

Abstract
We have previously shown that miR-486-5p is one of the most downregulated micro RNAs in lung cancer. The objective of the study was to investigate the role of miR-486-5p in the progression and metastasis of non-small-cell lung cancer (NSCLC). We evaluated miR-486-5p expression status on 76 frozen and 33 formalin-fixed paraffin-embedded tissues of NSCLC by quantitative reverse transcriptase PCR to determine its clinicopathologic significance. We then performed function analysis of miR-486-5p to determine its potential roles on cancer cell migration and invasion in vitro and metastasis in vivo. We also investigated the target genes of miR-486-5p in lung tumorigenesis. miR-486-5p expression level was significantly lower in lung tumors compared with their corresponding normal tissues (P<0.0001), and associated with stage (P =0.0001) and lymph node metastasis of NSCLC (P = 0.0019). Forced expression of miR-486-5p inhibited NSCLC cell migration and invasion in vitro and metastasis in mice by inhibiting cell proliferation. Furthermore, ectopic expression of miR-486-5p in cancer cells reduced ARHGAP5 expression level, whereas miR-486-5p silencing increased its expression. Luciferase assay demonstrated that miR-486-5p could directly bind to the 3′-untranslated region of ARHGAP5. The expression level of miR-486-5p was inversely correlated with that of ARHGAP5 in lung tumor tissues (P =0.0156). Reduced expression of ARHGAP5 considerably inhibited lung cancer cell migration and invasion, resembling that of miR-486-5p overexpression. miR-486-5p may act as a tumor-suppressor contributing to the progression and metastasis of NSCLC by targeting ARHGAP5. miR-486-5p would provide potential diagnostic and therapeutic targets for the disease.
Keywords: miR-486-5p, tumor-suppressor gene, lung cancer, ARHGAP5, therapy
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide, mainly because it is often diagnosed at advanced stage accompanied by extensive invasion and metastasis.1 Non-small-cell lung cancer (NSCLC) accounts for at least 80% of lung cancers.1 Therefore, investigations of the molecular mechanisms underlying progression and metastasis of NSCLC may help develop novel prognostic biomarkers and therapeutic targets for the malignancy, and thus are clinically important.
Micro RNAs (MiRNAs) are endogenous, ~22-nucleotide-long, non-coding RNAs.2 Individual miRNAs can target multiple distinct transcripts, and hence control a wide range of biological processes.3–5 Abnormal expressions of miRNAs have frequently been observed in various types of cancers.6 Importantly, downregulations of some miRNAs can motivate tumorigenesis by regulating several key pathways, including promoting cellular proliferation, evading apoptosis, stimulating angiogenesis and invasion and metastasis.7,8 Therefore, the miRNAs have functions of tumor suppressors in cancer development and progression.7,9 Using microarray to analyze primary lung tumor tissues for miRNA expressions, we have identified a set of 26 miRNAs whose abnormal expressions are associated with NSCLC.3,4 miR-486-5p is one of the most downregulated miRNAs in lung tumor tissues. We recently showed that analyzing expression levels of the miRNAs, particularly miR-486-5p, in sputum and plasma could provide a diagnostic approach for NSCLC.3–5
In the study, we aimed to evaluate the possible roles and related target genes of miR-486-5p in tumorigenesis of NSCLC. We found that the expression level of miR-486-5p was significantly lower in NSCLC tissues than in the corresponding normal lung tissues, and inversely associated with advanced stage and lymph node metastasis of NSCLC. Furthermore, enforced miR-486-5p expression restrained lung cancer cell migration and invasion in vitro and metastasis in vivo. In addition, ARHGAP5, a protumorigenic gene, was identified as a functional target of miR-486-5p. Therefore, miR-486-5p downregulation contributes to lung cancer progression and metastasis through regulating ARHGAP5.
RESULTS
Reduced expression of miR-486-5p is inversely associated with advanced stage and lymph node metastasis of NSCLC
We previously reported that miR-486-5p was underexpressed in NSCLC by using microarray analysis.3–5 To determine the clinicopathologic significance of the miR-486-5p aberration, we evaluated the expression level of miR-486-5p in 76 pairs of frozen NSCLC tissues and the corresponding normal lung tissues using quantitative reverse transcriptase PCR (qRT–PCR). MiR-486-5p expression was not significantly associated with age and gender of the patients and histological types of NSCLC (Table 1). However, the expression level was remarkably lower in NSCLC tissues than in their matched normal tissues (P<0.0001) (Figure 1a). Furthermore, the expression level of miR-486-5p in tumor tissues statistically decreased with increasing stage of NSCLC (P<0.0001) (Figure 1b). In addition, miR-486-5p expression was significantly lower in NSCLC that displayed lymph node metastasis than in NSCLC that did not have (P =0.0019) (Figure 1c). Moreover, the observations were confirmed in formalin-fixed paraffin-embedded (FFPE) specimens of 33 NSCLC tissues and the paired normal lung tissues (Supplementary Table 1). Therefore, the low miR-486-5p expression is closely related to the progression and metastasis of NSCLC.
Table 1.
Demographic and clinical characteristics of 76 NSCLC patients and the association with miR-486-5p expression in tumor tissue specimens
| Characteristics | Number of cases (%) | Median expression of miR-486-5p (mean±s.d.) | P |
|---|---|---|---|
| Age, year | |||
| ≥69 | 46 (60.5) | 0.03182±0.03206 | 0.18608 |
| <69 | 30 (39.5) | 0.02165±0.00276 | |
| Gender | |||
| Male | 56 (73.7) | 0.03083±0.04577 | 0.26550 |
| Female | 20 (26.3) | 0.02017±0.03556 | |
| Histology | |||
| Adenocarcinomas | 39 (51.3) | 0.02816±0.04354 | 0.19643 |
| Squamous cell carcinomas | 37 (48.7) | 0.02253±0.02902 | |
| Smokersa | |||
| Yes | 69 (90.8) | 0.02126±0.03763 | 0.16601 |
| No | 7 (9.2) | 0.03015±0.02010 | |
| T-status | |||
| 1a | 13 (17.1) | 0.03557±0.04555 | 0.34540 |
| 1b | 9 (11.8) | 0.03106±0.02678 | |
| 2 | 34 (44.8) | 0.03264±0.04135 | |
| 3 | 15 (19.7) | 0.03936±0.03584 | |
| 4 | 5 (6.6) | 0.03309±0.03276 | |
| N-statusb | |||
| No | 30 (39.5) | 0.02829±0.05080 | 0.00190 |
| Yes | 46 (60.5) | 0.01503±0.01916 | |
| Stage | |||
| I | 19 (25.0) | 0.07665±0.04979 | 0.00010 |
| II | 33 (43.4) | 0.02830±0.01585 | |
| III | 24 (31.6) | 0.01008±0.00746 | |
Abbreviation: NSCLC, non-small-cell lung cancer.
Smokers were defined as persons who had smoked a 20 pack year, or greater.
Lymph node status was classified as N0, N1, N2 and N3. In the present study, N1, N2 and N3 are considered to be ‘Yes’, whereas N0 is considered as ‘No’.
Figure 1.

Expression level of miR-486-5p is lower in lung tumor tissues than in the matched normal lung tissues and the reduced expression is associated with advanced clinical stage and lymph node metastasis of NSCLC. (a) miR-486-5p expression level was considerably lower in NSCLC tissues than in their matched normal tissues. The expression level of miR-486-5p was assessed in 76 pairs of primary NSCLC frozen tissues and the corresponding normal lung tissues using qRT–PCR. U6 small nuclear RNA was used as an internal control. (b) Low-level expression of miR-486-5p was associated with high tumor stage of NSCLC (P<0.001). (c) Low-level expression of miR-486-5p was related with lymph node metastasis of NSCLC (P =0.0019).
Ectopic overexpression of miR-486-5p restrains cell proliferation, migration and invasion of NSCLC cells
As a low level of miR-486-5p expression in NSCLC is a common molecular incident and correlated with advanced stage and metastasis of the disease, we hypothesize that ectopic expression of miR-486-5p in NSCLC can exert inhibitory effects on cell growth and invasion. To validate the hypothesis, we transfected a miR-486-5p mimic or scrambled sequence into A549 and H157 NSCLC cells, which had low basal levels of miR-486-5p in NSCLC cell lines (Supplementary Figure 1). Successful overexpression of miR-486-5p in the cells was confirmed by qRT–PCR. Interestingly, methylthiazol tetrazolium assay showed that forced expression of miR-486-5p could impair growth rate of the NSCLC cells (Figures 2a and b).
Figure 2.

MiR-486-5p suppresses NSCLC cell proliferation, migration and invasion. (a) Forced expression of miR-486-5p in H157 cells reduced cell growth determined by methylthiazol tetrazolium (MTT) assay. Cancer cells were plated in microtiter plates. On the following day, cells were transfected with miR-486-5p mimic or scrambled sequence, respectively. After 48 h, cell growth rate was determined by MTT. (b) Forced expression of miR-486-5p in A549 cells inhibited cell growth rate of NSCLC evaluated using MTT assay. (c) Ectopic expression of miR-486-5p in H157 cells restrained cell invasion and migration determined by Transwell assay. After transfection, cells were plated in a transwell. The cells that had migrated to the lower surface of the membrane were stained with crystal violet and counted under a light microscope. (d) Forced expression of miR-486-5p in A549 cells repressed cell invasion and migration determined by Transwell assay. All data were obtained from three independent experiments and shown as mean±s.d. and *P<0.05.
To explore the possible mechanism of overexpression of miR-486-5p underlying the inhibitory effect on cell growth, we performed 5-bromo-2′-deoxyuridine (BrdU) incorporation assay and apoptotic analysis. BrdU incorporation level was statistically lower in cells transfected with miR-486-5p mimic than in cells with scrambled sequence (P<0.05) (Supplementary Figure 2), suggesting that ectopic expression of miR-486-5p could reduce the proliferation of NSCLC cells. Annexin V fluorescein isothiocyanate (V-FITC) apoptotic assay showed that there was no significant difference of apoptotic rate between cells with miR-486-5p mimic and control cells (P>0.05) (Supplementary Figure 3). Therefore, ectopic expression of miR-486-5p might reduce cell growth of NSCLC mainly through inhibiting cell proliferation. Furthermore, miR-486-5p overexpression could suppress the migratory and invasive abilities of the NSCLC cells (H157 and A549) determined by Transwell assay (Figures 2c and d). Taken together, miR-486-5p might have tumor-suppressor function.
Overexpression of miR-486-5p inhibits NSCLC metastasis in vivo
To further investigate the role of miR-486-5p in tumorigenesis of NSCLC cells, we injected mice through tail vein with H460-bioluminescent cells that were transfected with miR-486-5p mimic or scrambled sequence control. The mice were monitored for tumor formation and metastasis by bioluminescence imaging. Positive imaging was observed in the lungs of the mice injected with H460 cells with scrambled sequence after 1 week (Figure 3a). Furthermore, additional positive signals outside lungs could be found in neck and abdomen of the mice in week 7 (Figure 3a). However, although positive signals were noticed in the lungs of the mice injected with H460 cells with miR-486-5p mimic in week 1, no positive imaging besides the lungs was found in week 7 (Figure 3b). We used mean photon counts of bioluminescence to evaluate malignant lesions in the mice as previously described.10 Mean photon counts of malignant nodules in the lungs of all mice inoculated with H460 cells with scrambled sequence was markedly higher than that seen in all mice injected with H460 cells with miR-486-5p mimic (P<0.05) (Supplementary Figure 4). In addition, mean photon counts of metastatic lesions was significantly higher in mice injected with H460 cells with scrambled sequence compared with mice injected with H460 cells with miR-486-5p mimic (P<0.05) (Supplementary Figure 5). Moreover, the average weight of primary lung tumors derived from H460 cells with scrambled sequence was significantly larger than that from H460 cells with miR-486-5p mimic (P = 0.008) (Supplementary Figure 6). Primary tumors in lungs generated from H460 cells transfected with scrambled sequence and the associated metastatic tumors in lymph nodes were confirmed by hematoxylin and eosin staining on tissue sections (Figures 3c and d). Therefore, miR-486-5p overexpression could inhibit lung tumorigenicity and metastasis in vivo.
Figure 3.

Forced expression of miR-486-5p restrains NSCLC metastasis in vivo. (a) Fluorescent imaging analysis showed that malignant lesions in the lungs of the mice injected with H460-luc2 cells transfected with scrambled sequence after 1 week, in the neck and abdomen after 7 weeks. Ten mice were used in the group. All 10 mice were monitored by using fluorescent imaging weekly for tumor formation. The figure only shows images that were taken at week 1 and week 7 from the same mouse. (b) Small positive imaging signal was obtained in the lungs of the mice injected with H460-luc2 cells transfected with miR-486-5p mimic after 1 week, and no positive signal in the neck and lower abdomen after 7 weeks. Ten mice were used in the group and were monitored using fluorescent imaging weekly. The figure only shows images that were taken from the same mouse at week 1 and week 7. More description about the data obtained from all 10 mice from each group was shown in Supplementary Figures 4–6. (c) The mice were killed and autopsied in week 8 after cancer cell injection. Serial sections were made from the lungs and metastatic tumor tissues and stained with hematoxylin and eosin (H&E). H&E staining confirms that the cancer cells transfected with scrambled sequence produced tumor in the lungs. (d) H&E staining shows that H460 cells with scrambled sequence created a metastatic tumor in lymph node.
Furthermore, we performed immunohistochemical analysis of Ki-67, a marker of proliferation, on tissue sections of tumors excised from mice. The number of Ki-67-positive cells was statistically lower in tumors created from cancer cells with miR-486-5p mimic than in tumors generated from cancer cells with scrambled sequence (Supplementary Figure 7A) (P =0.009). Moreover, we carried out the terminal nucleotidyl transferase-mediated nick end labeling assay for evaluating apoptosis.
Percentage of apoptotic cells was not statistically different between tumors created from cells with miR-486-5p mimic and those generated from cancer cells with scrambled sequence (Supplementary Figure 7B) (P =0.65). Therefore, the inhibition of in vivo tumorigenicity by forced miR-486-5p expression is likely attributed to decreased cell proliferation.
Protumorigenic factor ARHGAP5 is a direct target of miR-486-5p
To elucidate the mechanisms responsible for the tumor-suppressive abilities of miR-486-5p, we used bioinformatics analysis to identify its target genes. ARHGAP5 was identified as one of the candidate targets of miR-486-5p (Supplementary Table 2). MiR-486-5p can potentially bind to the 3′-untranslated region (UTR) of ARHGAP5 (Figure 4a). Previous studies showed that ARHGAP5 could inhibit RhoA activity and contribute to spreading and migration by enhancing cell protrusion, elongation and polarity.3–5 Furthermore, ARHGAP5 displayed a high expression level in aggressive tumors and had protumorigenic function in carcinogenesis.11,12 Therefore, we paid special attention to ARHGAP5 for deep investigation in the present study.
Figure 4.

ARHGAP5 is the target gene of miR-486-5p. (a) a miR-486-5p target site within 3′-UTR of ARHGAP5 was predicted by bioinformatic algorithms. (b) A luciferase reporter assay in A549 cells showed that luciferase activity controlled by ARHGAP5-3′-UTR was dramatically inhibited by ectopic miR-486-5p expression. (c) Top, H157 cells had a high expression level of miR-486-5p after transfection with miR-486-5p mimic. Bottom, AHGAP5 expression level was reduced in the H157 cells transfected with miR-486-5p mimics determined by western blotting with p190-B antibody. (d) Top, A549 cells had a high expression level of miR-486-5p after transfection with miR-486-5p mimic. Bottom, the level of ARHGAP5 protein expression was reduced in A549 cells transfected with the miR-486-5p mimic.
To determine whether ARHGAP5 could be regulated by miR-486-5p, we performed luciferase reporter assay. The luciferase activity of ARHGAP5-3′-UTR was reduced by ~60% in cells expressing miR-486-5p compared with those expressing the control (Figure 4b). We further transfected the cells with miR-486-5p mimic to produce overexpression of miR-486-5p in cancer cells and then measured ARHGAP5 expressions by using western blotting with p190-B antibody. Forced expression of miR-486-5p produced a decrease of ARHGAP5 expression (Figures 4c and d). Therefore, it is likely that miR-486-5p may bind to the 3′-UTR sequences of ARHGAP5, and might inhibit its expression through post-transcriptional regulation.
ARHGAP5 is involved in miR-486-5p-induced suppression of NSCLC cell proliferation, migration and invasion
To further explore the functions of ARHGAP5 in lung tumorigenesis, we used specific small interfering RNAs against ARHGAP5 (si-ARHGAP5) to reduce expression of ARHGAP5 in NSCLC cells. As shown in Figure 5a, si-ARHGAP5 dramatically reduced ARHGAP5 expression. Methylthiazol tetrazolium and colony formation assays showed that cell growth and proliferation were significantly repressed becauae of downregulation of ARHGAP5 (Figure 5b). Furthermore, Wound healing and Transwell assays indicated that ARHGAP5 downregulation inhibited NSCLC cell migration and invasion (Figures 5c and d), which, resembled the inhibitory effects of miR-486-5p on the cancer cells. Therefore, ARHGAP5 might be protumorigenic factor in the development and progression of NSCLC.
Figure 5.

Reduced expression of ARHGAP5 inhibits cell proliferation, migration and invasion. (a) Top, the transcription level of ARHGAP5 was significantly inhibited by si-ARHGAP5 in A549 cells as determined by qRT–PCR. Bottom, ARHGAP5 protein was repressed by the small interfering RNA at 48 h in A549 cells as determined by western blotting with p190-B antibody. (b) Top, methylthiazol tetrazolium assay showed that cell proliferation was decreased in A549 cancer cells transfected with si-ARHGAP5 compared with cells with scrambled sequence. Bottom, Colony formation assay demonstrated that cell growth of A549 cells was repressed due to downregulation of ARHGAP5. (c) Wound-healing assay indicated that ARHGAP5 downregulation inhibited cell migration and invasion of A549 cells. (d) Transwell assay showed that knockdown of ARHGAP5 restrained cell migration and invasion of H157 cells.
To determine whether deregulation of ARHGAP5 by miR-486-5p involved in cell migration and invasion, we transfected NSCLC cells with miR-486-5p inhibitor and si-ARHGAP5. Compared with control cell group, the cells transfected with miR-486-5p inhibitor displayed higher expression of ARHGAP5, whereas the cells with the cotransfection of both miR-486-5p inhibitor and si-ARHGAP5 exhibited lower ARHGAP5 expression (Figures 6a and b). Interestingly, the cells transfected with miR-486-5p inhibitor displayed higher migration- and invasion potential compared with the cells transfected with both miR-486-5p inhibitor and si-ARHGAP5 (Figures 6c and d). The observations suggest that the effects of miR-486-5p downregulation on the promotion of cancer cell migration and invasion could be diminished by si-ARHGAP5. Therefore, ARHGAP5 may have an important role in the cell migration and invasion of NSCLC mediated by miR-486-5p.
Figure 6.

ARHGAP5 is involved in miR-486-5p-induced suppression of cell migration and invasion of NSCLC. (a) H358 cells treated with miR-486-5p inhibitor and si-ARHGAP5 exhibited reduced ARHGAP5 expression, whereas H358 cells treated with miR-486-5p inhibitor had increased ARHGAP5 expression determined by western blotting with p190-B antibody. (b) H226 cells transfected with miR-486-5p inhibitor and si-ARHGAP5 had reduced ARHGAP5 expression, whereas H226 cells treated with miR-486-5p inhibitor showed elevated ARHGAP5 expression. (c) H358 cells treated with miR-486-5p inhibitor and si-ARHGAP5 had reduced migration and invasion potential, whereas H358 cells treated with miR-486-5p inhibitor displayed increased potential of migration and invasion determined by Transwell assay. (d) H226 cells treated with miR-486-5p inhibitor and si-ARHGAP5 had reduced migration and invasion potential, whereas H226 cells treated with miR-486-5p inhibitor showed elevated potential of migration and invasion.
Upregulation of ARHGAP5 is inversely associated with downregulation of miR-486-5p in clinical specimens of NSCLC
To further investigate clinical significance of ARHGAP5 expression, we examined ARHGAP5 expression by using immunohistochemical analysis on FFPEs of 54 NSCLC with p190-B antibody. As shown in Figure 7a, p190-B exhibited negative or weak staining in alveolar epithelial cells and bronchial epithelial cells of normal lung tissue. In contrast, p190-B displayed positive staining in tumor tissues with different intensity (Figures 7b and c). Furthermore, p190-B expression was positively correlated with tumor, nodes and metastasis-classification (TNM) stage and lymph node metastasis of NSCLC (All P<0.001) (Supplementary Table 3). Taken together, ARHGAP5 is frequently overexpressed in NSCLC and the elevated expression is positively associated with the progression of the disease.
Figure 7.
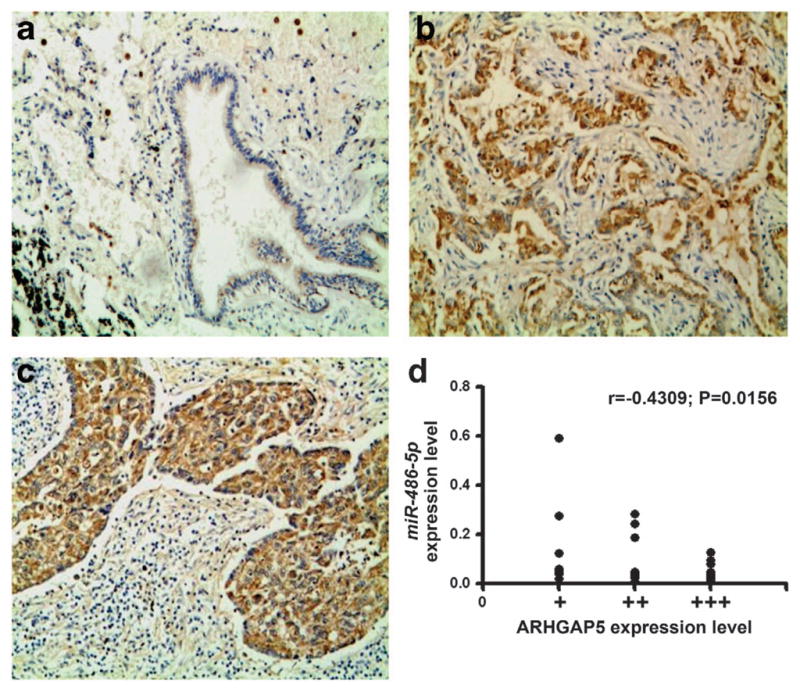
ARHGAP5 is overexpressed in tumor tissues and the overexpression is inversely correlated with miR-486-5p. (a) Bronchial epithelial cells of a normal lung tissue displayed negative or weak staining of p190-B antibody. (b) A tissue sample of lung adeno-carcinoma showed strong immunohistochemistry staining. (c) A tissue sample of squamous cell carcinoma of lung had positive staining. (d) A scatter diagram showed an inverse correlation between miR-486-5p and ARHGAP5 expressions in the same set of NSCLC tissue specimens.
To explore the relationship between miR-486-5p and ARHGAP5 in clinical specimens, we compared ARHGAP5 expression data from immunohistochemistry analysis with results of miR-486 expression level from qRT–PCR analysis on FFPE specimens of 33 NSCLC tissues. There was an inverse correlation between miR-486-5p and ARHGAP5 expressions in the specimens (P =0.0156) (Figure 7d). Therefore, the findings further signify that ARHGAP5 upregulation might result from suppression of miR-486-5p in NSCLC.
DISCUSSION
Reduced miR-486-5p expression is a frequent molecular event in human malignances.5,13–15 We have showed that miR-486-5p is repressed in early-stage NSCLC by using microarray analysis.3–5 We further demonstrated that analyzing expression of miR-486-5p in sputum and plasma specimens could provide a diagnostic approach for the early detection of lung cancer.3–5 Oh et al.16 recently found that miR-486-5p might target OLFM4 anti-apoptotic factor, and hence had important function in the progression and metastasis of gastric cancer. However, the exact mechanism of miR-486-5p dysregulation in NSCLC remains unknown. In the present study, we first found that miR-486-5p was frequently downregulated in lung tumor tissues and the reduced miR-486-5p expression was closely related to advanced stage and lymph node metastasis of NSCLC. Furthermore, we demonstrated that miR-486-5p overexpression could suppress NSCLC cell proliferation, migration and invasion in vitro and metastasis in vivo. In addition, we identified protumorigenic ARHGAP5 as a target of miR-486-5p. Therefore, miR-486-5p could be a novel tumor-suppressor miRNA, and its downregulation might contribute to lung cancer progression and metastasis through regulating ARHGAP5 function.
Elucidating the molecular mechanism(s) of lymph node metastasis is a critical issue, as lymph node metastasis is a fundamental factor in the determination of the clinical staging and prognosis of NSCLC. It is widely accepted that cellular adhesion, motility and invasion are required for the spreading of tumor cells from their primary tumor to lymph nodes in the process of metastasis. Interestingly, ARHGAP5-encoded protein is an important regulator of RhoA,17,18 which is a prototypical member of the Rho GTPase family. Rho GTPase can regulate many cellular processes, particularly including cellular adhesion, motility and polarity.18–23 Furthermore, upregulation of ARHGAP5 in cancers can contribute to invasive and metastatic behavior. For example, ARHGAP5 upregulation has protumorigenic functions that enhance tumor cell migration in a variety of cancers, such as breast tumor,24–26 hepatocellular carcinoma and melanoma.16,27 In consistent with the previous findings, the results obtained from our current study demonstrated that ARHGAP5 expression was significantly increased in NSCLC tissues, and the high expression level was correlated with a more aggressive behavior of the disease. Moreover, reduced ARHGAP5 expression restrains cell growth, proliferation, migration and invasion of NSCLC. Therefore, upregulation of ARHGAP5 has a vital tumorigenic role in carcinogenesis of NSCLC. In addition, our functional analysis showed that overexpression of miR-486-5p inhibited ARHGAP5 expression and conferred inhibition of cell proliferation and migration, which was parallel to small interfering RNA-mediated knockdown of ARHGAP5. The observations would bring a new insight about the essential mechanisms of regulating the RhoA pathway in the progression and metastasis of NSCLC. Importantly, these findings reveal that ARHGAP5 is a biologically significant target gene of miR-486-5p. Furthermore, the miR-486-5p/ARHGAP5 pathway might be a previously unrecognized regulator involved in lung tumor progression and lymph node metastasis of NSCLC. Therefore, miR-486-5p may serve as a potential target for therapeutic intervention against invasive and metastatic NSCLC.
There are several possible reasons for the observation of downregulation of miR-486-5p in tumor tissues. For instance, miR-486-5p is located on chromosome 8p11.21, one of the most frequent genomic deletion regions that contain potential tumor-suppressor genes in various types of tumors, such as NSCLC.17,18 Allelic loss of the genomic region may be responsible for the downregulation of miR-486-5p. Furthermore, miR-486-5p is located in a CpG island on chromosome 2q35. Epigenetic silencing through DNA methylation and/or histone deacetylation may also lead to miR-486-5p downregulation. Nevertheless, further studies are required to evaluate causes of miR-486-5p dysgulation in carcinogenesis of NSCLC.
ARHGAP5 is identified as a target of miR-486-5p. However, the antioncogenic properties of miR-486-5p downregulation might not solely be explained by its ability to regulate the single gene alone, because a single miRNA could regulate numerous genes in tumorigenesis.19 Indeed, using bioinformatic prediction analysis, we identified at least eight other potential targets of miR-486-5p, including some cancer-related genes. For example, OLFM4 was recently proposed as a biologically relevant miR-486-5p target in the context of gastric cancer.16 Furthermore, miR-486-5p has been shown to target PTEN in muscle cells.20 PTEN is a major negative regulator of the PI3-kinase pathway, which regulates growth, survival and proliferation.21 PTEN has been identified as a tumor-suppressor that is mutated in a large number of cancers, including NSCLC.22,23 Therefore, we cannot exclude the possibility that these candidate targets for miR-486-5p other than ARHGAP5 could be involved in tumor-suppressive function of miR-486-5p. We are exploring the correlation between miR-486-5p and other target candidates and determining whether miR-486-5p can biologically regulate the potential targets in a different study. On the other hand, bioinformatic analysis suggests that the ARHGAP5 may be targeted by more than 10 different miRNAs (Supplementary Table 4), implying that other miRNAs may also act to mediate function of ARHGAP5 in lung tumorigenesis. For example, miR-15a is one of the miRNAs that are predicted as candidates to regulate ARHGAP5. Interestingly, miR-15a has previously been identified as a tumor-suppressor in chronic lymphocytic leukemia, multiple myeloma and acute myeloid leukemia by regulating cell proliferation.28 Therefore, future studies to identify additional novel targets of miR-486-5p and other miRNAs that can also regulate ARHGAP5 will allow us to have deep understanding mechanisms underlying the development and progression of NSCLC.
In conclusion, reduced miR-486-5p expression frequently exists in lung tumor, and is closely associated with progression and metastasis of NSCLC. MiR-486 could act as a tumor-suppressor in the development and progression of NSCLC through targeting ARHGAP5. With more understanding its function, miR-486-5p may be used as a potential metastatic/prognostic marker and therapeutic target in treatment of NSCLC.
MATERIALS AND METHODS
Surgical tissue specimens
The study protocol was approved by the Institutional Review Boards of Tumor Hospital of Hebei Medical University and University of Maryland School of Medicine. The frozen surgical tumor and corresponding normal lung tissues of 76 patients with NSCLC were obtained. Demographic and clinical characteristics of the patients are shown in Table 1. The 76 NSCLC patients consist of 20 females and 56 males, ages 54–83 years (median, 69 years). Thirty-nine patients were diagnosed with adenocarcinoma and 37 with squamous cell carcinoma. Nineteen patients had stage I disease, 33 patients had stage II disease, and 24 patients had stage III disease. Furthermore, FFPE sections of lung tumor and corresponding normal lung tissues of 54 NSCLC patients were also collected (Supplementary Table 3). All variants, including age, sex, stage and lymph node metastasis, were obtained from clinical and pathologic records. None of the patients had received preoperative adjuvant chemotherapy or radiotherapy.
Cell culture
Human NSCLC cell lines (H460, A549, H1944, H358, H157, H1792, H226, H292, and H522) were obtained from the American Type Culture Collection. Cells were maintained at 37 °C in a humidified air atmosphere containing 5% carbon dioxide in RPMI1640 (MGC-803, HGC-27), F12 (AGS) or Dulbecco’s Modified Eagle’s Media (HEK293T) supplemented with 10% fetal bovine serum.
RNA extraction and quantitative real-time PCR
Total RNA from cultured cells and frozen tissue specimens was extracted using a mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA) according to the manufacturer’s instructions. Total RNA from 4 μm-thick FFPE sections was isolated using a RecoverAll Total Nucleic Acid Isolation Kit (Ambion) optimized for FFPE samples according to the manufacturer’s instructions. qRT–PCR assays were carried out to detect miRNA expression using Taqman miRNA assays (Applied Biosystems, Foster City, CA, USA) as described in our previous study.3–5 U6 small nuclear RNA was used as an internal control. Furthermore, ARHGAP5 transcript expression was measured by using qRT–PCR with forward primer 5′-(CATCTGTTTTTGGCCAACCT)-3′ and reverse primer 5′-(GTGGAGGAGCCA CAATGTTT)-3′. To determine ARHGAP5 transcript expression level, β-Actin was used as an internal control. Experiments were repeated at least three times.
Enforcing or reducing expressions of miR-486-5p in NSCLC cells
To force expression of miR-486-5p in cancer cells, cells were transfected with precursor molecules mimicking miR-486-5p (Ribobio Co., Guangzhou, China) or scrambled sequence by using LipoFectamine 2000 (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions. To reduce expression of miR-486-5p, an inhibitor of miR-486 or negative inhibitor control (Ambion) was transfected into cancer cells by using HiPerFect transfection reagent (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Experiments were repeated at least three times.
Small interfering RNA silencing of ARHGAP5
Sequences of small interfering RNA specifically against ARHGAP5 (si-ARHGAP5) are 5′-AGAUCAUAAUAUCAAUCUATT-3′. Transfections were performed using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s protocol with si-ARHGAP5 (Genephma, Shanghai, China) or scrambled sequences. At least three independent experiments were carried out.
Bioinformatics
Two software programs, TargetScan 5.2 (Release 5.2, June 2011; http://www.targetscan.org/) and miRecords (http://mirecords.biolead.org/), were used to predict the potential target genes of miR-486-5p.
ARHGAP5 gene 3′-UTR luciferase reporter assay
To create 3′-UTR luciferase reporter construct of ARHGAP5, 1757-bp sequences from putative miR-486-5p-binding sites were synthesized and ligated into the pGL3-REPORT vector (Promega, Shanghai, China). The following primers were used to amplify the 3′-UTR of ARHGAP5: 5′-CCG ACGCGTATGCTTGTGGCTAAAGTGAGT-3′ and 5′-CCGCTCGAGAAACAGTGAAACCTTCCAGTAA-3′. The amplified fragment was cloned into pGL3 luciferase report vector at Mlu I and Xho I sites. The sequence of plasmid (pGL3-ARHGAP5) was confirmed by DNA sequencing. Cancer cells (5 × 104 per well) were seeded in a 24-well plate the day before transfection, and then co-transfected with firefly luciferase-3′-UTR (pGL3-ARHGFAP5, 500 ng) and pRL-TK vector (Promega) along with miR-486-5p mimics or control (Ribobio Co.). After 2 days, firefly luciferase and Renilla luciferase were measured by using synergy HT microplate reader (Biotek, Beijing, China) with the Dual-Glo Luciferase assay system (Promega). Luciferase activities were normalized to Renilla luciferase activity. Experiments were repeated at least three times.
Methylthiazol tetrazolium assay
Methylthiazol tetrazolium assay was performed as previously described.29,30 Briefly, after transfection, cells were plated in 96-well plates, and the cells viability was assessed in ten replicates. The experiments were done at least three times.
Colony formation assay
For colony formation assays, after 24-hour post-transfection, the cells were diluted and replated in six-well plates. After 10 days, visible colonies were fixed with methanol, stained with crystal violet, counted and normalized to the control group. The experiments were performed at least three times.
BrdU incorporation assay
Cells were plated on coverslips. BrdU (10 μM) was added to the culture medium for 12 h. Cells were fixed in 4% paraformaldheyde for 10 min, washed with phosphate-buffered saline, and incubated with HCl 2 N for 2 min. Immunocytofluorescence was performed on cells with mouse anti BrdU antibody (Dako, Carpinteria, CA, USA), and the fluorochrome conjugated secondary antibody against mouse Ig (Invitrogen). DAPI was used to counterstain the nuclei. Immunostained cells were analyzed under fluorescent microscope (Leica, Solms, Germany). Results were expressed as the ratio of BrdU- to DAPI-positive cells. The experiments were performed at least three times.
Annexin V apoptosis assay
Cells were stained with annexin V fluorescein isothiocyanate (V-FITC) and propidium iodide (PI) using the Annexin V-FITC Kit (Beckman Colter, Boulevard Brea, CA, USA) for flow cytometric analysis. The apoptotic index was calculated as the percentage of annexin V+/PI-cells.
Wound-healing assay
To determine cell migration, cells were seeded in six-well plates and incubated to generate confluent cultures. Wounds were scratched in the cell monolayer using a 200 micropipette tip. The cells were rinsed with phosphate-buffered saline. The migration of the cells at the edge of the scratch was monitored at time 0, 24 and 48 h, respectively. The cells were stained and photographed. At least three independent experiments were carried out.
Transwell assay
To determine cell invasion and migration, after transfection, cells were plated in medium without serum in the top chamber of a transwell (Corning, Horseheads, NY, USA). The bottom chamber contained standard medium with 10% fetal bovine serum. After 24-h incubation, the cells that had migrated to the lower surface of the membrane were fixed with formalin, stained with crystal violet and photographed under microscope. Cell numbers were counted under a light microscope at × 400 magnification. Experiments were carried out at least three times.
In vivo metastatic assay
H460-luc2, a luciferase expressing NSCLC cell line stably transfected with firefly luciferase gene (luc2) was obtained from Caliper Life Sciences, Inc. (Hopkinton, MA, USA). 5 × 106 H460-luc2 cells transfected with miR-486-5p mimic or scrambled sequence using LipoFectamine 2000 (Invitrogen). The cancer cells were injected via the tail vein into ten athymic Swiss mice per group, respectively. D-Luciferin (Xenogen Co., Alameda, CA, USA) was injected into animals at a dosage of 150 mg/kg body weight for luciferin in vivo imaging by using IVIS 200 Imaging System (Xenogen Corp.) as described in our previous work.30 We monitored in vivo tumor formation by bioluminescence imaging in 1, 2, 3, 4, 5, 6 and 7 weeks after injection of the H460-luc2 cells, because bioluminescence is a sensitive and cost-effective approach to quantify in vivo tumors that are not grossly measurable.10 Furthermore, we used mean photon counts of bioluminescence to evaluate malignant lesions in ten mice per group as previously described.10,30 In addition, mice were killed at 8 weeks for full autopsy. All tumors were excised, weighed, harvested, fixed and embedded. The average weight of tumors was calculated and compared between the groups. Tissue specimens were also stained by hematoxylin and eosin for histological study.
Western blot
Total proteins (100 μg) extracted from cell lines and tissues were analyzed by SDS–polyacrylamide gel electrophoresis and were transferred electrophoretically to nitrocellulose membrane. To evaluate expression of ARHGAP, blots were blocked with 5% non-fat milk in Tris-Buffered Saline and Tween 20, and incubated with a primary rabbit monoclonal antibody, p190-B (Epitomics, Inc., Hangzhou, China). The monoclonal p190-B antibody was produced by immunizing animals with a synthetic peptide corresponding to residues in human p190-B RhoGAP. Furthermore, the specificity of the antibody for p190-B RhoGAP was confirmed by the company (Epitomics). In addition, the antibody produced a single clean band of 190 kDa in Western blot analysis of surgical human lung tissue specimens (Supplementary Figure 8). Antibody for β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a control. The blots were then reprobed with secondary antibody and visualized by the ECL system (GE Healthcare Life Sciences, Piscataway, NJ, USA).
Immunohistochemistry assay
Immunohistochemistry staining for determining ARHGAP5 expression on human lung tissue specimens was done on FFPEs using p190-B antibody (Epitomics). As a negative control, primary antibody was replaced by phosphate-buffered saline. All sections were examined and scored independently by two investigators without any knowledge of the clinicopathological data of the patients. At least five fields per slide were randomly chosen for analysis of immunohistochemistry staining. The immunohistochemistry staining was evaluated according to the intensity of reactivity using a four-tier system: 0, no staining (−); 1, weak staining ( + ); 2, moderate staining ( + + ); and 3, strong staining ( + + + ).
To assess cell proliferation status on tissue sections of tumors excised from mice, the mouse-anti-human Ki-67 monoclonal antibody (Dako, Glostrup, Denmark) was applied to determine nuclear expression of Ki-67 as described in our previous report.31
In situ detection of apoptosis by terminal nucleotidyl transferase-mediated nick end labeling assay
In situ terminal nucleotidyl transferase-mediated nick end labeling assay was carried out on 4-μm thick sections of tumor specimens from mice for evidence of the damaged DNA characteristic of apoptotic cells as previously described.32 Cells for apoptosis were identified as dark brown nuclei under a microscope. The number of apoptotic cells was counted from a minimum of five fields with >1000 cells/field.
Statistical analysis
The differences of in vitro results between groups were analyzed by using Student t-test when there were only two groups, or evaluated by one-way analysis of variance when there were more than two groups. We used mean photon counts of bioluminescence to evaluate malignant lesions in all 10 mice per group as previously described.29,30 To evaluate differences of tumor metastasis of mice between two groups, statistical significance of differences of photon counts was assessed by two-sided Student’s t-tests. Difference of miR-486-5p expressions between tumor tissues and normal lung tissues of human subjects was calculated by a two-tailed independent samples t-test. Spearman’s correlation analysis was used to determine associations between miR-486-5p expression and clinical characteristics of the NSCLC patients, and correlation between miR-486-5p and ARHGAP5 expressions. In all cases, a P-value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by Grants 81101482 from the National Natural Science Foundation of China and Grants C2011206001 from the Natural Science Foundation of Hebei province, China (to LX) and American Cancer Society-Research Scholar Grant in Basic, Preclinical, Clinical and Epidemiology Research-115154AF109040, National Cancer Institute (NCI)-R01CA161837-01, an exploratory research grant from the Maryland Stem Cell Research Fund, and VA-Merit grant-I01 CX000512-01 (to FJ).
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Tarver T. Cancer facts & figures 2012. American Cancer Society (ACS) J Consumer Health Internet. 2012;16:366–367. [Google Scholar]
- 2.Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 3.Yu L, Todd NW, Xing L, Xie Y, Zhang H, Liu Z, et al. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int J Cancer. 2010;127:2870–2878. doi: 10.1002/ijc.25289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xing L, Todd NW, Yu L, Fang H, Jiang F. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod Pathol. 2010;23:1157–1164. doi: 10.1038/modpathol.2010.111. [DOI] [PubMed] [Google Scholar]
- 5.Shen J, Liu Z, Todd NW, Zhang H, Liao J, Yu L, et al. Diagnosis of lung cancer in individuals with solitary pulmonary nodules by plasma microRNA biomarkers. BMC Cancer. 2011;11:374. doi: 10.1186/1471-2407-11-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 7.Saito Y, Jones PA. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle. 2006;5:2220–2222. doi: 10.4161/cc.5.19.3340. [DOI] [PubMed] [Google Scholar]
- 8.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 9.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188–6196. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins DE, Oei Y, Hornig YS, Yu SF, Dusich J, Purchio T, et al. Bioluminescent imaging (BLI) to improve and refine traditional murine models of tumor growth and metastasis. Clin Exp Metastasis. 2003;20:733–744. doi: 10.1023/b:clin.0000006815.49932.98. [DOI] [PubMed] [Google Scholar]
- 11.Chakravarty G, Hadsell D, Buitrago W, Settleman J, Rosen JM. p190-B RhoGAP regulates mammary ductal morphogenesis. Mol Endocrinol. 2003;17:1054–1065. doi: 10.1210/me.2002-0428. [DOI] [PubMed] [Google Scholar]
- 12.Gen Y, Yasui K, Zen K, Nakajima T, Tsuji K, Endo M, et al. A novel amplification target, ARHGAP5, promotes cell spreading and migration by negatively regulating RhoA in Huh-7 hepatocellular carcinoma cells. Cancer Lett. 2009;275:27–34. doi: 10.1016/j.canlet.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 13.Tan X, Qin W, Zhang L, Hang J, Li B, Zhang C, et al. A 5-microRNA signature for lung squamous cell carcinoma diagnosis and hsa-miR-31 for prognosis. Clin Cancer Res. 2011;17:6802–6811. doi: 10.1158/1078-0432.CCR-11-0419. [DOI] [PubMed] [Google Scholar]
- 14.Bansal A, Lee IH, Hong X, Anand V, Mathur SC, Gaddam S, et al. Feasibility of mcroRNAs as biomarkers for Barrett’s Esophagus progression: a pilot cross-sectional, phase 2 biomarker study. Am J Gastroenterol. 2011;106:1055–1063. doi: 10.1038/ajg.2011.37. [DOI] [PubMed] [Google Scholar]
- 15.Ragusa M, Majorana A, Statello L, Maugeri M, Salito L, Barbagallo D, et al. Specific alterations of microRNA transcriptome and global network structure in colorectal carcinoma after cetuximab treatment. Mol Cancer Ther. 2010;9:3396–3409. doi: 10.1158/1535-7163.MCT-10-0137. [DOI] [PubMed] [Google Scholar]
- 16.Oh HK, Tan AL, Das K, Ooi CH, Deng NT, Tan IB, et al. Genomic loss of miR-486 regulates tumor progression and the OLFM4 antiapoptotic factor in gastric cancer. Clin Cancer Res. 2011;17:2657–2667. doi: 10.1158/1078-0432.CCR-10-3152. [DOI] [PubMed] [Google Scholar]
- 17.Midorikawa Y, Yamamoto S, Tsuji S, Kamimura N, Ishikawa S, Igarashi H, et al. Allelic imbalances and homozygous deletion on 8p23. 2 for stepwise progression of hepatocarcinogenesis. Hepatology. 2009;49:513–522. doi: 10.1002/hep.22698. [DOI] [PubMed] [Google Scholar]
- 18.Jiang F, Yin Z, Caraway NP, Li R, Katz RL. Genomic profiles in stage I primary non small cell lung cancer using comparative genomic hybridization analysis of cDNA microarrays. Neoplasia. 2004;6:623–635. doi: 10.1593/neo.04142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 20.Small EM, O’Rourke JR, Moresi V, Sutherland LB, McAnally J, Gerard RD, et al. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc Natl Acad Sci USA. 2010;107:4218–4223. doi: 10.1073/pnas.1000300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 22.Teng DH, Hu R, Lin H, Davis T, Iliev D, Frye C, et al. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res. 1997;57:5221–5225. [PubMed] [Google Scholar]
- 23.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23. 3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 24.Heckman-Stoddard BM, Vargo-Gogola T, McHenry PR, Jiang V, Herrick MP, Hilsenbeck SG, et al. Haploinsufficiency for p190B RhoGAP inhibits MMTV-Neu tumor progression. Breast Cancer Res. 2009;11:R61. doi: 10.1186/bcr2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sordella R, Classon M, Hu KQ, Matheson SF, Brouns MR, Fine B, et al. Modulation of CREB activity by the Rho GTPase regulates cell and organism size during mouse embryonic development. Dev Cell. 2002;2:553–565. doi: 10.1016/s1534-5807(02)00162-4. [DOI] [PubMed] [Google Scholar]
- 26.McHenry PR, Sears JC, Herrick MP, Chang P, Heckman-Stoddard BM, Rybarczyk M, et al. P190B RhoGAP has pro-tumorigenic functions during MMTV-Neu mammary tumorigenesis and metastasis. Breast Cancer Res. 2010;12:R73. doi: 10.1186/bcr2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakahara H, Mueller SC, Nomizu M, Yamada Y, Yeh Y, Chen WT. Activation of beta1 integrin signaling stimulates tyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J Biol Chem. 1998;273:9–12. doi: 10.1074/jbc.273.1.9. [DOI] [PubMed] [Google Scholar]
- 28.Gao SM, Xing CY, Chen CQ, Lin SS, Dong PH, Yu FJ. miR-15a and miR-16-1 inhibit the proliferation of leukemic cells by down-regulating WT1 protein level. J Exp Clin Cancer Res. 2011;30:110. doi: 10.1186/1756-9966-30-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan T, Li R, Todd NW, Qiu Q, Fang HB, Wang H, et al. Up-regulation of 14-3-3zeta in lung cancer and its implication as prognostic and therapeutic target. Cancer Res. 2007;67:7901–7906. doi: 10.1158/0008-5472.CAN-07-0090. [DOI] [PubMed] [Google Scholar]
- 30.Mei YP, Liao JP, Shen J, Yu L, Liu BL, Liu L, et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene. 2012;31:2794–2804. doi: 10.1038/onc.2011.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Zhang Z, Li R, Ang KK, Zhang H, Caraway NP, et al. Overexpression of S100A2 protein as a prognostic marker for patients with stage I non small cell lung cancer. Int J Cancer. 2005;116:285–290. doi: 10.1002/ijc.21035. [DOI] [PubMed] [Google Scholar]
- 32.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.