| PtenIEKO | 39 | 2€ | 1λ | 3 | 1 |
| PtenIEKO; Tgfbr2IEKO | 43 | 4μ,£.¥ | 35δ,&.¥ | 4 | 49 |
Control = wild-type mice, matched strain background
P = 0.54, PtenIEKO (2/39) vs. Control (0/20), Fisher’s exact test
P = 1.0, PtenIEKO (1/39) vs. Control (0/20), Fisher’s exact test
P = 0.30, PtenIEKO; Tgfbr2IEKO (4/43) vs. Control (0/20), Fisher’s exact test
P = 0.68, PtenIEKO; Tgfbr2IEKO (4/43) vs. PtenIEKO (2/39), Fisher’s exact test
P < 0.0001, PtenIEKO; Tgfbr2IEKO (35/43) vs. Control (0/20), Fisher’s exact test
P < 0.0001, PtenIEKO; Tgfbr2IEKO (35/43) vs. PtenIEKO (1/39), Fisher’s exact test
N=2 mice developed one adenocarcinoma and one hyperplastic lesion with dysplasia
Tgfbr2 inactivation in the context of Pten loss promotes intestinal tumor formation
The inactivation of TGFBR2 and loss of PTEN are common concurrent molecular events observed in CRC, raising the possibility that deregulation of these pathways may cooperate to promote the formation of intestinal cancer.(19, 20, 37) This possibility led us to generate mice that were deficient for both Pten and Tgfbr2 in the intestinal epithelium (PtenIEKO;Tgfbr2IEKO). Tgfbr2 inactivation in the context of Pten loss significantly shortened the life span of these mice to a median survival of 36 weeks (Figure 1, PtenIEKO;Tgfbr2IEKO mice compared to Tgfbr2IEKO mice, PtenIEKO mice or Control mice, P < 0.0001 for all comparisons). A majority of PtenIEKO; Tgfbr2IEKO mice (N=37/43) developed gross lesions, a significant increase in the number of tumor-bearing PtenIEKO;Tgfbr2IEKO mice as compared to Tgfbr2IEKO mice, PtenIEKO mice or Control mice (Table 1, P < 0.0001 for all comparisons). A total of 49 adenocarcinomas in PtenIEKO; Tgfbr2IEKO mice were confirmed by pathology. The majority of tumors were mucinous adenocarcinomas with transmural effacement by neoplastic glands and large lakes of tumor-produced mucin. In some cases, mucin lakes extended through the serosa and into the serosal lymphatics. (Figure 2A) Mucinous adenocarcinomas occurred in the small intestine (N=16, 32.7%), cecum (N=11, 22.4%), colon (N=13, 26.5%), and cecal junctions (N=9, 18.4%). (Table 2) Alcian blue staining further confirmed mucin production in the adenocarcinomas (Supplemental Figure S1). Confirmation of the expected gene status of the normal intestinal mucosa and tumors of the various genotypes was carried out using PCR-based assays that assess for cre-mediated recombination of the floxed alleles. (Supplemental Figure S2) (33, 38)
Figure 1. Inactivation of Tgf-β signaling and loss of Pten lead to markedly reduced survival.
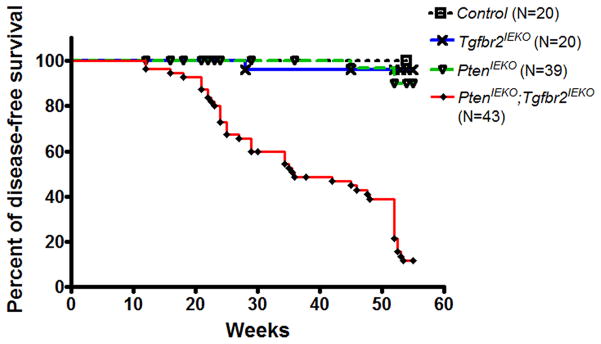
Kaplan-Meier analysis reveals a median survival of 36 weeks in PtenIEKO;Tgfbr2IEKO mice (N=43). A significant decrease in the survival of PtenIEKO;Tgfbr2IEKO mice was observed compared to Tgfbr2IEKO mice, PtenIEKO mice or Control mice (P < 0.0001, log-rank test, for all comparisons). There was no significant difference in survival when the Tgfbr2IEKO mice or PtenIEKO mice were compared to Control mice (P > 0.1 for both comparisons, log-rank test). Of note, all mice that survived to 54 weeks of age were sacrificed at that age.
Figure 2. Inactivation of Tgf-β signaling and loss of Pten lead to formation of invasive adenocarcinomas.

(A) Low-power image of a representative colonic mucinous adenocarcinoma from a PtenIEKO;Tgfbr2IEKO mouse (H&E, original magnification: 20X). There is transmural effacement by neoplastic glands and large lakes of tumor-produced mucin. In some cases, mucin lakes extend through the serosa and into the serosal lymphatics. The inset was taken from the boxed region in (A) at higher magnification and the arrow points to intravascular neoplastic cells within the primary colonic mucinous adenocarcinoma (H&E, original magnification: 200X). (B) Low-power image of a liver section reveals intravascular neoplastic cells (H&E, original magnification: 100X). The inset was taken from the boxed region in (B) at higher magnification and the arrow points to the intravascular neoplastic cells (H&E, original magnification, 200X). (C) Low-power image of a mesenteric metastasis arising from a PtenIEKO;Tgfbr2IEKO mouse (H&E, original magnification: 40X). The inset was taken from the boxed region in (C) at higher magnification to show the neoplastic glands and mucin production (H&E, original magnification: 200X). (D) Low-power image of Alcian blue stained section of the same mesenteric metastasis shown in panel C (Alcian blue stain, original magnification: 40X). The inset was taken from the boxed region in (D) at higher magnification to show the mucin production in this metastasis (Alcian blue stain, original magnification: 200X). (E) E-cadherin immunostaining of the same mesentery metastasis shown in (C) (E-cadherin IHC, original magnification: 40X). The inset was taken from the boxed region in (E) at higher magnification to demonstrate strong expression of E-cadherin, consistent with the epithelial origin of this metastasis (original magnification: 200X). (F) PCR genotyping results demonstrate the recombination of the Tgfbr2flx allele in peritoneal metastasis (PT met1 and PT met2) and mesentery metastases (MSE met1 and MSE met2). The positive control DNA is from the small intestine mucosa of a Tgfbr2IEKO mouse, and the negative control DNA is from the lymph nodes of a PtenIEKO;Tgfbr2IEKO mouse that did not have grossly evident metastatic disease.
Table 2.
Anatomic distribution of adenocarcinomas in PtenIEKO;Tgfbr2IEKO mice
| Genotype | Number of ACA | Small Intestine | Cecum | Colon | Cecal Junction |
|---|---|---|---|---|---|
| PtenIEKO; Tgfbr2IEKO | 49 | 16 (32.7%) | 11(22.4%) | 13(26.5%) | 9(18.4%) |
Notably, 8.1% of the tumor-bearing PtenIEKO;Tgfbr2IEKO mice were found to have metastases within the adjacent mesentery, pancreas, and peritoneal cavity (body wall, uterus, and prostate). A single mouse did have metastasis to the liver with intravascular neoplastic cells. (Figure 2B–C) Thus, metastases were confirmed histologically and appeared to occur both via intravascular and transcoelomic routes. The metastases were also assessed with Alcian blue staining, E-cadherin immunostaining, and by PCR for the recombined Tgfbr2flx alleles to confirm that they were derived from the primary mucinous adenocarcinomas. (Figure 2D–F)
Thus, inactivation of Tgfbr2 with loss of Pten in the intestinal epithelium results in the formation of adenocarcinomas in the small intestine, cecum, and colon. The presence of metastases in some of the mice suggests that loss of TGF-β signaling contributes to ametastatic phenotype in the context of Pten loss.(39)
Signaling pathway deregulation in the tumors arising in the PtenIEKO;Tgfbr2IEKO mice
We first confirmed that the neoplastic cells in the PtenIEKO;Tgfbr2IEKO mice did not express Pten by IHC. (Figure 3B) Since PTEN exerts tumor suppressive functions by inhibiting the PI3K-AKT pathway, we analyzed the activation status of the PI3K-AKT pathway by IHC using antibodies specific for phospho-AKT Serine 473 (Ser473). We observed neoplastic epithelial cells in PtenIEKO;Tgfbr2IEKO mice had increased nuclear phosphorylated Akt Ser473 compared to the normal intestinal epithelium. (Figure 3C)
Figure 3. Signaling pathway deregulation in PtenIEKO;Tgfbr2IEKO tumors.

(A) Low-power image of a representative intestinal adenocarcinoma from a PtenIEKO;Tgfbr2IEKO mouse (H&E, original magnification: 20X). The boxed region in (A) is shown at higher magnification in the adjacent photomicrographs of immunostained tissue. Neoplastic epithelial cells in PtenIEKO;Tgfbr2IEKO mice show (A) decreased PTEN expression by PTEN immunostaining and (B) increased Akt activation by phospho-Akt (Ser473) immunostaining. The invasive front of a representative adenocarcinoma shows (C) membrane-bound β-catenin. (A–C, original magnification 200X). (D) Upper level, analysis of activation of Akt by immunoblotting of lysates from normal mucosa of wild-type (WT) control and Tgfbr2IEKO mice (left panel); in normal mucosa of PtenIEKO mice, normal mucosa and tumors from PtenIEKO;Tgfbr2IEKO mice (right panel). β-actin was used as a loading control. Middle level and lower level, relative Akt phosphorylation levels at Thr308 and Ser473 sites as determined by densitometry, normalized to total Akt and further normalized to β-actin. Note a significant decrease in relative Akt phosphorylation levels at Thr308 and Ser473 sites in tumors tissue (T) compared to normal tissue (N) from PtenIEKO;Tgfbr2IEKO mice. (* p<0.05 for both relative pAkt T308 and S473, Mann-Whitney test) (E) Upper level, analysis of activation of ERK1/2 by immunoblotting of lysates from normal mucosa of wild-type Control and Tgfbr2IEKO mice (left panel); in normal mucosa of PtenIEKO mice, normal mucosa and tumors from PtenIEKO;Tgfbr2IEKO mice (right panel). β-actin was used as a loading control. Lower level, relative ERK1/2 phosphorylation levels as determined by densitometry, normalized to total ERK1/2 and further normalized to β-actin. Note a significant increase in relative ERK1/2 phosphorylation levels in tumor tissue (T) compared to normal tissue (N) from PtenIEKO;Tgfbr2IEKO mice. (* p<0.05, Mann-Whitney test).
Next, we assessed the phosphorylation status of Akt at both Thr308 and Ser473 in the normal intestinal mucosa and tumors of the mice by immunoblotting. (Figure 3E) We detected total Akt but not phospho-Akt in the normal mucosa of the wild-type control mice, which is consistent with previous reports.(35) Of interest, a modest increase in phospho-Akt (Ser473), but not Thr308, was detected in the normal mucosa of the Tgfbr2IEKO mice, indicating that inactivation of TGF-β signaling leads to phosphorylation of Akt at Ser473 in the setting of intact Pten. This is presumably secondary to indirect effects of loss of Tgfbr2 and was unexpected in light of prior studies in epithelial cells that showed that TGF-β signaling can induce Akt phosphorylation at Ser473.(40) Increased Akt phosphorylation levels at both Thr308 and Ser473 sites were observed in the normal mucosa of Pten-deficient mice compared to wild-type mice. However, we observed less phosphorylated Akt in the tumors from the PtenIEKO;Tgfbr2IEKO mice compared to the normal mucosa, even though total Akt levels were increased in the tumors. (Figure 3E, right panel) These results raise the possibility of negative feedback regulation occurring in the tumors and suggest that a reduction in PI3K activity is selected for during intestinal cancer formation.
We also assessed the activation status of the MAPK signaling pathway in the normal intestinal mucosa and tumors because this signaling pathway is often induced in human CRC and can be regulated by TGF-β.(41) We found that the MAPK pathway, as measured by phospho-ERK1/2, was activated at a basal state of the normal mucosa of the control mice and the Tgfbr2IEKO mice. (Figure 3F, left panel) In contrast to increased Akt phosphorylation, less MAPK activation was observed in the majority of normal mucosa samples of the Pten-deficient mice, regardless of theTgfbr2 status, and there was also more inter-individual variation in the PtenIEKO mice compared to the control mice. (Figure 3F, right panel) Despite there being less ERK activation in the normal intestines of the mice lacking Pten, we observed increased ERK1/2 phosphorylation in the tumors when compared to normal mucosa in the PtenIEKO;Tgfbr2IEKO mice, indicating that the MAPK pathway is upregulated in the PtenIEKO;Tgfbr2IEKO tumors. To determine if somatic oncogenic Kras mutations are responsible for the increased MAPK pathway activation in the tumors from the PtenIEKO;Tgfbr2IEKO mice, we sequenced the tumors for Kras mutations. However, we did not detect mutations in Kras (codons 12, 13 or 61) in any of PtenIEKO;Tgfbr2IEKO tumors assayed (N=5).
The intestinal tumors in the PtenIEKO;Tgfbr2IEKO mice arise independently of the canonical Wnt-β-catenin pathway
Given the high frequency of APC and CTNNB1 mutations and subsequent Wnt pathway activation in human CRC and the fact that loss of Tgfbr2 or Pten alone does not appear to be sufficient to initiate intestinal tumor formation,(26) we assessed the activation state of the Wnt signaling pathway in the tumors arising in the PtenIEKO;Tgfbr2IEKO mice. In addition, the Wnt pathway was of interest in light of conflicting data regarding its role in initiating tumor formation in the setting of Pten deficiency.(35, 36, 42, 43) The activation state of the Wnt signaling pathway was assessed by determining the nuclear localization status of β-catenin. (Figure 3D) The membrane-bound localization of β-catenin was observed in 75% of the PtenIEKO tumors analyzed (N=4) and in 100% of the PtenIEKO;Tgfbr2IEKO tumors analyzed (N=10). We only observed nuclear localization of β-catenin in one adenoma from a PtenIEKO mouse (Supplemental Figure S4), which we presume was secondary to a somatic event in the tumor with subsequent activation of Wnt signaling. We also analyzed the levels of Wnt target genes, Axin2 and Cd44, by qRT-PCR.(44–46) We did not observe a significant up-regulation of these Wnt target genes in the tumors arising in PtenIEKO;Tgfbr2IEKO mice as compared to the normal intestinal epithelial tissue from the PtenIEKO, Tgfbr2IEKO or PtenIEKO;Tgfbr2IEKO mice. (P = 0.45 for Axin2; P = 0.33 for Cd44, Supplemental Figure S5) Overall, these results suggest that the canonical Wnt-β-catenin pathway is not activated in these mice.
Inactivation of Tgfbr2 and Pten loss do not cooperate to induce tumor formation through effects on genomic stability
Both PTEN and TGF-β signaling have been implicated in the control of genomic stability. (47) Thus, we assessed the tumors for aneuploidy using microarray comparative genomic hybridization (array CGH) (N=3). However, we found no chromosomal gains or losses in the tumors. (Supplemental Figure S6) These results demonstrate that these tumors do not have gross chromosomal abnormalities representative of chromosomal instability and also do not reveal a common locus that is altered somatically in the tumors. The identification of a commonly deleted or amplified locus would have indicated a possible somatic event that was cooperating with loss of TGFBR2 and PTEN to drive tumor formation in the PtenIEKO;Tgfbr2IEKO mice.
Deregulation of cell cycle control proteins in PtenIEKO;Tgfbr2IEKO mouse tumors
In light of the increased MAPK pathway signaling in the PtenIEKO;Tgfbr2IEKO tumors, we assessed tumor cell proliferation (by Ki-67 immunostaining) to determine if enhanced proliferation is one of the biological effects driving tumor formation in the PtenIEKO;Tgfbr2IEKO mice. (Figure 4A) We observed that a substantial portion of the neoplastic cells in the PtenIEKO;Tgfbr2IEKO mice were positive for Ki67 staining (average percent positive cells/tumor = 50.0% ± 26%). As both PI3K-AKT and TGF-β pathways have been implicated in the induction of apoptosis, we also assessed apoptosis in tumors from the PtenIEKO and PtenIEKO;Tgfbr2IEKO mice by cleaved caspase-3 immunostaining. (Figure 4B) We found neoplastic glands in the PtenIEKO;Tgfbr2IEKO mice had very little cleaved caspase-3 staining. We also assessed cleaved caspase-3 by immunoblotting and observed decreased cleaved caspase-3 protein level in the tumors arising in the PtenIEKO;Tgfbr2IEKO mice compared to the normal mucosa in the other mouse cohorts. (Supplemental Figure S7) These results suggest that increased proliferation and decreased apoptosis contribute to the formation of the tumors in the PtenIEKO;Tgfbr2IEKO mice.
Figure 4. Assessment of proliferation, apoptosis and cyclin dependent kinase inhibitors in normal tissue and PtenIEKO;Tgfbr2IEKO tumors.

(A) Representative photomicrograph of Ki67 immunostained tumor from PtenIEKO;Tgfbr2IEKO mice. (B) Representative photomicrograph of cleaved caspase-3 immunostained tumor from PtenIEKO;Tgfbr2IEKO mice. Rare stained cells are present and stained cellular debris is noted within the lumen of neoplastic glands (arrow). (C) Assessment of p15Ink4b (Cdkn2b), p21Cip1 (Cdkn1a) and p27Kip1(Cdkn1b) mRNA by qRT-PCR in matched normal mucosa and tumors from PtenIEKO;Tgfbr2IEKO mice. (Mann-Whitney test, * p=0.01 for p15Ink4b(Cdkn2b), 0.0033 for p21Cip1(Cdkn1a), and 0.015 for p27Kip1(Cdkn1b)) (D) Assessment of p15Ink4b, p21Cip1 and p27Kip1 protein levels by immunoblotting lysates from normal mucosa of various genotypes, and normal mucosa and tumors from the PtenIEKO;Tgfbr2IEKO mice. GAPDH was used as a loading control. Normalized protein levels of p15Ink4b, p21Cip1 and p27Kip1 were determined by densitometry, normalized to GAPDH. Note a significant decrease in normalized levels of p15Ink4b, p21Cip1 and p27Kip1 in tumor tissue (T) compared to normal tissue (N) from PtenIEKO;Tgfbr2IEKO mice. (* p<0.05, Mann-Whitney test, N=3)
Cyclin dependent kinase (CDK) inhibitors p15INK4B, p21CIP1 and p27KIP1 are well-documented target genes of the TGF-β signaling pathways.(48–50) They play an essential role in the control of the cell cycle and proliferation and their expression is often decreased in cancer.(51–53) Therefore, we assessed the levels of Cdkn2b/p15Ink4b, Cdkn1a/p21Cip1 and Cdkn1b/p27Kip1 mRNA by qRT-PCR in the matched normal and tumor tissue. (Figure 4C) Expression of all three CDK inhibitors genes was significantly lower in the tumor samples compared to the matched normal epithelium (for all comparisons, P < 0.02). Furthermore, we analyzed the protein levels by immunoblot. (Figure 4D) Expression of p15Ink4b, p21Cip1 and p27Kip1 proteins was detected in the normal mucosa of various genotypes regardless of Pten and Tgfbr2 status. Comparison of protein levels between normal and tumor tissue from PtenIEKO;Tgfbr2IEKO mice revealed significantly decreased expression of p15INK4B, p21Cip1 and p27Kip1 in the tumor tissue (T) compared to normal tissue (N) (* P < 0.05, Mann-Whitney test, N=3). These results reveal a marked decrease in expression of CDK inhibitors in tumors, suggesting this is a tumor-promoting biological consequence resulting from the cooperation of inactivation of Tgfbr2 and loss of Pten.
Reconstitution of PTEN and TGFBR2 cooperate to induce p21Cip1 expression in human colon cancer cells
In order to assess the causal role of PTEN and TGFBR2 in the regulation of the CDK inhibitors, we reconstituted PTEN and/or TGFBR2 in the SNU-C4 colon cancer cell line, a human CRC cell line with both mutant PTEN and TGFBR2.(54) We then evaluated the SNU-C4 cells for p21CIP1 promoter activation using the p21Pδ2.1-luc reporter, a p21CIP1 promoter containing the minimal TGF-β response elements (55). As shown in Figure 5, reconstitution of TGFBR2 in SNU-C4 cells induced expression of the p21Pδ2.1-luc reporter by 4-fold compared to control vector. Similarly, reconstitution of PTEN alone induced a 3-fold increase in luciferase activity. Reconstitution with both wild-type TGFBR2 and PTEN further induced the expression of p21Pδ2.1-luc by 10-fold. To confirm the specificity of the cooperation between TGFBR2 and PTEN on the transcriptional activation of p21CIP1, we assessed the effect of reconstituted PTEN and TGFBR2 on activating the 3TP-lux luciferase reporter, which assesses TGF-β pathway activation. Reconstitution of the SNU-C4 cells with PTEN alone did not induce 3TP-lux activity. (Figure 5, Supplemental Figure S8) Furthermore, there was no further increase in the luciferase activity of the 3TP-lux reporter in TGFBR2 and PTEN co-transfected cells compared to TGFBR2-transfected alone cells, indicating the additive effect of PTEN and TGFBR2 on p21CIP1 transcription activation is specific for p21CIP1. These results suggest deregulated cell cycle proteins are likely downstream mediators through which inactivation of TGFBR2 and loss of PTEN cooperate to promote tumor formation and progression.
Figure 5. Reconstitution of TGFBR2 and PTEN induce p21CIP1 expression in colon cancer cells.
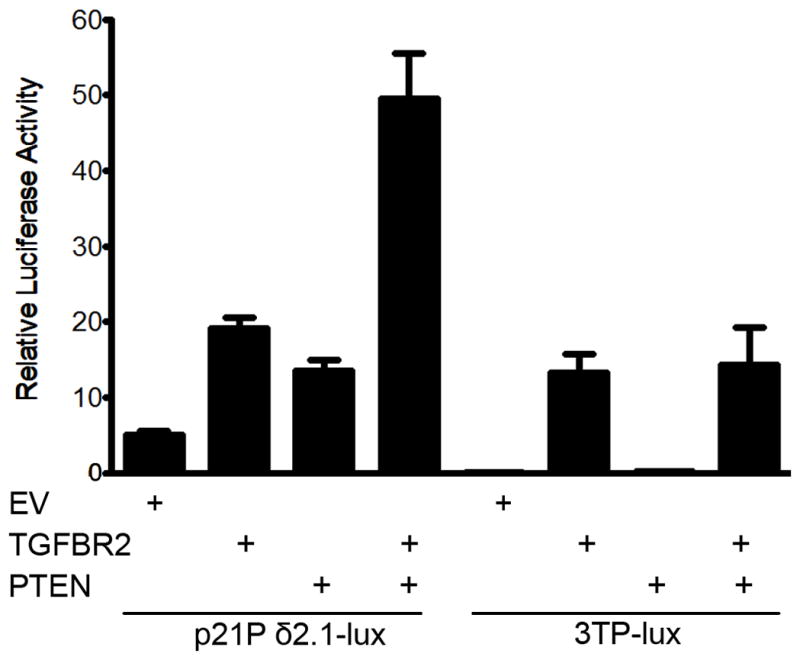
Stimulation of p21Pδ2.1 reporter luciferase activity in the colon cancer cell line SNU-C4 transfected with empty vector (EV), TGFBR2, PTEN, or both TGFBR2 and PTEN. Transfected SNU-C4 cells were treated with TGF-β (2 ng/ml). Histograms represent relative luciferase activity normalized to Renilla luciferase reporter activity. The error bars represent the standard error from three independent experiments. The 3TP-lux luciferase reporter was used to confirm the specificity of the cooperation between TGFBR2 and PTEN on the activation of p21CIP1 luciferase reporter. Reconstitution of the SNU-C4 cells with PTEN alone did not induce 3TP-lux activity. Furthermore, there was no increase in the luciferase activity of the 3TP-lux reporter in TGFBR2 and PTEN co-transfected cells compared to TGFBR2-transfected alone cells, indicating the specificity of the additive effect of PTEN and TGFBR2 on p21CIP1 expression.
Discussion
It is widely appreciated that the majority of CRC develop as a consequence of the progressive accumulation of genetic and epigenetic alterations in evolving clones of tumor cells.(9) The Cancer Genome Atlas Network (TCGA) recently completed whole exome sequencing of 224 CRCs and found components in the TGF-β and PTEN/PI3K/AKT pathways were frequent mutational targets.(26) A major challenge emerging from cancer genome sequencing projects, like the TCGA study described above, is how to determine which of the mutations found in the tumors are “drivers” of cancer formation and which are “passengers”. Some of these mutations have been studied in cancer cell lines, but their relevance to in vivo tumor initiation and progression is not clear. Consequently, we generated the PtenIEKO;Tgfbr2IEKO mice to study the effect of deregulation of these two pathways on cancer formation in vivo.
Deregulation of the PTEN/PI3K-AKT pathway has been associated with the growth and progression of CRC.(56, 57) Loss of PTEN appears to be the most common mechanism for deregulating the PI3K pathway, but little is known regarding the specific mechanisms through which PTEN loss mediates its effects in vivo on colon cancer formation. Our studies and previously published studies now identify potential functional consequences of loss of PTEN in driving tumor formation in the intestines, and demonstrate the importance of gene cooperation in the formation of CRC. (35, 42, 58) Using mice that lack Pten in the intestinal epithelium, we found phosphorylation of Akt at both Ser473 and Thr308 was increased in the normal intestinal epithelial cells of Pten-deficient mice. This finding is consistent with the role of PTEN as a negative regulator of PI3K-AKT activity.(15, 59) However, the loss of PTEN alone had weak tumor promoting effects in the intestines, which suggests that for effective tumorigenesis, deregulation of the PI3K pathway requires the cooperation of other deregulated pathways. Our results differ from those of a recent published study in which mice expressing a dominantly active Pik3ca transgene spontaneously developed metastatic intestinal cancer.(60) Potential explanations for the common spontaneous development of tumors in their mouse model are the use of the Pi3kca* transgene, which is a dominant active form of Pi3kca p110α, and the use of mice in a different genetic background than the mice in our studies.
In light of the modest effect of PTEN loss on intestinal tumor formation and because mutational inactivation of TGFBR2 is often concurrent with loss of PTEN in human CRC(9), we proposed that the TGF-β and PI3K pathways may cooperate to drive CRC formation. The results of this study in which we observed a substantial increase in adenocarcinoma formation in mice lacking both Pten and Tgfbr2 provide support for this idea. However, it is worth noting that the majority of tumor-bearing mice developed 1–2 adenocarcinoma, indicating low tumor multiplicity in these mice. Therefore, we propose that additional cooperating events are needed for the initiation of intestinal tumors in the PtenIEKO;Tgfbr2IEKO mice. Along these lines, it is interesting that there is no evidence of Wnt pathway activation in the tumors in this mouse model. The recently identified “hypermutated class” of CRCs found by TCGA also often lack APC mutations(26), which raises the possibility that the PtenIEKO;Tgfbr2IEKO mouse model may recapitulate features of this newly described class of CRC. Further studies of this class of human CRC may provide insight into the nature of the additional somatic events occurring in the PtenIEKO;Tgfbr2IEKO mouse tumors and will also allow a determination of how well the PtenIEKO;Tgfbr2IEKO mice model the hypermutated class of CRC.
After observing cooperation between inactivation of Tgfbr2 and loss of Pten on intestinal tumor formation, we investigated potential mechanisms that could be driving this effect. One potential mechanism revealed by our studies is the deregulation of CDK inhibitor expression. Inactivation of Tgfbr2 and Pten loss resulted in marked suppression of cyclin dependent kinase inhibitors in the tumor cells. Consistent with loss of CDK inhibitor expression, we observed increased proliferation in these tumors, which has also been observed in MSI CRCs.(61) These results suggest that inhibition of CRC development by PTEN and TGFBR2 is due in part to the collaborative regulation of cell cycle proteins. Consistent with this possibility, we observed that reconstitution of PTEN and TGFBR2 had an additive effect on inducing p21CIP1 expression in human cancer cells.
Our studies suggest that the loss of CDK inhibitors is a significant mechanism through which PTEN and TGFBR2 inactivation promotes intestinal cancer formation. We have not yet identified a mechanism through which inactivation of Tgfbr2 and loss of Pten may be affecting expression of CDK inhibitors, but candidate mechanisms include alterations in Pik3r1 activity or in activin signaling.(56, 62–65) Furthermore, there are additional candidate biological effects through which Tgfbr2 inactivation and PTEN loss may promote intestinal tumor formation. For instance, we observed the loss of senescence markers p19Arf and Dcr2 in PtenIEKO;Tgfbr2IEKO tumors. (Supplemental Figure S9) Senescence functions as a barrier to oppose tumorigenesis and can be triggered by a variety of mechanisms including the activation of oncogenic pathways or the loss of PTEN.(66–68) AKT activation has been shown to induce senescence through mechanisms involving p27Kip1 in prostate cancer.(69) Thus, it is possible that Tgfbr2 inactivation may help tumor cells escape Pten-loss induced senescence, which could be another cooperative mechanism that drives the tumor progression in the PtenIEKO;Tgfbr2IEKO mice. Further studies are needed to assess this possibility.
In summary, we have generated an in vivo mouse model to assess whether cooperation between two frequent events found in human colon cancer, TGFBR2 mutations and PTEN loss, plays a functional role in the molecular pathogenesis of CRC. Our results provide evidence that the cooperative interactions between inactivation of TGFBR2 and loss of PTEN converge on deregulating CDK inhibitors to promote tumorigenesis in CRC. These findings raise the possibility that agents targeted at the cyclin:CDK enzyme complexes may be particularly effective in CRCs that carry TGFBR2 mutations and lack PTEN expression. Furthermore, our mouse model recapitulates molecular features seen in a subset of CRC (“hypermutated class” of CRC) and has the potential for use as a pre-clinical model for CRC to test novel therapeutic targets.
Materials and Methods
Generation and characterization of Villin-Cre; Ptenflx/flx;Tgfbr2flx/flx mice
The generation of the following genetically engineered mice has been previously described: Villin-Cre;Tgfbr2flx/flx (Tgfbr2IEKO) and Ptenflx/flx (33, 38). The Tgfbr2IEKO and Ptenflx/flx mice were mated to generate the following genotypes: Villin-Cre; Ptenflx/flx;Tgfbr2wt/wt (PtenIEKO), and Villin-Cre;Ptenflx/flx;Tgfbr2flx/flx (PtenIEKO;Tgfbr2IEKO). The floxed Tgfbr2 (Tgfbr2flx/flx) and Villin-Cre mice were maintained on C57BL/6J background. Prior to generating the mice with the multiple mutant genotypes, Ptenflx/flx mice were backcrossed onto a C57BL/6J background for at least five generations to obtain mice that are >90% C57BL/6J. Thus, the background strain of experimental mice was predominantly C57BL/6J with a small proportion of 129Sv. Mice were fed ad libitum with a standard rodent diet. Mice were harvested at 54 weeks of age, or earlier if moribund. The studies were approved by the local IACUC.
Mouse tissue processing
Mouse tissues were handled as previously described.(33, 34). Briefly, the small intestine, cecum, and colon were removed, opened, flushed with cold PBS and then examined grossly for tumors. Grossly visible tumors were measured and collected for histopathology. All major organs were examined for gross lesions, and mesenteric lymph nodes, lungs, liver were collected and evaluated histologically in all the mice. Gross lesions suspected to be metastases were confirmed histologically by S.K. The tissues were either snap-frozen in liquid nitrogen and stored at −80 C for RNA and protein extractions or fixed in 10% neutral buffered formalin (Fisher Scientific, Pittsburgh, PA) for 3–7 days, transferred to 70% ethanol, embedded in paraffin, and cut into 4 μm sections for H&E staining or immunostaining.
Immunoblotting
Protein lysates were obtained using standard methods and then resolved by SDS-PAGE gels and transferred to PVDF membranes (Thermo Scientific, Rockford, IL). Densitometric quantification of immunoblots was performed using the ImageJ 1.43 software. The following primary antibodies were used: rabbit anti-p44/42 total MAPK (ERK1/2) (1:1000, #9102 Cell Signaling, Danvers, MA), rabbit anti-phospho-p44/42 MAPK (phospho-ERK1/2) (Thr202/Tyr204, 1:2000, #4370 Cell Signaling), rabbit anti-pAKT (Ser473, 1:1,000, #9271 Cell Signaling), rabbit anti-pAKT (Thr308, 1:1,000, #9275 Cell Signaling), rabbit anti-total AKT(1:1,000, #9297 Cell Signaling), rabbit p15INK4B (1:1,000, #4822 Cell Signaling), rabbit p21Cip1 (1:1,000, SC-469 Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-p27Kip1 (1:1,000, kindly provided by Dr. Matthew Fero), rabbit anti-p19Arf (1:1,000, PC435 Calbiochem®, EMD Biosciences, Billerica, MA), rabbit anti-Dcr2 (1:1,000, Ab2019, Abcam, Cambridge, MA), goat anti-β-Actin (1:5,000, SC-1616 Santa Cruz Biotechnology). The following secondary antibodies were used: goat anti-rabbit IgG-HRP (1:5,000, #SC-2004, Santa Cruz Biotechnology), donkey anti-goat IgG-HRP (1:5,000, #SC-2020, Santa Cruz Biotechnology).
Immunohistochemistry (IHC) staining
Hemotoxylin and Eosin (H&E) and Alcian blue staining, Ki67, β-catenin, and E-cadherin immunohistochemical assays were performed in the Experimental Histopathology Core at the Fred Hutchinson Cancer Research Center following routine laboratory protocol. Specific protocols are available upon request.
Comparative genomic hybridization
Acquired gains and losses of chromosomal material were assessed by microarray comparative genomic hybridization (array CGH). Three tumors from three individual mice were analyzed. Matched non-neoplastic reference DNA was obtained from the spleen from each mouse to minimize detection of constitutional copy number variants. Analysis was performed as previously described.(34)
Sequencing analysis of Kras gene mutation
Genomic DNA was isolated from normal intestinal mucosa and tumors using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Amplification of Kras exon1&2 was performed by polymerase chain reaction (PCR) as previously described.(70)
Real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR)
mRNA was extracted from snap-frozen tissues using TRIzol (Life Technologies, Grand Island, NY) following the manufacturer’s instructions. cDNA was generated using standard procedures using oligodT primers (Life Technologies). TaqMan gene expression assays (Assays-on-Demand; Applied Biosystems, Foster City, CA) for p15Ink4b (Cdkn2b, Mm00483241_m1), p21Cip1 (Cdkn1a, Mm00432448_m1), p27Kip1(Cdkn1b, Mm00438168_m1) and β-glucuronidase (Gusb, Mm01197698_m1) were used for qRT-PCR. The assays were performed using the Bio-Rad CFX96 real-time PCR systems (Bio-Rad, Hercules, CA). The results of the qRT-PCR assays were normalized to β-glucuronidase (Gusb). Statistical analysis was performed using the GraphPad Prism version 4.00 software. The Mann-Whitney test was used for comparisons of quantitative results from the qRT-PCR assays. A P value of <0.05 was regarded as significant.
Tissue culture
The human colon cancer cell line SNU-C4 was generously provided by the Korean Cell Line Bank (Dr. Ja-Lok Ku, Seoul, Korea) and grown in RPMI 1640 (Life Technologies) supplemented with 10% FBS (HyClone, Logan, UT).
Plasmid transfection experiments
Transfections were performed using FuGene9 (Roche, Indianapolis, IN) following the manufacturer’s protocol. The pCMV5-TGFBR2 expression vector, which carries HA tagged wild-type TGFBR2, was kindly provided by Joan Massagué (Memorial Sloan-Kettering Cancer Center, New York, NY). An expression vector containing the human PTEN open reading frame pORF9-hPTEN was purchased from Invivo Gen (San Diego, CA).
Luciferase reporter assay
SNU-C4 cells were transiently transfected with the p21Pδ2.1 reporter (kindly provided by Xiaofan Wang, Duke University, Durham, NC) or p3TP-lux reporter (kindly provided by Joan Massagué, Memorial Sloan-Kettering Cancer Center, New York, NY) concomitantly with the pRL-TK reporter construct (Promega, Madison, WI), which expresses Renilla luciferase. Subsequently, the cells were treated with TGF-β1 (2 ng/ml), and luciferase activity was evaluated 48h after transfection using the Dual Luciferase Reporter Assay System (Promega) with a Veritas luminometer (Turner Biosystems, Sunnyvale, CA). Luciferase activities were normalized based on Renilla luciferase activity.
Supplementary Material
Acknowledgments
Support for these studies was provided by the NIH (RO1CA115513, P30CA15704, UO1CA152756, U54CA143862, and P01CA077852 WMG), a Burroughs Wellcome Fund Translational Research Award for Clinician Scientist (WMG), and an Interdisciplinary Training in Cancer Research Grant (T32 CA080416 SMM).
Abbreviations
- CRC
colorectal cancer
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- qRT-PCR
real-time quantitative reverse transcription polymerase chain reaction
- TGF-β
transforming growth factor-beta
- Tgfbr1
transforming growth factor-beta type I receptor
- Tgfbr2
transforming growth factor-beta type II receptor
- Dcr2
a decoy receptor of the TNF-related factor TRAIL
Footnotes
Conflicts of Interest: No conflicts of interest exist for any of the authors.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–70. doi: 10.1016/s0092-8674(00)81333-1. Epub 1996/10/18. [DOI] [PubMed] [Google Scholar]
- 3.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256(5057):668–70. doi: 10.1126/science.1350108. Epub 1992/05/01. [DOI] [PubMed] [Google Scholar]
- 4.Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer research. 1994;54(22):5953–8. Epub 1994/11/15. [PubMed] [Google Scholar]
- 5.Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML. Beta-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer research. 1999;59(7):1442–4. Epub 1999/04/10. [PubMed] [Google Scholar]
- 6.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–32. doi: 10.1056/NEJM198809013190901. Epub 1988/09/01. [DOI] [PubMed] [Google Scholar]
- 7.Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, et al. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436(7052):792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 8.Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut. 2011 doi: 10.1136/gut.2009.206250. Epub 2010/10/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135(4):1079–99. doi: 10.1053/j.gastro.2008.07.076. Epub 2008/09/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50(1):113–30. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu XQ, Rajput A, Geng L, Ongchin M, Chaudhuri A, Wang J. Restoration of transforming growth factor-beta receptor II expression in colon cancer cells with microsatellite instability increases metastatic potential in vivo. The Journal of biological chemistry. 2011;286(18):16082–90. doi: 10.1074/jbc.M111.221697. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nature Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4(4):257–62. doi: 10.1016/s1535-6108(03)00248-4. Epub 2003/10/31. [DOI] [PubMed] [Google Scholar]
- 14.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature reviews Cancer. 2009;9(8):550–62. doi: 10.1038/nrc2664. Epub 2009/07/25. [DOI] [PubMed] [Google Scholar]
- 15.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. The Journal of biological chemistry. 1998;273(22):13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 16.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100(4):387–90. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 17.Naguib A, Cooke JC, Happerfield L, Kerr L, Gay LJ, Luben RN, et al. Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study: associations with clinicopathological and dietary factors. BMC Cancer. 11:123. doi: 10.1186/1471-2407-11-123. Epub 2011/04/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, et al. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. The American journal of pathology. 2002;161(2):439–47. doi: 10.1016/S0002-9440(10)64200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL. Transforming Growth Factor beta Enhances Epithelial Cell Survival via Akt-dependent Regulation of FKHRL1. Mol Biol Cell. 2001;12(11):3328–39. doi: 10.1091/mbc.12.11.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fearon ER. Molecular genetics of colorectal cancer. Annual review of pathology. 6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 21.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature reviews Molecular cell biology. 2012;13(5):283–96. doi: 10.1038/nrm3330. Epub 2012/04/05. [DOI] [PubMed] [Google Scholar]
- 22.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 23.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. The Journal of clinical investigation. 2010;120(3):681–93. doi: 10.1172/JCI40535. Epub 2010/03/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Massague J, Blain SW, Lo RS. TGF[beta] Signaling in Growth Control, Cancer, and Heritable Disorders. Cell. 2000;103(2):295. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. doi: 10.1038/nature11252. Epub 2012/07/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grady W, Rajput A, Myeroff L, Liu D, Kwon K-H, Willis J, et al. Mutation of the type II transforming growth factor-β receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer research. 1998;58:3101–4. [PubMed] [Google Scholar]
- 28.Grady W, Myeroff L, Swinler S, Rajput A, Thiagalingam S, Lutterbaugh J, et al. Mutational inactivation of transforming growth factor β receptor type II in microsatellite stable colon cancers. Cancer research. 1999;59:320–4. [PubMed] [Google Scholar]
- 29.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nature reviews Cancer. 2006;6(7):506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 30.Grady W, Markowitz SD. TGF-β Signaling Pathway and Tumor Suppression. In: Derynck R, Miyazano K, editors. The TGF-β Family. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2008. pp. 889–938. [Google Scholar]
- 31.Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska AE, Wirth PS, et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer research. 2004;64(14):4687–92. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- 32.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends in cell biology. 2001;11(11):S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 33.Munoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, et al. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer research. 2006;66(20):9837–44. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- 34.Trobridge P, Knoblaugh S, Washington MK, Munoz NM, Tsuchiya KD, Rojas A, et al. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology. 2009;136(5):1680–8. e7. doi: 10.1053/j.gastro.2009.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marsh V, Winton DJ, Williams GT, Dubois N, Trumpp A, Sansom OJ, et al. Epithelial Pten is dispensable for intestinal homeostasis but suppresses adenoma development and progression after Apc mutation. Nature genetics. 2008;40(12):1436–44. doi: 10.1038/ng.256. [DOI] [PubMed] [Google Scholar]
- 36.Byun DS, Ahmed N, Nasser S, Shin J, Al-Obaidi S, Goel S, et al. Intestinal epithelial-specific PTEN inactivation results in tumor formation. American journal of physiology. 2011;301(5):G856–64. doi: 10.1152/ajpgi.00178.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chittenden TW, Howe EA, Culhane AC, Sultana R, Taylor JM, Holmes C, et al. Functional classification analysis of somatically mutated genes in human breast and colorectal cancers. Genomics. 2008;91(6):508–11. doi: 10.1016/j.ygeno.2008.03.002. Epub 2008/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32(2):148–9. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 39.Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res. 8(5):629–42. doi: 10.1158/1541-7786.MCR-10-0139. Epub 2010/05/13. [DOI] [PubMed] [Google Scholar]
- 40.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. The Journal of biological chemistry. 2000;275(47):36803–10. doi: 10.1074/jbc.M005912200. Epub 2000/09/02. [DOI] [PubMed] [Google Scholar]
- 41.Gomes LR, Terra LF, Wailemann RA, Labriola L, Sogayar MC. TGF-beta1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer. 12:26. doi: 10.1186/1471-2407-12-26. Epub 2012/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langlois MJ, Roy SA, Auclair BA, Jones C, Boudreau F, Carrier JC, et al. Epithelial phosphatase and tensin homolog regulates intestinal architecture and secretory cell commitment and acts as a modifier gene in neoplasia. Faseb J. 2009;23(6):1835–44. doi: 10.1096/fj.08-123125. [DOI] [PubMed] [Google Scholar]
- 43.He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nature genetics. 2004;36(10):1117–21. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- 44.Wielenga VJ, Smits R, Korinek V, Smit L, Kielman M, Fodde R, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. The American journal of pathology. 1999;154(2):515–23. doi: 10.1016/S0002-9440(10)65297-2. Epub 1999/02/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Molecular and cellular biology. 2002;22(4):1184–93. doi: 10.1128/MCB.22.4.1184-1193.2002. Epub 2002/01/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, Sancho E, et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Molecular and cellular biology. 2006;26(22):8418–26. doi: 10.1128/MCB.00821-06. Epub 2006/09/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glick AB, Weinberg WC, Wu I-H, Quan W, Yuspa SH. TGF-β1 suppresses genomic instability downstream of a G1 arrest by a p53 and Rb independent pathway. Cell. 1996 submitted. [PubMed] [Google Scholar]
- 48.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nature medicine. 2002;8(10):1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 49.Massague J. TGFbeta in Cancer. Cell. 2008;134(2):215–30. doi: 10.1016/j.cell.2008.07.001. Epub 2008/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moses H, Yang E, Pietenpol J. TGF-β stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63:245–7. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- 51.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nature reviews Cancer. 2008;8(4):253–67. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 52.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nature reviews Cancer. 2009;9(6):400–14. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee MH, Yang HY. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell Mol Life Sci. 2001;58(12–13):1907–22. doi: 10.1007/PL00000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin KH, Park YJ, Park JG. PTEN gene mutations in colorectal cancers displaying microsatellite instability. Cancer letters. 2001;174(2):189–94. doi: 10.1016/s0304-3835(01)00691-7. Epub 2001/11/02. [DOI] [PubMed] [Google Scholar]
- 55.Datto MB, Yu Y, Wang XF. Functional analysis of the transforming growth factor beta responsive elements in the WAF1/Cip1/p21 promoter. The Journal of biological chemistry. 1995;270(48):28623–8. doi: 10.1074/jbc.270.48.28623. Epub 1995/12/01. [DOI] [PubMed] [Google Scholar]
- 56.Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61(20):7426–9. Epub 2001/10/19. [PubMed] [Google Scholar]
- 57.Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, et al. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis. 2002;23(1):201–5. doi: 10.1093/carcin/23.1.201. Epub 2002/01/05. [DOI] [PubMed] [Google Scholar]
- 58.Byun DS, Ahmed N, Nasser S, Shin J, Al-Obaidi S, Goel S, et al. Intestinal epithelial-specific PTEN inactivation results in tumor formation. Am J Physiol Gastrointest Liver Physiol. 301(5):G856–64. doi: 10.1152/ajpgi.00178.2011. Epub 2011/08/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95(1):29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 60.Leystra AA, Deming DA, Zahm CD, Farhoud M, Olson TJ, Hadac JN, et al. Mice expressing activated PI3K rapidly develop advanced colon cancer. Cancer research. 2012;72(12):2931–6. doi: 10.1158/0008-5472.CAN-11-4097. Epub 2012/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grady WM, Willis JE, Trobridge P, Romero-Gallo J, Munoz N, Olechnowicz J, et al. Proliferation and Cdk4 expression in microsatellite unstable colon cancers with TGFBR2 mutations. International journal of cancer. 2006;118(3):600–8. doi: 10.1002/ijc.21399. [DOI] [PubMed] [Google Scholar]
- 62.Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16(6):463–74. doi: 10.1016/j.ccr.2009.10.016. Epub 2009/12/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panopoulou E, Murphy C, Rasmussen H, Bagli E, Rofstad EK, Fotsis T. Activin A suppresses neuroblastoma xenograft tumor growth via antimitotic and antiangiogenic mechanisms. Cancer Res. 2005;65(5):1877–86. doi: 10.1158/0008-5472.CAN-04-2828. Epub 2005/03/09. [DOI] [PubMed] [Google Scholar]
- 64.Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126(3):654–9. doi: 10.1053/j.gastro.2004.01.008. Epub 2004/02/28. [DOI] [PubMed] [Google Scholar]
- 65.Bauer J, Sporn JC, Cabral J, Gomez J, Jung B. Effects of activin and TGFbeta on p21 in colon cancer. PloS one. 2012;7(6):e39381. doi: 10.1371/journal.pone.0039381. Epub 2012/07/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen CN, Lin JJ, Chen JJ, Lee PH, Yang CY, Kuo ML, et al. Gene expression profile predicts patient survival of gastric cancer after surgical resection. J Clin Oncol. 2005;23(29):7286–95. doi: 10.1200/JCO.2004.00.2253. [DOI] [PubMed] [Google Scholar]
- 67.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 68.Young LC, Listgarten J, Trotter MJ, Andrew SE, Tron VA. Evidence that dysregulated DNA mismatch repair characterizes human nonmelanoma skin cancer. The British journal of dermatology. 2008;158(1):59–69. doi: 10.1111/j.1365-2133.2007.08249.x. [DOI] [PubMed] [Google Scholar]
- 69.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer cell. 2008;14(2):146–55. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ise K, Nakamura K, Nakao K, Shimizu S, Harada H, Ichise T, et al. Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene. 2000;19(26):2951–6. doi: 10.1038/sj.onc.1203600. Epub 2000/06/29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.