

Abstract
Many tumor suppressor genes (TSGs) are silenced through synergistic layers of epigenetic regulation including abnormal DNA hypermethylation of promoter CpG islands, repressive chromatin modifications and enhanced nucleosome deposition over transcription start sites. The protein complexes responsible for silencing of many of such TSGs remain to be identified. Our previous work demonstrated that multiple silenced TSGs in colorectal cancer cells can be partially reactivated by DNA demethylation in cells disrupted for the DNA methyltransferases 1 and 3B (DNMT1 and 3B) or by DNMT inhibitors (DNMTi). Herein, we used proteomic and functional genetic approaches to identify additional proteins that cooperate with DNMTs in silencing these key silenced TSGs in colon cancer cells. We discovered that DNMTs and the core components of the NuRD (Mi-2/nucleosome remodeling and deacetylase) nucleosome remodeling complex, chromo domain helicase DNA-binding protein 4 (CHD4) and histone deacetylase 1 (HDAC1) occupy the promoters of several of these hypermethylated TSGs and physically and functionally interact to maintain their silencing. Consistent with this, we find an inverse relationship between expression of HDAC1 and 2 and these TSGs in a large panel of primary colorectal tumors. We demonstrate that DNMTs and NuRD cooperate to maintain the silencing of several negative regulators of the WNT and other signaling pathways. We find that depletion of CHD4 is synergistic with DNMT inhibition in reducing the viability of colon cancer cells in correlation with reactivation of TSGs, suggesting that their combined inhibition may be beneficial for the treatment of colon cancer. Since CHD4 has ATPase activity, our data identify CHD4 as a potentially novel drug target in cancer.
Keywords: NuRD, DNMTs, tumor suppressor genes, silencing, colon cancer, WNT signaling
INTRODUCTION
Cancer often results from a combination of activation of oncogenes and loss of function of tumor suppressor genes (TSGs). In many cases, the activity of TSGs is not lost by mutation, but rather these genes are silenced through epigenetic mechanisms which include DNA methylation, histone modifications and nucleosome remodeling, that often act in concert to provide transcriptional repression.1-3 The resultant chromatin landscape can include histone hypoacetylation and methylation of H3K9, H3K27 and H4K20.2 Hypermethylation of promoter CpG islands often occurs in conjunction with this repressive chromatin environment and frequently coincides with nucleosome deposition over transcription start sites, leading to occlusion of transcription factor binding sites and impedance of transcription initiation.4-6
The mechanism of transcriptional repression mediated by the DNA methylation machinery involves with both methylated DNA and the DNA methyltransferase (DNMT) proteins.7 Methylated DNA is recognized by methyl-CpG binding domain proteins (MBDs) such as MBD2 and MeCP2, which guide protein complexes with chromatin remodeling and/or histone modifying activity, including the SIN3A, CoREST, SWI-SNF and NuRD (Mi-2/nucleosome remodeling and deacetylase) complex, to specific sequences in the genome.8-11 The subunits of the NuRD complex include the helicase-like ATPases CHD3/4 (chromo domain helicase DNA-binding protein 3/4), histone deacetylases HDAC1/2, the metastasis-associated proteins MTA1/2/3 and histone chaperone proteins RBBP4/7 and GATAD2A/B, and methyl-DNA binding proteins MBD2/3, of which MBD2 recruits NuRD to hypermethylated sequences to reposition nucleosomes.11-13 In addition to its own transcriptional repression activity, the DNMTs can also function as scaffolds to recruit other repressor proteins. For example, DNMTs can cooperate with the ATP-dependent chromatin remodeler LSH in transcriptional repression.14 Conversely, proteins that interact with or modify histones may recruit the DNA methylation machinery to aid in transcriptional regulation. Several Polycomb Group complex constituents, such as EZH2 and CBX7, may recruit DNMTs to cooperate in stable silencing of Polycomb Group-target genes.15,16 DNA-binding transcriptional factors such as the oncogenic fusion protein PML-RARα can recruit DNMTs, Polycomb Group proteins and the NuRD complex to induce transcriptional repression of the TSG RARβ.17,18
Given that TSGs are often inactivated by synergistic layers of epigenetic regulation, effective reactivation of silenced TSGs may require inhibition of multiple epigenetic processes. Many silenced TSGs that control critical regulatory pathways in colorectal tumors, including the secreted frizzled-related protein (SFRP) family, which encode for potent inhibitors of the WNT signaling pathway, and the tissue inhibitor of metalloproteinase 3 (TIMP3), which inhibits metastasis, are partially reactivated in the colorectal carcinoma cell line HCT116 cells that are hypomorphic for the maintenance DNA methyltransferase DNMT1 (~10% expression) and deleted for the de novo methyltransferase DNMT3B (DKO) or by drugs that both inhibit and deplete DNMTs such as 5-aza-2′-deoxycytidine (DAC), in association with promoter demethylation.19 All these results suggest that DNMTs have a major role in the maintenance of TSG silencing. Our earlier work demonstrated that HDACi trichostatin A (TSA) and DNMT inhibitors (DNMTi) synergistically reactivate many of the above-mentioned TSGs when combined.20,21 We previously found that TSGs, which are only partially DNA methylated and not fully silenced, but expressed at low levels, are induced by TSA treatment alone, whereas more fully DNA methylated and silenced genes cannot be reactivated by TSA alone.20,21 However, all of these TSGs can be partially reactivated by DNMTi and fully reactivated by combining DNMTi and HDACi, suggesting that DNMTs and yet to be identified HDAC(s) cooperate in the maintenance of TSG silencing.
In the present study, we have used two independent approaches to identify the protein complexes that cooperate with DNMTs in repression of above-mentioned TSGs in colorectal cancer (CRC) cell lines. We demonstrate a novel cooperation between DNMTs and the chromatin remodeling complex NuRD, which maintains the aberrant silencing of key TSGs including SFRPs and TIMP3. As such, our findings demonstrate that these TSGs are silenced by three synergistic layers of epigenetic regulation. Our work also identifies HDAC1 and 2 as the relevant drug targets among the larger HDAC family and identifies CHD4 as a potentially novel therapeutic target.
RESULTS
DNMT inhibition and knockdown of HDAC1 and 2 synergize in reactivating TSGs
We previously conducted a genomic screen for genes upregulated by DAC and TSA in the human CRC cell line RKO.21 The genes upregulated by the combined DAC and TSA treatment include TIMP2, TIMP3 and SFRP1. Further work revealed that three other members of the SFRP gene family (SFRP2, SFRP4 and SFRP5) are also methylated and silenced in RKO and HCT116 cells.22 Interestingly, the expression of TIMP3 and SFRP1/2/4/5 is restored in HCT116 DKO cells in which two DNMTs (DNMT1 and DNMT3B) are genetically disrupted and DNA methylation is almost depleted. This result suggested that the epigenetic silencing of these TSGs largely relies on the DNMTs and/or DNA methylation.22
In the present study, to further characterize the molecular mechanism of the cooperation between DNA hypermethylation and histone deacetylation in TSG silencing, we selected SFRPs and TIMPs as our guide genes as they are defined DNA hypermethylated genes in RKO and HCT116 cells. According to their different responses to TSA, we divided these genes into two groups. Group 1 genes (SFRP1 and TIMP2) could be partially reactivated by treatment with TSA alone, whereas group 2 genes (TIMP3, SFRP2 and SFRP5) could not be activated by TSA alone (Figure 1a). Both groups could be reactivated in a synergistic fashion by the combined treatment (Figure 1a). We set out to identify the TSA-sensitive HDAC(s) responsible for this epigenetic silencing. RKO cells were transfected with siRNA pools targeting 11 class I and II HDACs and cultured in the absence or presence of DAC. We gathered data for TSG reactivation for those siRNA pools that induced >70% knockdown of the target HDACs (Figure 1a; Supplementary Figure S1A). Based on our cutoff, there was some basal expression of group 1 genes SFRP1 and TIMP2. Concordantly, group 1 but not group 2 TSGs could be partially reactivated by TSA treatment alone but also by depletion of HDAC1, which was enhanced further by concomitant knockdown of HDAC2 (Figure 1a: group 1). DAC treatment in combination with depletion of HDAC1 resulted in a strongly increased reactivation of both group 1 and group 2 TSGs tested, and this was enhanced even further when HDAC2 was simultaneously knocked down, indicating a major role for these two HDACs in the silencing of our selected TSGs (Figure 1a: group 2; Supplementary Figure S1B). All siRNAs targeting HDAC1 and HDAC2 potently knocked down their target mRNA, and each HDAC1 siRNA reactivated TSGs, arguing against off-target effects (Supplementary Figures S1C and D). However, as 70% knockdown of some HDACs may be insufficient to result in a loss-of-function phenotype, we cannot exclude the possibility that other HDACs may also cooperate with DNMTs to mediate epigenetic TSG silencing.
Figure 1.
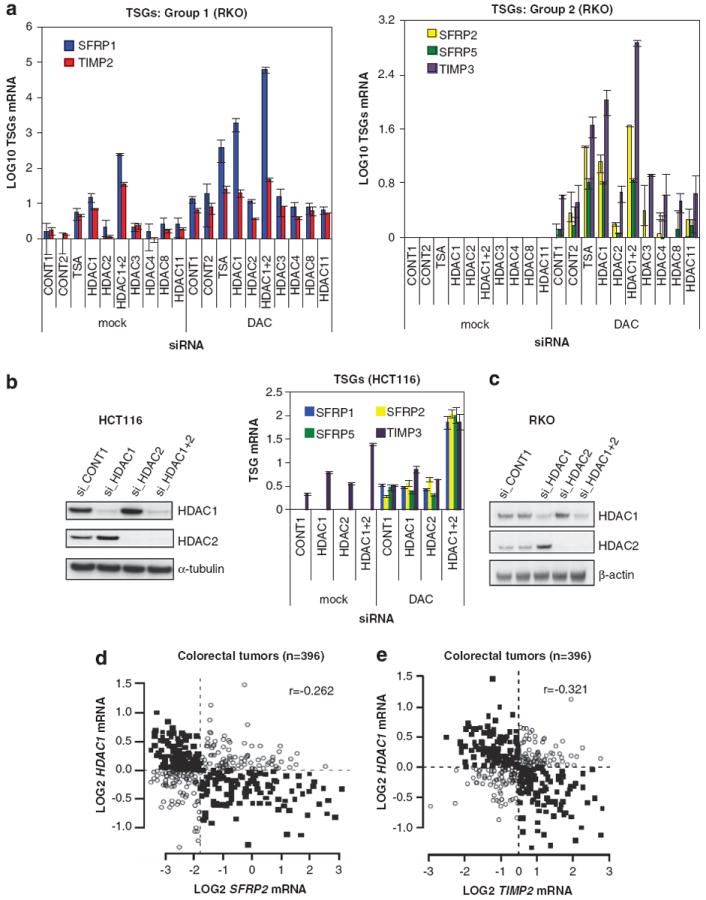
DNMT inhibition and knockdown of HDAC1 and 2 synergize in reactivating silenced TSGs. (a) DNMT inhibition and knockdown of HDAC1 and 2 synergize in reactivation of TSGs. RKO cells were transfected with scrambled siRNAs (CONT1 and 2) or siRNA pools targeting HDAC1-11, split and then treated with or without 1 μm DAC. Only the HDAC siRNA pools that induced >70% knockdown were included in the analysis. RKO cells were also treated with 300 nm TSA in the absence and presence of DAC. Expression of indicated TSGs was measured by QRT-PCR and Log10 transformed, using the lowest Ct value measured (see Materials and methods). Error bars denote s.d. See also Supplementary Figure S1A. (b) Depletion of HDAC1 and 2 enhances DAC-induced reactivation of TSGs in HCT116 cells. HCT116 cells were transfected with CONT1, HDAC1 and/or HDAC2 siRNA pools, split and treated with or without 100 nm DAC. Knockdown was verified by analyzing HDAC1 and HDAC2 protein expression by western blotting, α-tubulin serves as a loading control (left panel). Expression of indicated TSGs was measured by QRT-PCR (right panel). Error bars denote s.d. (c) HDAC1 and HDAC2 siRNA pools induce depletion of HDAC1 and HDAC2 protein levels. RKO cells were transfected with scrambled, HDAC1 and/or HDAC2 siRNA pools. HDAC1 and HDAC2 protein expression was analyzed by western blotting, β-actin serves as a loading control. (d, e) Inverse correlation of HDAC1 and expression of TSGs. Correlation plots of HDAC1 and SFRP2 (d) or TIMP2 (e) were drawn using gene expression data sets of 396 colorectal tumors.25 Expression levels are indicated as Log2 ratios against a colon cancer reference pool. Median expression levels are indicated by the dashed lines. Solid square symbols represent discordant binary expression (low and high levels) and open circles indicate concordant expression between HDAC1 and TSGs. See also Table 1.
We also examined the same panel of TSGs in HCT116 cells, except TIMP2, which is not DNA hypermethylated and is basally expressed.21 Similarly to RKO cells, the depletion of both HDAC1 and HDAC2 in HCT116 cells acted in synergy with DAC in the reactivation of the four TSGs tested (Figure 1b). We note that, although their combined depletion also enhanced DAC-induced TSG reactivation, knockdown of HDAC1 or HDAC2 alone was not sufficient to reactivate TSGs in HCT116 cells (Figure 1b). In this regard, and consistent with previous studies23 this may be explained, in part, by our finding of a reciprocal compensatory mechanism linking HDAC1 and HDAC2. Thus, knockdown of HDAC1 leads to induction of HDAC2 protein levels and vice versa (Figures 1b and c).23,24 Alternatively, the threshold of HDAC1/2 proteins to maintain TSG silencing may be different across different cancer cell lines. We conclude that depletion of HDAC1 and 2 acts in a synergistic fashion with DAC to reactivate our selected TSGs in CRC cells.
An inverse relationship between HDAC1 and 2 expression and TSGs
Next, we studied expression of HDAC1 and 2 and our panel of TSGs in 396 early stage primary CRCs.25 Among the seven TSGs we examined, we found a statistically significant inverse relationship between HDAC1 expression and five TSGs and a similar inverse relationship between HDAC2 expression and three TSGs (Table 1; Figures 1d and e). These data demonstrate a strong inverse relationship between HDAC1 and to lesser extent HDAC2 and expression of key TSGs, in concordance with our finding that HDAC1 has a significant role in TSG silencing in RKO cells. Interestingly, HDAC1 is overexpressed in many cancers and this increased expression was associated with poor clinical outcome.26,27 We hypothesize that HDAC1 overexpression may contribute to tumorigenesis by repression of key TSGs.
Table 1.
Inverse correlation of HDAC1 and 2 and expression of TSGs
| Gene | n |
HDAC1
|
HDAC2
|
||
|---|---|---|---|---|---|
| r | P-value | r | P-value | ||
| SFRP1 | 396 | −0.183 | 2.48e–04 | −0.137 | 6.2e–03 |
| SFRP2 | 396 | −0.262 | 1.17e–07 | −0.054 | 0.28 |
| SFRP5 | 396 | −0.102 | 4.17e–02 | −0.063 | 0.21 |
| TIMP2 | 396 | −0.321 | 6.25e–11 | −0.237 | 1.83e–06 |
| TIMP3 | 396 | −0.193 | 1.10e–04 | −0.051 | 0.31 |
| CDKN2A | 396 | 0.033 | 0.52 | −0.132 | 8.65e–03 |
| MLH1 | 396 | 0.015 | 0.76 | 0.274 | 2.95e–08 |
Abbreviations: HDAC1, histone deacetylase 1; SFRP, secreted frizzled-related protein; TIMP, tissue inhibitor of metalloproteinase; TSGs, tumor suppressor genes. A statistical significant correlation (highlighted in bold) was found between HDAC1 and 2 and indicated TSGs in gene expression data sets of 396 colorectal tumors25 using a Pearson correlation (r) analysis.
HDAC1 requires the NuRD complex for silencing a subset of TSGs HDACs lack substrate specificity for their targets as they do not discriminate between individual lysine residues.28 However, the specificity of HDACs can be guided by association with other proteins. HDAC1 and 2 are core subunits of the repressor complexes such as CoREST and NuRD, which, as discussed previously, can be targeted to methylated DNA. We investigated the involvement of these complexes in TSG silencing by knocking down their essential components. All siRNA pools induced >60% knockdown of their targets (Figure 2a, left). Knockdown of CHD3 or RCOR1 failed to enhance DAC-induced re-expression of three TSGs (Figure 2a, right). However, the CHD4 siRNA pool potently reactivated these silenced TSGs in combination with DAC treatment (Figure 2a; Supplementary Figure S2A). We found three individual CHD4 siRNAs, each of which depleted CHD4 mRNA, but not HDAC1 mRNA, that were able to reactivate TSGs (Supplementary Figures S2B–D). Thus, NuRD is a major suppressor of these three TSGs. Next, we determined whether overexpression of mouse Hdac1, which is not targeted by the human-specific HDAC1#1 siRNA, was able to reconstitute TSG repression in RKO cells depleted for HDAC1 or CHD4. RKO cells were transfected with scrambled siRNA, human HDAC1#1 siRNA or CHD4 siRNA pool, split, treated with DAC and transduced with plasmids overexpressing GFP or mouse Hdac1. Indeed, overexpression of mouse Hdac1 restores the repression of three selected TSGs in RKO cells depleted for human HDAC1 (Figure 2b). Importantly, HDAC1 appears to require the NuRD complex for silencing of these three TSGs, since overexpression of mouse Hdac1 could not restore TSG suppression in cells depleted for CHD4 and treated with DAC (Figure 2b). In summary, these data show that HDAC1/2 functionally cooperate with DNMTs in the silencing of a subset of TSGs, a process in which the NuRD complex plays a major role.
Figure 2.
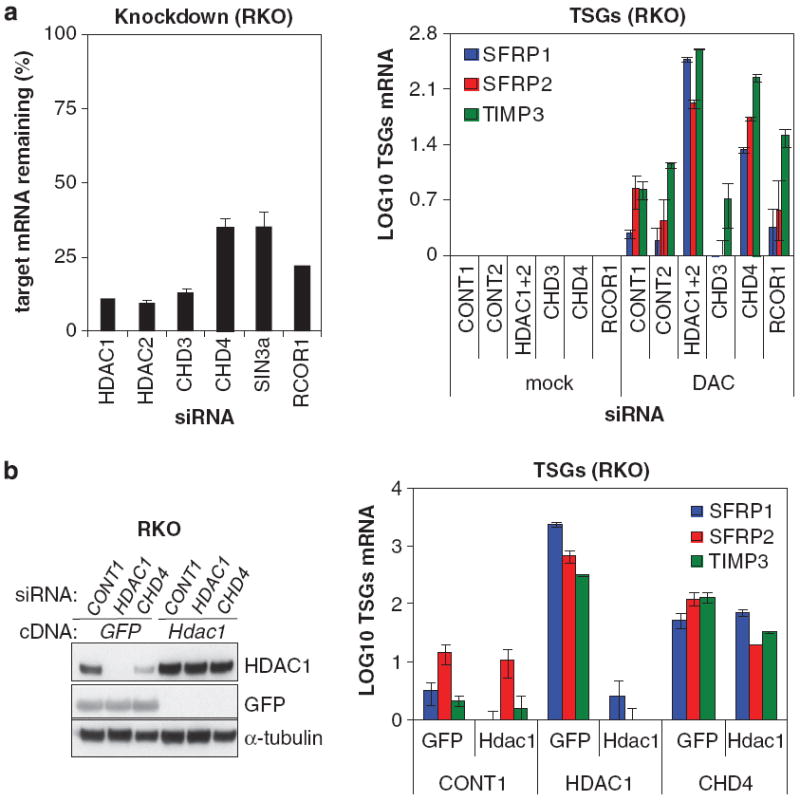
HDAC1 requires the NuRD complex for silencing of TSGs. (a) DNMT inhibition and knockdown of CHD4 induce reactivation of TSGs. RKO cells were transfected with scrambled (CONT1 and 2), HDAC1, HDAC2, CHD3, CHD4 and RCOR1 siRNA pools, split and treated with or without 1 μm DAC. The knockdown abilities of the siRNA pools are depicted as the relative mRNA remaining compared with scrambled siRNA pools (left) Expression of indicated TSGs was analyzed by QRT-PCR and Log10 transformed (right). Error bars denote s.d. (b) HDAC1 requires CHD4 for silencing of TSGs. RKO cells were transfected with CONT1, human-specific HDAC1#1 and CHD4 siRNA pool, split, treated with 1 μm DAC and transduced with plasmids overexpressing GFP or wild-type Hdac1. Expression of mouse Hdac1, human HDAC1 and GFP was analyzed by western blotting with an antibody recognizing GFP and an antibody recognizing both human and mouse HDAC1, α-tubulin serves as a loading control (left panel). Expression of indicated TSGs was determined by QRT-PCR and Log10 transformed (right panel). Error bars denote s.d.
DNMTs physically interact with the NuRD complex
In addition to our siRNA screen, we took a biochemical approach to identify novel protein complexes that cooperate with DNMTs in TSG silencing. Endogenous DNMT1-containing protein complexes were isolated from HCT116 cells by immunoprecipitation and DNMT1-interacting partners were identified by mass spectrometry. Besides known DNMT1 interactors including PCNA, PARP1 and G9a, we detected many components of the NuRD complex: CHD4, MBD3, MTA1/2, GATAD2A/B, suggesting that DNMT1 interacts with NuRD (Figure 3a; Supplementary Table S1).29-31 We confirmed these interactions in reciprocal endogenous co-immunoprecipitation experiments. The NuRD subunits CHD4, HDAC1/2 and MTA1/2 co-immunoprecipitated with DNMT1 (Figure 3b, left). Conversely, DNMT1 co-immunoprecipitated with these NuRD subunits (Figure 3b, right). We also tested whether DNMT3B can associate with NuRD. Because HCT116 cells express very low levels of DNMT3B, we transiently expressed a FLAG-tagged DNMT3B construct in HCT116 DNMT3B KO cells and found that seven NuRD subunits co-immunoprecipitated with exogenous DNMT3B (Figure 3c). Finally, we were also able to find physical interactions between DNMT1 and NuRD in RKO cells (Supplementary Figure S3). Together, these data demonstrate the existence of two novel DNMT–NuRD interactions: DNMT1–NuRD and DNMT3B–NuRD.
Figure 3.
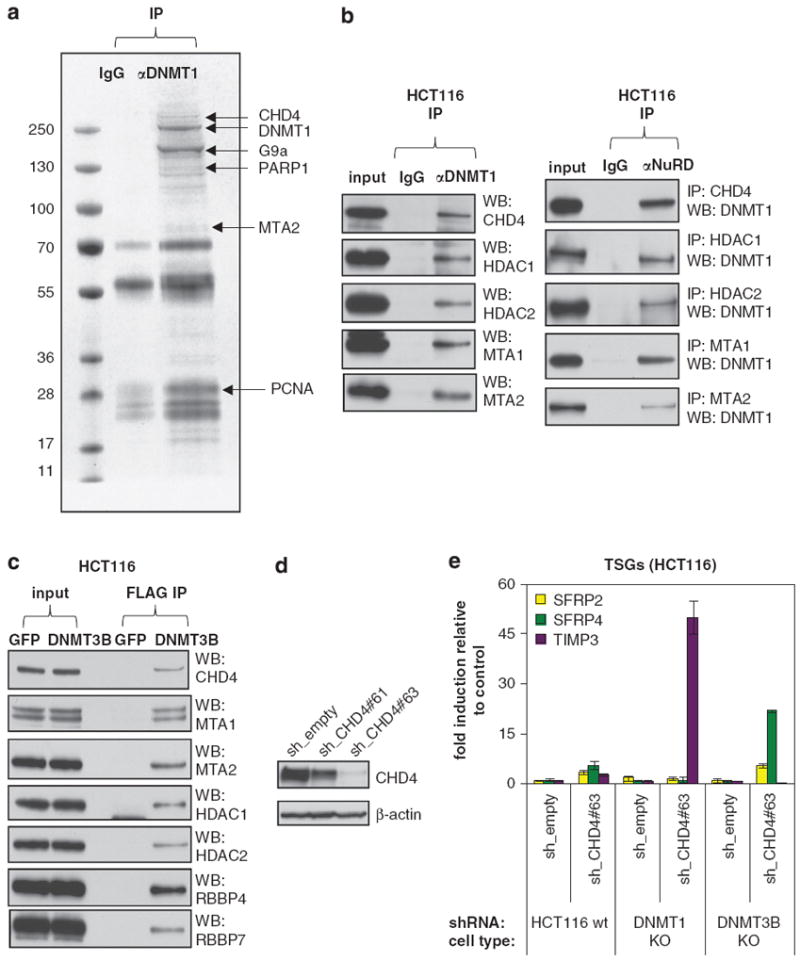
Physical and functional interactions between DNMTs and NuRD. (a) Purification and mass spectrometric analysis of polypeptides associating with DNMT1 identifies NuRD. Endogenous DNMT1 was immunoprecipitated from HCT116 cells, resolved by SDS–PAGE and stained. IgG served as a negative control. Protein bands were retrieved and analyzed by mass spectrometry. The bands are labeled with the identified proteins. See also Supplementary Table S1. (b) Physical interaction between DNMT1 and the NuRD complex. Nuclear extracts from HCT116 cells were immunoprecipitated with antibodies against either DNMT1 (left) or NuRD complex subunits (right). Immunoprecipitates were immunoblotted using the indicated antibodies. (c) Physical interaction between DNMT3B and the NuRD complex. Nuclear extracts from HCT116 DNMT3B KO cells, transfected with vectors overexpressing FLAG-tagged GFP or DNMT3B, were immunoprecipitated with an anti-FLAG M2 affinity gel. The eluted immunoprecipitates were immunoblotted using the indicated antibodies. (d) CHD4 shRNAs induce CHD4 protein depletion. HCT116 cells were transduced with an empty vector or shRNAs targeting CHD4. CHD4 protein levels were analyzed by western blotting, β-actin serves as a loading control. (e) Functional cooperation between CHD4 and either DNMT1 or DNMT3B in silencing TSGs. Wild-type, DNMT1 hypomorphic or DNMT3B −/− HCT116 cells were transduced with an empty vector or a functional shRNA targeting CHD4. Expression of indicated TSGs was analyzed by QRT-PCR and is represented as fold induction over empty vector. Error bars denote s.d.
Differential cooperation between NuRD and DNMTs in the maintenance of TSG silencing
As discussed above, the SFRP family members and TIMP3 are partially reactivated in HCT116 DKO cells.22,32 Because both methylated DNA and DNMT proteins are largely depleted in DKO cells, one important question is whether NuRD can functionally cooperate with DNMT proteins in addition to methylated DNA. To address this question, we took advantage of the HCT116 DNMT1 hypomorphic cells (DNMT1 KO) or DNMT3B KO cells in which one of these two DNMT proteins is largely depleted but the DNA methylation pattern and the silencing status of TSGs are well maintained.19 We then tested whether disruption of the NuRD complex in these two DNMT mutant cell lines can abolish the silencing of our selected TSGs. We analyzed the expression of these TSGs in CHD4 knockdown of the three isogenic HCT116 cell lines (Figures 3d and e). Although we did not observe the synergy of CHD4 KD and DNMT depletion in the reactivation of SFRP1/5 (Supplementary Figure S4), SFRP2 and SFRP4 were strongly enhanced in reactivation by CHD4 depletion in DNMT3B KO but not in DNMT1 KO cells. In contrast, TIMP3 was strongly enhanced in reactivation by CHD4 knockdown in DNMT1 KO cells but not in DNMT3B KO cells. Collectively, these data demonstrate that in addition to its role as a methylated DNA reader, the NuRD complex can cooperate with DNMT1 or DNMT3B protein to maintain the silencing of different subset of TSGs.
DNMTs and NuRD occupy promoters of TSGs
We subsequently investigated whether the NuRD complex and DNMT proteins directly associates with promoters of TSGs by analyzing their occupancy at TSG promoters with chromatin immunoprecipitation (ChIP). We found that DNMT1, DNMT3B, HDAC1 and CHD4 occupy the promoters of all six TSGs tested in both HCT116 and RKO cells (Figures 4a and b). As the ChIP signal of CHD4 is much stronger than the ChIP signals of other proteins, the occupancy of DNMTs to these TSGs is more evident in Supplementary Figure S5 which is the expanded view of Figures 4a and b.
Figure 4.
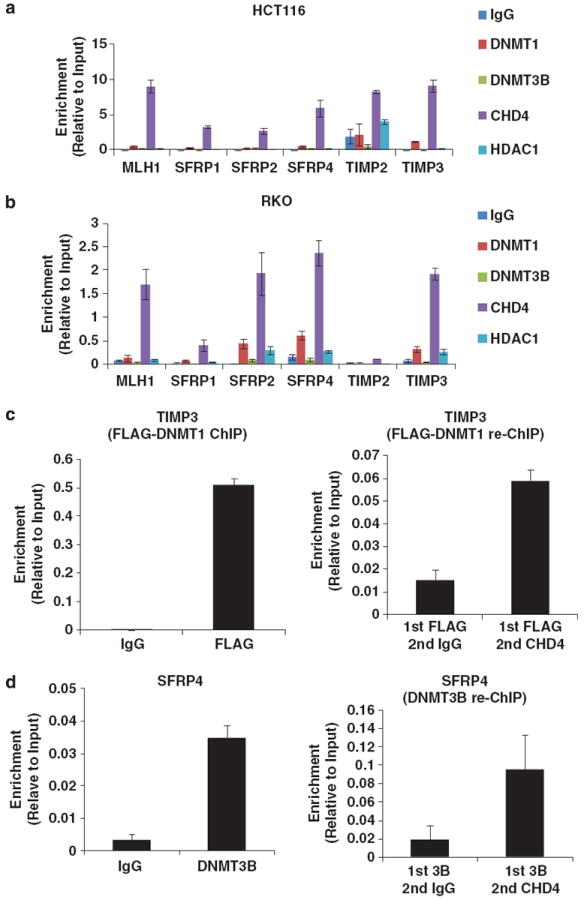
DNMTs and NuRD associate with the promoters of TSG. (a, b) DNMT1, DNMT3B, HDAC1 and CHD4 associate with the promoters of TSGs in HCT116 and RKO cells. Occupancy of DNMT1, DNMT3B, HDAC1 and CHD4 in the proximal promoter region of indicated TSGs in HCT116 cells (a) and RKO cells (b). Results are presented as percentage of input. Rabbit IgG served as a negative control. (c) DNMT1 and the NuRD complex exists in the same protein complex on the TIMP3 promoter. Re-ChIP experiment was performed in an HCT116 DNMT1 hypomorphic cell line stably expressing FLAG epitope-tagged DNMT1. (d) DNMT3B and the NuRD complex exists in the same protein complex on the SFRP4 promoter. Re-ChIP experiment was performed in HCT116 cells. Error bars denote s.d.
We next used re-ChIP to examine whether DNMT–NuRD interactions occur directly on chromatin surrounding the promoters of our selected TSGs. In the attempt to establish the DNMT re-ChIP assay, we noticed that the elution efficiency of traditional re-ChIP elution buffer (10 mm DTT) is extremely low. Therefore, for the DNMT re-ChIP, we developed a new protocol using immunogen peptide to elute the chromatin complex. We note that this re-ChIP protocol is not sensitive enough to detect all DNMT–NuRD complexes sitting on their target promoters as the elution efficiency of antigen peptide competition is only about 10% compared with the standard SDS elution buffer (data not shown). Even so, we are able to detect the existence of the DNMT1–NuRD complex on the TIMP3 promoter and the existence of the DNMT3B–NuRD complex on the SFRP4 promoter, as shown in Figures 4c and d. Given the fact that the sensitivity of this re-ChIP assay is low and the amount of re-ChIPed DNA is generally extremely low, we cannot rule out the possibility that DNMT–NuRD complexes also associate with other TSGs. Taken together, our data not only demonstrate that DNMTs and NuRD physically interact and occupy promoters of TSGs to maintain stable gene silencing, but also support the idea that these two DNMT–NuRD complexes possibly have different specificities in the epigenetic silencing of TSGs.
Cooperation of DNMTs and NuRD maintains the silencing of genes encoding for WNT antagonists
Next, we set out to identify genes that are epigenetically silenced by the cooperation between NuRD and DNMTs by analyzing the transcriptomes of cells depleted for CHD4 or HDAC1 and treated with DAC. Using RNA-seq, we found a population of silenced transcripts including SFRP1 and 2, TIMP2 and 3 that were reactivated by DNMT inhibition and knockdown of HDAC1 or CHD4 (Figures 5a–d). We used the reactivation dynamics of these positive control TSGs as cutoff criteria and found that knockdown of HDAC1 enhanced 407 transcripts in DAC-induced reactivation and knockdown of CHD4 enhanced 1674 transcripts in DAC-induced reactivation (Supplementary Dataset S1A and B). Importantly, 40% of genes reactivated by both HDAC1 siRNAs and DAC treatment were also reactivated by both CHD4 siRNAs and DAC, suggesting that NuRD and DNMTs cooperate to maintain the silencing of these genes (Supplementary Dataset S1C). Using Ingenuity Pathway Analysis, we found that genes reactivated by DAC treatment and CHD4 or HDAC1 depletion are strongly linked to development, in agreement with NuRD being a key determinant in cellular differentiation and development (Table 2).12 In addition, these target genes were linked to genetic disorders, gastrointestinal disease and cancer with the WNT/β-catenin pathway being significantly enriched among the HDAC1 target genes (Table 2). Indeed, in both data sets, we found, besides the SFRPs, many other WNT inhibitors, including WNT inhibitory factor 1 (WIF1), Dickkopf 1 and 3 (DKK1 and 3), VANG-like 2 (VANGL2) and SRY-box 9 and 17 (SOX 9 and 17), which are all commonly silenced by promoter hypermethylation in CRC33-35 (Supplementary Dataset S1). We tested a few of these genes (DKK1, DKK3 and VANGL2) by QRT-PCR and confirmed their reactivation in CHD4 or HDAC1 knockdown cells simultaneously treated with DAC (Figures 5e and f). Collectively, these data suggest that DNMTs and NuRD cooperate to maintain silencing of several hundred of genes including multiple negative regulators of the WNT pathway.
Figure 5.
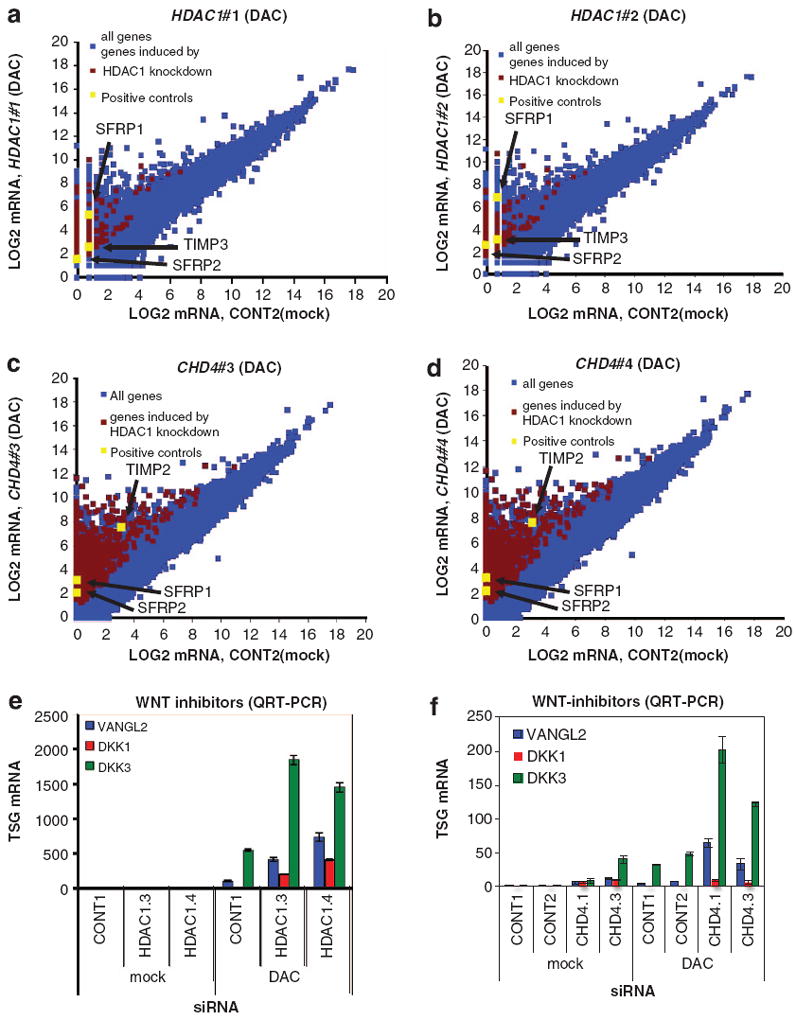
Inhibition of the DNMT–NuRD complex leads to reactivation of silenced genes encoding for WNT antagonists. (a, b) Whole transcriptome analysis of HDAC1 knockdown cells treated with DAC. Depicted are scatter plots of Log2-transformed RNA-seq reads of RKO cells transfected with HDAC1#1 (a) or HDAC1#2 siRNA (b) and treated with DAC in comparison with untreated scrambled (CONT2) siRNA-transfected cells from a single experiment. The stripes are caused by the addition of 1 to all values to avoid negative Log2 values. Genes synergistically reactivated by HDAC1 KD and DAC treatment were selected by these criteria: (1) transcripts that could only be reactivated by both HDAC1#1 and HDAC1#2 siRNA and DAC treatment but not by scrambled siRNA and DAC treatment and (2) transcripts that were >2-fold induced by scrambled siRNA and DAC treatment and >2-fold further enhanced by both HDAC1#1 and HDAC1#2 siRNAs. Transcripts enhanced in reactivation by HDAC1 knockdown are highlighted in red with the positive controls SFRP1, SFRP2 and TIMP3 depicted in yellow. (c, d) Whole transcriptome analysis of CHD4 knockdown cells treated with DAC. Depicted are scatter plots of Log2-transformed RNA-seq reads of RKO cells transfected with CHD4#3 (c) or CHD4#4 siRNA (d) and treated with DAC in comparison with untreated scrambled siRNA- (CONT2) transfected cells from three independent biological replicate experiments. Genes synergistically reactivated by CHD4 KD and DAC treatment were selected by these criteria: (1) transcripts that could only be reactivated by both CHD4#3 and CHD4#4 siRNA and DAC treatment but not by scrambled siRNA and DAC treatment and (2) transcripts that were 42-fold induced by scrambled siRNA and DAC treatment and >2-fold further enhanced by CHD4#3 and CHD4#4 siRNAs. Transcripts enhanced in reactivation by CHD4 knockdown are highlighted in red with the positive controls SFRP1, SFRP2 and TIMP2 depicted in yellow. (e, f) Reactivation of WNT inhibitors by HDAC1 or CHD4 depletion and DNMT inhibition. RKO cells were transfected with scrambled, HDAC1 (e) or CHD4 (f) siRNAs and treated with or without 1 μm DAC. Expression of indicated WNT inhibitors was analyzed by QRT-PCR. Error bars denote s.d.
Table 2.
Ingenuity analysis of transcripts that are synergistically reactivated by DAC
|
Genes reactivated by CHD4 KD and DAC
|
Genes reactivated by HDAC1 KD and DAC
|
||
|---|---|---|---|
| Name | P-value | Name | P-value |
| Diseases and disorders | |||
| Genetic disorders | 7.79E–23 to 1.20E–03 | Genetic disorders | 5.52E–06 to 1.32E–02 |
| Cancer | 1.04E–15 to 1.37E–03 | Cancer | 7.33E–05 to 1.32E–02 |
| Gastrointestinal disease | 1.50E–14 to 1.05E–03 | Cardiovascular disease | 7.69E–05 to 1.32E–02 |
| Skeletal and muscular disorders | 1.10E–13 to 8.87E–04 | Skeletal and muscular disorders | 1.02E–04 to 1.32E–02 |
| Neurological disease | 4.91E–13 to 1.20E–03 | Neurological disease | 1.08E–04 to 7.64E–03 |
| Physiological function | |||
| Tissue development | 7.13E–18 to 1.09E–03 | Nervous system development and function | 7.52E–07 to 1.32E–02 |
| Embryonic development | 1.68E–13 to 1.10E–03 | Embryonic development | 1.36E–06 to 1.32E–02 |
| Organ development | 1.68E–13 to 8.22E–04 | Organ development | 1.36E–06 to 1.32E–02 |
| Organismal development | 1.68E–13 to 8.87E–04 | Organismal development | 1.36E–06 to 1.32E–02 |
| Tissue morphology | 3.06E–13 to 1.26E–03 | Tissue development | 1.36E–06 to 1.32E–02 |
| Top canonical pathways | |||
| Calcium signaling | 2.04E–08 | Axonal guidance signaling | 2.69E–07 |
| Hepatic fibrosis/hepatic stellate cell activation | 2.99E–08 | Role of bone cells in rheumatoid arthritis | 6.8E–04 |
| Atherosclerosis signaling | 2.57E–05 | Role of immune cells in rheumatoid arthritis | 7.03E–04 |
| Cellular effects of sildenafil (viagra) | 6.65E–05 | LPS/IL-1 mediated inhibition of RXR function | 1.39E–03 |
| Role of osteoblasts, osteoclasts and chondrocytes in rheumatoid arthritis | 7.22E–05 | Wnt/β-catenin signaling | 1.92E–03 |
Abbreviations: CHD4, chromo domain helicase DNA-binding protein 4; DAC, 5-aza-2′-deoxycytidine; DNMT, DNA methyltransferase; HDAC1, histone deacetylase 1; LPS, lipopolysaccharide; IL-1, interleukin 1; NuRD, Mi-2/nucleosome remodeling and deacetylase. DNMT–NuRD target genes are linked to gastrointestinal disease and are enriched for components of the WNT signaling pathway. Transcripts enhanced in DAC-induced reactivation by CHD4 or HDAC1 knockdown were analyzed by ingenuity pathway analysis (see Materials and Methods for cutoff criteria). The diseases and disorders, physiological functions and top canonical pathways that are significantly enriched are shown.
Knockdown of the NuRD components CHD4 and HDAC1 in combination with DNMT inhibition leads to synthetic sickness, in correlation with reactivation of TSGs
Two genes have a synthetic sickness interaction if their combined inactivation leads to a lower than expected fitness.36 It is known that tumor cells can tolerate the incomplete depletion of DNMT1 protein by DAC and that overexpression of SFRPs leads to apoptosis.22 Given that NuRD and DNMTs cooperate to target many WNT pathway inhibitors, we tested whether knockdown of the NuRD catalytic subunit CHD4 could sensitize cells to DAC treatment. To test this, CHD4 knockdown RKO cells were treated with increasing concentrations of DAC and incubated with a pro-fluorescent peptide, containing a caspase 3 and 7 DEVD recognition motif, which releases a DNA dye upon cleavage by caspases 3 and 7, resulting in green fluorescent staining of nuclear DNA. Cells were followed for 6 days with time-lapse imaging and TSG reactivation was analyzed in parallel. Depletion of CHD4 strongly potentiated DAC-induced cell death, in association with reactivation of five TSGs (Figure 6). We found similar results in CHD4-depleted and DAC-treated RKO and HCT116 cells in long-term proliferation assays. RKO cells with CHD4 knockdown proliferated slower, even in the absence of DAC treatment, and sensitized cells to DAC treatment, with induced reactivation of four TSGs (Supplementary Figures S6A–C). In HCT116 cells, CHD4 depletion only marginally affected cell growth in the absence of DAC but synergistically inhibited proliferation when combined with DAC, in association with TSG reactivation (Supplementary Figures S6D–F). Collectively, these findings demonstrate that depletion of the NuRD component CHD4 sensitizes CRC cells to DAC treatment and that this occurs temporally with reactivation of silenced TSGs.
Figure 6.
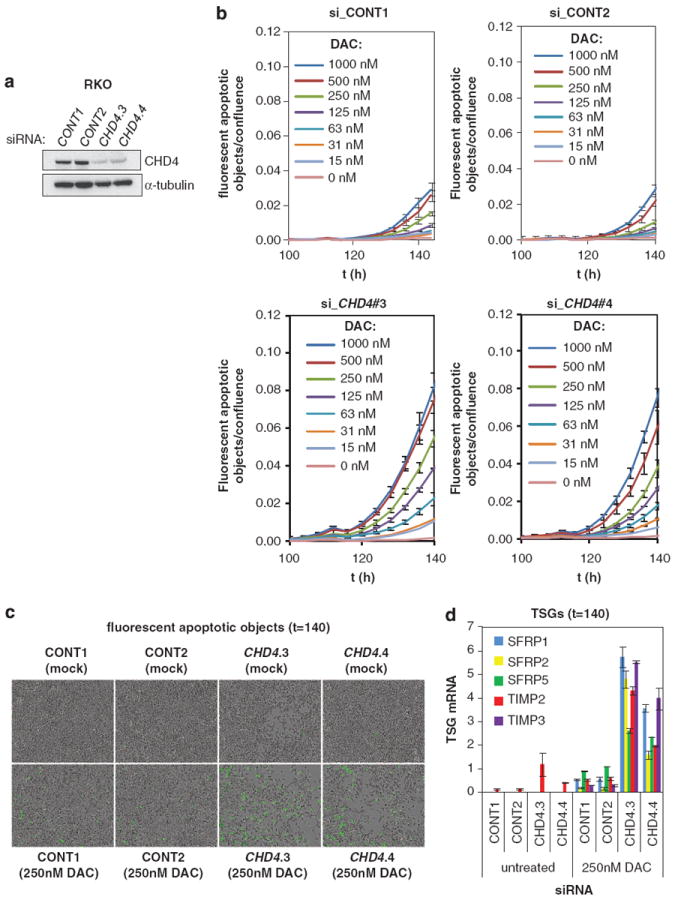
DAC and knockdown of CHD4 synergize in cell death induction, in correlation with reactivation of TSGs. (a) CHD4 siRNAs induce depletion of CHD4. RKO cells were transduced with scrambled (CONT1-2) or CHD4 siRNAs. CHD4 knockdown was verified by examining CHD4 protein levels, α-tubulin serves as a loading control. (b) Knockdown of CHD4 sensitizes RKO cells to DAC treatment. RKO cells from (a) were untreated or treated with indicated concentrations of DAC. Kinetic measures of the number of caspase-3/7-positive cells, recorded over time and plotted as fluorescent objects, divided by confluence are shown. n = 3 wells per data point shown, error bars denote s.d. (c, d) Knockdown of CHD4 induces synthetic sickness lethality with DNMTi in association with TSG reactivation. RKO cells from (a, b) were untreated or treated with 250 nm DAC and analyzed for caspase 3 and 7 activation by time-lapse microscopy after 140 h (c) and analyzed for TSG reactivation by QRT-PCR (d). Error bars denote s.d.
DISCUSSION
It is well known that the combined treatment of DNMTi and HDACi in cancer cells reactivates completely silenced TSGs more effectively than each of these drugs alone.20,21,37 One potential complication in the use of HDACi and DNMTi in cancer therapy is their limited target selectivity and off-target effects.38,39 As such, effective epigenetic therapy with these agents may benefit from identifying the relevant drug targets within these larger enzyme families to allow the development of more selective inhibitors. Through a genetic screen, we discovered that HDAC1/2 are the major TSA-sensitive HDACs, which cooperate with DNMTs to maintain the epigenetic silencing of multiple TSGs in colon cancer cells. We also find an inverse relationship between HDAC1 and to lesser extent HDAC2 mRNA levels and expression of two key TSG families in colorectal tumors. These data have potential clinical relevance as they provide evidence for HDAC1/2 as the relevant drug targets within the larger HDAC family. Our data thus make the case for the development of selective HDAC1/2 inhibitors, which may be useful in colon cancer treatment when combined with DNMTi.
How does HDAC1/2 cooperate with DNMTs/DNA methylation to maintain TSG silencing? To answer this question, we took both genetic and biochemical approaches specifically looking for HDAC1/2 containing complexes that are necessary for the maintenance of TSG silencing. Interestingly, both approaches identified the same target, the NuRD complex which is a known reader for methylated DNA. Our finding that NuRD can interact with not only methylated DNA but also DNMT proteins expands our understanding of the cooperation between NuRD and the DNA methylation machinery. In line with this, we demonstrate that DNMTs and NuRD not only physically co-occupy the promoters of a subset of TSGs but also functionally cooperate to maintain their silencing, suggesting that DNA hypermethylation works in conjunction with nucleosome remodeling and histone deacetylation to orchestrate epigenetic gene silencing. Interestingly, our genetic interaction experiment in HCT116 DNMT mutant cell lines showed that depletion of one DNMT or CHD4 is not sufficient to reactivate TSGs such as TIMP3 and SFRP2/4, which can be reactivated only when both CHD4 and DNMTs are depleted. This result suggested that DNMTs and NuRD can cooperate in both a complex-dependent model and a complex-independent model.
Previously, inhibition of the NuRD complex alone has been suggested as an option for cancer treatment.12 Deficiency of Mbd2 suppressed intestinal tumorigenesis in a mouse model of colon cancer.40 Interestingly, a follow-up study proposed that Mbd2 loss may lead to attenuation of Wnt signaling by upregulation of a Wnt antagonist, Lect241 which is in support of our finding that multiple WNT inhibitor genes are silenced by the cooperation between NuRD and DNMTs. Furthermore, we show that depletion of the NuRD complex subunit CHD4 leads to synthetic sickness when combined with DAC treatment. Thus, one potential mechanism for the observed synthetic sickness interaction between DNMT and NuRD inhibition might be inhibition of the WNT pathway by the activation of WNT inhibitor genes, leading to cell death. This is supported by our previous findings that in colon cancer cells with hypermethylated SFRPs, restoration of SFRPs attenuates WNT signaling and induces cell apoptosis.22 It must be noted, however, that at present, we cannot exclude alternative mechanisms for the synthetic sickness lethality. For example, DNMTs and NuRD have been shown to be involved in the DNA damage response, G1/S cell-cycle transition and assembly of heterochromatin formation, which can also affect cell viability when inhibited.42-45 As such, our study identifies combined inhibition of DNMTs and the NuRD complex as a potential novel therapeutic strategy. We note that CHD4 has ATPase activity, which is required for nucleosome sliding and remodeling. As inhibitors of specific ATPases have been successfully developed, CHD4 is a potentially drug-able target for cancer therapy.46 Moreover, corresponding to enhanced tumor cell death, our synthetic sickness lethality experiments suggest that combined inhibition of all these epigenetic mechanisms induces more efficient reactivation of TSGs than inhibition of each of these mechanisms alone.
While our studies have uncovered important new roles for components of the NURD complex in cancer-specific gene silencing with promoter DNA hypermethylation, many important future extensions of the work are needed. First, our work suggested that increased expression level of HDAC1/2 has a negative correlation with the expression of TSGs suppressed by the NuRD complex. As both HDAC1 and HDAC2 are also components of other repressor complexes, it would be interesting to examine the expression level of multiple NuRD-specific subunits such as CHD4 in human CRC and whether HDAC1/2 can mediate TSG silencing in the context of other complexes. Second, HDAC1 and HDAC2 can be redundant for some biological processes but they can also have specific roles in other contexts. Although our results suggest that HDAC1 seems to have a more dominant role than HDAC2 in the silencing of five TSGs, we cannot rule out that HDAC2 may also have an important role in the maintenance of TSG silencing. Future investigation of genes specifically silenced by HDAC1 or HDAC2 would provide valuable information for the application of more specific HDAC inhibitors in cancer therapy.
MATERIALS AND METHODS
Compounds
DAC (decitabine, Sigma-Aldrich, St Louis, MO, USA) was dissolved in 50% acetic acid/dH2O and stored at −80 °C. TSA (Sigma-Aldrich) was dissolved in DMSO and stored at −80 °C.
Plasmids
shRNAs sequence can be found in Supplementary Table S2. pQCXIP vectors (Promega, Madison, WI, USA) expressing GFP, wild-type or mutant Hdac1 were kind gifts of Drs M Holzel and JH Dannenberg.24
Cell culture and viral transduction
RKO cells were from ATCC, all other cell lines were from the laboratory of Dr R Bernards, and were maintained in medium recommended by ATCC. HCT116 cell lines deleted for DNMT1 or DNMT3B have been previously generated.19 Retroviral and lentiviral transductions were performed as previously described.47
Cell proliferation assays
Growth curves were performed according to the standard 3T3 protocol in the absence or presence of 1 μm DAC using the Casy Cell Counter (Scharfe System, GmbH, Reutlingen, Germany) or TC10 Cell Counter (Bio-Rad, Hercules, CA, USA). In all, 2.0 × 105 of RKO cells or 0.75 × 105 HCT116 cells were plated in 6-well dishes. In the case of cell depletion, all cells remaining were re-plated.
Time-lapse imaging and apoptosis assay
RKO cells (1500 per well) were reverse transfected in 384-well plates. DAC (15–1000 nm) and 1:1000 CellPlayer 5 mm NucView Caspase-3/7 Apoptosis reagent (Essen BioScience, Ann Arbor, MI, USA) were added every subsequent day when medium was refreshed. Four wells per condition were imaged (phase-contrast and fluorescence) with a 4-h interval for 6 days using the IncuCyte FLR (Essen BioScience). Apoptosis was quantified by dividing the confluence of fluorescent apoptotic cells by total confluence as determined by the IncuCyte software (build 2011A rev2, Essen BioScience).
siRNA transfections
5 × 105/well RKO or 2 × 105/well HCT 116 cells were reverse transfected with 50 nm siRNAs (Dharmacon/Thermo Fischer Scientific, Waltham, MA, USA) in 6-well format with Lipofectamine 2000 (Invitrogen/Life Technologies, Carlsbad, CA, USA). For TSG reactivation assays, transfected cells were split at day 3 and either untreated or treated with 100 nm DAC (HCT116) or 1 μm DAC (RKO) for 2 days. For reconstitution experiments with siRNAs targeting HDAC1 and CHD4, transfected RKO cells were washed next day with MEM containing 1 μm DAC. Cells were split in MEM with 1 μm DAC at day 3, transduced on 2 consecutive days with pQXCIP vectors overexpressing GFP, wild-type or Y303F mutant Hdac1 and stringently selected with 4 μg puromycin o/n. Cells were processed at day 5 or 6 for TSG expression analysis.
RNA isolation and cDNA synthesis
RNA was isolated using TRIzol (Invitrogen) and converted to cDNA using Superscript II (Invitrogen). QRT-PCRs were run on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using FastStart Universal SYBR Green Master mix (Roche, Applied Science, Penzberg, Germany). mRNA levels were normalized by using at least two housekeeping genes: GAPDH, PGK, RPL4 or ACTIN. QRT values measured above cycle 35 were considered as 0. For RKO cells, the data were Log10 transformed and standardized using the lowest Ct value measured. Primer Sequences are listed in Supplementary Table S2.
RNA-seq
RNA was extracted with TRIzol and processed using the TruSeq protocol (Illumina, San Diego, CA, USA) on an Ilumina Hiseq2000 according to manufacturer’s protocol. RNA-seq reads (18 × 107 to 27 × 107 50bps single-end reads per sample) were mapped to the human reference genome (build 37) using TopHat (v.1.3.0, Johns Hopkins University, Baltimore, MD, USA / University of California-Berkeley, Berkeley, CA, USA).48 HTSeq-count (v. 0.5.3p3, EMBL Heidelberg, Heidelberg, Germany) was used to generate a list of total number of uniquely mapped reads (12 to 18 × 107 pairs of reads per sample) for each gene present in the Gene Transfer Format (GTF) file (Ensembl release 64). Sequence counts were normalized for sequence efficiency and 1 was added to all values to avoid negative values after Log2 transformation.
ChIP experiments
ChIP protocol was described previously49 and the details are provided in Supplementary materials. The ChIP primer sequences are in Supplementary Table S2.
Western blotting and co-immunoprecipitation
Western blotting and co-immunoprecipitation were described previously.49 Antibody information and conditions are provided in Supplementary Table S3.
Endogenous DNMT1 immunoprecipitation and tandem mass spectrometry analysis
Endogenous DNMT1 was immunoprecipitated as described previously.49 The gel was stained with SimplyBlue (Invitrogen). In-gel digestion of protein bands, mass spectrometry analysis of peptides, and protein identification were performed as described earlier50,51 except trypsin was used for digestion and a human IPI proteome database was used for protein identification.
Statistical analysis
The relationship between HDAC1 and 2 expression levels and indicated TSGs was determined in gene expression data sets of previously published 396 primary colorectal tumor samples.25 Expression levels are indicated as Log2 ratios against a colon cancer reference pool. Expression levels of indicated TSGs were correlated (Pearson correlation) with levels of HDAC1 and 2.
Supplementary Material
Acknowledgments
We thank JH Dannenberg for the gift of Hdac1 plasmids. We thank JH Dannenberg, P Kumar and members of the Bernards, Baylin laboratory for discussions and Kathy Bender and H. Liu for manuscript preparation. This work was supported by a grant from the Netherlands Organization for Scientific Research (NWO) to RB, and by grants ES011858 National Institute of Environmental Health Sciences (NIEHS) CA043318 National Cancer Institute (NCI) to SB. The views presented in this article do not necessarily reflect those of the US Food and Drug Administration.
Footnotes
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Ting AH, McGarvey KM, Baylin SB. The cancer epigenome—components and functional correlates. Genes Dev. 2006;20:3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 4.Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee TH. DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc. 2010;132:1782–1783. doi: 10.1021/ja910264z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin JC, Jeong S, Liang G, Takai D, Fatemi M, Tsai YC, et al. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell. 2007;12:432–444. doi: 10.1016/j.ccr.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 8.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 9.Lunyak VV, Burgess R, Prefontaine GG, Nelson C, Sze SH, Chenoweth J, et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- 10.Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat Genet. 2003;34:187–192. doi: 10.1038/ng1158. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23:62–66. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 14.Myant K, Stancheva I. LSH cooperates with DNA methyltransferases to repress transcription. Mol Cell Biol. 2008;28:215–226. doi: 10.1128/MCB.01073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 16.Mohammad HP, Cai Y, McGarvey KM, Easwaran H, Van Neste L, Ohm JE, et al. Polycomb CBX7 promotes initiation of heritable repression of genes frequently silenced with cancer-specific DNA hypermethylation. Cancer Res. 2009;69:6322–6330. doi: 10.1158/0008-5472.CAN-09-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–1082. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- 18.Morey L, Brenner C, Fazi F, Villa R, Gutierrez A, Buschbeck M, et al. MBD3, a component of the NuRD complex, facilitates chromatin alteration and deposition of epigenetic marks. Mol Cell Biol. 2008;28:5912–5923. doi: 10.1128/MCB.00467-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 20.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31:141–149. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 23.Jurkin J, Zupkovitz G, Lagger S, Grausenburger R, Hagelkruys A, Kenner L, et al. Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle. 2011;10:406–412. doi: 10.4161/cc.10.3.14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre Jetal. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29:2586–2597. doi: 10.1038/emboj.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salazar R, Roepman P, Capella G, Moreno V, Simon I, Dreezen C, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29:17–24. doi: 10.1200/JCO.2010.30.1077. [DOI] [PubMed] [Google Scholar]
- 26.Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14:1669–1677. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 27.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–176. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 28.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Reale A, Matteis GD, Galleazzi G, Zampieri M, Caiafa P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene. 2005;24:13–19. doi: 10.1038/sj.onc.1208005. [DOI] [PubMed] [Google Scholar]
- 30.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 31.Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3:1709–1723. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ying Y, Tao Q. Epigenetic disruption of the WNT/beta-catenin signaling pathway in human cancers. Epigenetics. 2009;4:307–312. doi: 10.4161/epi.4.5.9371. [DOI] [PubMed] [Google Scholar]
- 34.Aleman A, Adrien L, Lopez-Serra L, Cordon-Cardo C, Esteller M, Belbin TJ, et al. Identification of DNA hypermethylation of SOX9 in association with bladder cancer progression using CpG microarrays. Br J Cancer. 2008;98:466–473. doi: 10.1038/sj.bjc.6604143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park M, Moon RT. The planar cell-polarity gene stbm regulates cell behaviour and cell fate in vertebrate embryos. Nat Cell Biol. 2002;4:20–25. doi: 10.1038/ncb716. [DOI] [PubMed] [Google Scholar]
- 36.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 37.Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 39.Wozniak RJ, Klimecki WT, Lau SS, Feinstein Y, Futscher BW. 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene. 2007;26:77–90. doi: 10.1038/sj.onc.1209763. [DOI] [PubMed] [Google Scholar]
- 40.Sansom OJ, Berger J, Bishop SM, Hendrich B, Bird A, Clarke AR. Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat Genet. 2003;34:145–147. doi: 10.1038/ng1155. [DOI] [PubMed] [Google Scholar]
- 41.Phesse TJ, Parry L, Reed KR, Ewan KB, Dale TC, Sansom OJ, et al. Deficiency of Mbd2 attenuates Wnt signaling. Mol Cell Biol. 2008;28:6094–6103. doi: 10.1128/MCB.00539-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyl-transferase rather than DNA demethylation. Proc Natl Acad Sci USA. 1994;91:11797–11801. doi: 10.1073/pnas.91.25.11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010;190:741–749. doi: 10.1083/jcb.201001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ, et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol. 2010;190:731–740. doi: 10.1083/jcb.200912135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huss M, Wieczorek H. Inhibitors of V-ATPases: old and new players. J Exp Biol. 2009;212(Pt 3):341–346. doi: 10.1242/jeb.024067. [DOI] [PubMed] [Google Scholar]
- 47.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, et al. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clements EG, Mohammad HP, Leadem BR, Easwaran H, Cai Y, Van Neste L, et al. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 2012;40:4334–4346. doi: 10.1093/nar/gks031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu LR, Zhu Z, Chan KC, Issaq HJ, Dimitrov DS, Veenstra TD. Improved titanium dioxide enrichment of phosphopeptides from HeLa cells and high confident phosphopeptide identification by cross-validation of MS/MS and MS/MS/MS spectra. J Proteome Res. 2007;6:4150–4162. doi: 10.1021/pr070152u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.