

Abstract
Hexavalent chromium, CrVI, is a heavy metal endocrine disruptor, known as a mutagen, teratogen, and a group A carcinogen. Environmental contamination with CrVI, including drinking water, has been increasing in more than 30 cities in the United States. CrVI is rapidly converted to CrIII intracellularly, and CrIII can cause DNA strand breaks and cancer or apoptosis through different mechanisms. Our previous study demonstrated that lactational exposure to chromium results in a delay or arrest in follicle development and a decrease in steroid hormone levels in F1 female rats, both of which are mitigated (partial inhibition) by vitamin C. The current study tested the hypothesis that lactational exposure to CrIII accelerates follicle atresia in F1 offspring by increasing reactive oxygen species (ROS) and decreasing cellular antioxidants. Results showed that lactational exposure to CrIII dose-dependently increased follicular atresia and decreased steroidogenesis in postnatal day 25, 45, and 65 rats. Vitamin C mitigated or inhibited the effects of CrIII at all doses. CrIII increased hydrogen peroxide and lipid hydroperoxide in plasma and ovary; decreased the antioxidant enzymes (AOXs) GPx1, GR, SOD, and catalase; and increased glutathione S-transferase in plasma and ovary. To understand the effects of CrVI on ROS and AOXs in granulosa (GC) and theca (TC) cell compartments in the ovary, ROS levels and mRNA expression of cytosolic and mitochondrial AOXs, such as SOD1, SOD2, catalase, GLRX1, GSTM1, GSTM2, GSTA4, GR, TXN1, TXN2, TXNRD2, and PRDX3, were studied in GCs and TCs and in a spontaneously immortalized granulosa cell line (SIGC). Overall, CrVI downregulated each of the AOXs; and vitamin C mitigated the effects of CrVI on these enzymes in GCs and SIGCs, but failed to mitigate CrVI effects on GSTM1, GSTM2, TXN1, and TXN2 in TCs. Thus, these data for the first time reveal that lactational exposure to CrIII accelerated follicular atresia and decreased steroidogenesis in F1 female offspring by altering the ratio of ROS and AOXs in the ovary. Vitamin C is able to protect the ovary from CrIII-induced oxidative stress and follicle atresia through protective effects on GCs rather than TCs.
Keywords: Chromium, Ovary, Atresia, Vitamin C, Reactive oxygen species, Antioxidants, Free radicals
Introduction
Hexavalent chromium (CrVI) has been used in more than 50 different industries throughout the world. It has many uses in metal plating, leather tanning, and the production of textiles, dyes, and pigments; in the metallurgical and chemical industries; in Cr-based catalytic converters of automobiles; in heat resistance; and as an antirust agent in water cooling [1]. Owing to increased use and improper disposal of CrVI waste in the environment, CrVI levels in the water, soil, and air continue to increase [2]. Significant contamination with CrVI has been found in drinking water sources in California [3], Texas [4], and more than 30 cities in the United States [5]. Occupational exposure to CrVI is found among approximately half a million industrial workers in the United States and several millions worldwide [3]. CrVI is known to cause various health problems, including lung cancer [6] and infertility in men and women and developmental problems in children [7]. Women working in dichromate manufacturing industries and tanneries and living around Cr-contaminated areas have high levels of Cr in blood and urine and encounter gynecological illness, premature abortion, postnatal hemorrhage, and birth complications [8–11]. Because Cr can pass through the placental barrier and can be transported through mother’s milk to offspring [12], it can cause adverse effects on the developing embryos/fetuses and newborn children. CrVI-induced developmental defects include postimplantation loss and reduced fetal weight [13–15]. Although CrVI is known to adversely affect reproductive health in women [16], the actual mechanism of reproductive toxicity is not clearly understood.
At physiological pH, CrVI exists in the form of an oxyanion with an overall −2 charge that resembles sulfate and phosphate; and CrVI is taken up by cells via the anionic transport system [17,18]. This transport system, along with intracellular reduction reactions, allows Cr to accumulate in cells at concentrations that are much higher than the extracellular levels [19,20]. Once CrVI enters the cell, it is reduced to trivalent Cr (CrIII) by various intracellular reductants, predominantly vitamin C and glutathione (GSH), as both exist in high-millimolar concentrations [21]. Vitamin C and GSH are highly abundant in the brain [22], liver [23], lungs [24,25], and ovary [26,27]. Epithelial lining fluid in the lung contains 100-fold higher GSH than in the plasma [28]. Eventually, all the CrVI that enters a cell is reduced to CrIII [21]. During the intracellular reduction process of CrVI to CrIII, a whole spectrum of reactive oxygen species (ROS) (i.e., superoxide ( ), hydrogen peroxide (H2O2), and hydroxyl radical (•OH)) is produced [29], with the depletion of antioxidant enzymes (AOXs) such as superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase, resulting in oxidative stress that damages all components of the cell, including proteins, lipids, and DNA [30]. Copper/zinc SOD (SOD1) and manganese SOD (SOD2) are the two major intracellular enzymes that inactivate superoxide radicals. SOD1 is present in both cytoplasmic and nuclear compartments, whereas SOD2 is localized to mitochondria [31]. The expression and activity of SOD1 is directly related to oocyte quality and is considered to be a biomarker in assisted reproductive techniques [32]. ROS such as ( and H2O2 are the products of univalent and bivalent reduction of oxygen (O2), respectively. SODs detoxify into H2O2. H2O2 is then converted to water and molecular O2 by catalase and GPx. Oxidative stress has been proposed as a major pathway of Cr toxicity [29].
Mitochondria are the major intracellular source of ROS generated during cellular respiration. Mitochondrial ROS production is associated with the activation of apoptotic and necrotic cell death [33]. Therefore, a tightly regulated balance exists between ROS production and AOX defense systems in the mitochondria. Mitochondrial peroxiredoxin (PRDX)-3 is an important reducer of mitochondrial H2O2. Mitochondria-specific AOXs, such as PRDX3, thioredoxin (TXN)-2, and TXN reductase (TXNRD)-2, provide a major line of defense against mitochondrial ROS [34]. PRDX3 depletion in cells leads to an increase in H2O2 levels in mitochondria [35]. An age-related decrease in the mRNA expression of the mitochondrial AOXs PRDX3 and TXN2 is reported in the mouse ovary [36], with a parallel increase in oxidative stress. Moreover, mitochondria incorporate, as well as recycle vitamin C, transported through glucose transporters, thus playing a critical role in redox homeostasis [37]. Our previous in vitro study indicated that CrVI-induced apoptosis of granulosa cells (GCs) is mediated through increased mitochondrial translocation of BAX, BAD, ERK1/2, and p53 [38].
Gonadotropins, growth factors, and steroid hormones play differential and dual roles in regulating cell survival and cell death pathways in GC and theca cells (TCs). For an example, administration of luteinizing hormone and follicle-stimulating hormone (FSH) to cultured preovulatory follicles induces an antiapoptotic response in GCs, which overrides the proapoptotic response in TCs due to caspase-3 and −7 activation [39]. Regulation of AOXs and redox homeostasis in the ovary is also a hormone-mediated process [40]. Therefore, it is imperative to understand the differential regulation of ROS and AOX enzymes in follicular GC and TC compartments. We hypothesize that lactational exposure to CrIII induces follicular atresia in F1 offspring by altering the ratio of ROS and antioxidants, which is mitigated by vitamin C supplementation. This hypothesis was tested by the following objectives: (i) to determine the effect of CrIII on follicular atresia of F1 offspring; (ii) to understand the effects of CrIII on oxidative stress and alteration of AOXs in the ovary; (iii) to determine the effect of CrVI treatment on the regulation of mRNA expression of the cytosolic and mitochondrial AOXs SOD1, SOD2, catalase, GLRX1, glutathione S-transferase (GST) M1, GSTM2, GSTA4, glutathione reductase (GR), TXN1, TXN2, TXNRD2, and PRDX3 in GCs and TCs in vitro; and (iv) to determine the intervention potential of vitamin C on the adverse effects of CrIII in the ovary. We used primary cultures of rat GCs and TCs. GC responses to CrVI treatment were also compared with those in a spontaneously immortalized rat granulosa cell line (SIGC) [41] to evaluate the suitability of SIGCs as a model system to study heavy metal-induced oxidative stress on GC. As noted above, CrVI is reduced to CrIII by antioxidants within cells, resulting in the more stable form of CrIII in cells and body fluids, including breast milk [42,43]. Therefore, whereas lactating mothers in these studies were dosed with CrVI, during the treatment period the F1 female pups received CrIII through the mothers’ milk. In the following sections, reference is made to CrVI for in vitro treatments and in vivo dosing of mothers and to CrIII as the oxidation state of Cr delivered to female pups through their mother’s milk.
Materials and methods
Chemicals
Potassium dichromate (a hexavalent chromium compound, CrVI) was purchased from Sigma–Aldrich (St. Louis, MO, USA; Cat. No. 483044). Purity of the chemical was 99.99% based on trace metals analysis performed by the manufacturer. The TUNEL assay kit (Cat. No. 11684817910) and protease and phosphatase inhibitor tablets were purchased from Roche Applied Science (Germany). The H2O2 (Cat. No. 600050), lipid hydroperoxide (LPO; Cat. No. 705002), SOD (Cat. No. 706002), catalase (Cat. No. 707002), GPx (Cat. No. 703102), GST (Cat. No. 703302), and GR (Cat. No. 703202) assay kits were purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). ELISA kits for estimating serum hormones, estradiol-17β (E2; Cat. No. EIA2693), testosterone (T; Cat. No. EIA1559), progesterone (P4; Cat. No. EIA1561), and FSH (Cat. No. EIA1288) were purchased from DRG International (Springfield, NJ, USA). Trizol reagent (Cat. No. 15596026) was purchased from Ambion Life Sciences (Austin, TX, USA). RNeasy kit (Cat. No. 74104), RNAprotect reagent (Cat. No. 76526), reverse transcription kit (Cat. No. 205311), SybrGreen polymerase chain reaction (PCR) kit (Cat. No. 204143), and proteinase K (Cat. No. 19131) were from Qiagen (Valencia, CA, USA). Primary antibody for FSH receptor was purchased from Sigma–Aldrich (Cat. No. F3929). Enhanced chemiluminescence substrate was from Pierce Biotechnology (Rockford, IL, USA). Fetal bovine serum was from HyClone Chemicals (Logan, UT, USA). Unless otherwise mentioned all other chemicals were purchased from Sigma–Aldrich.
Animals
Timed pregnant Sprague–Dawley rats were purchased from Charles River Laboratories and maintained in AALAC-approved animal facilities with a 12-h light/12-h dark regime at 23–25 °C and provided with a Teklad 6% mouse/rat diet and water ad libitum. Animal use protocols were performed in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and were in accordance with the standards established by the Guiding Principles in the Use of Animals in Toxicology and approved by the Institutional Animal Care and Use Committee of Texas A&M University.
In vivo dosing and experimental design
Lactating rats were divided into the following groups: (1) control, rats receiving regular drinking water; (2) CrVI treatment, rats receiving three doses of potassium dichromate dissolved in drinking water, namely, (2a) 50 ppm CrVI, (2b) 100 ppm CrVI, and (2c) 200 ppm CrVI; (3) CrVI+vitamin C treatment, rats receiving three doses of CrVI with vitamin C (500 mg/kg body wt/day) supplementation through gavage, namely, (3a) 50 ppm CrVI+vitamin C, (3b) 100 ppm CrVI+vitamin C, and (3c) 200 ppm CrVI+vitamin C. On the day of birth, male pups were removed, and the litters were culled to 4 female pups per mother rat; thus each treatment group consisted of 5 mothers and 20 F1 female pups. In cases in which fewer than 4 female pups were delivered, additional pups were fostered from other litters.
In all experimental groups, the lactating mother rats received CrVI treatment in drinking water from the day of parturition to day 21 postpartum. Three age groups were chosen to study the effects of Cr on the ovary to represent prepubertal age (PND 25), peripubertal age (PND 45), and postpubertal age (PND 65). Previous findings showed that F1 rats of mothers (F0) exposed to a high dose of CrVI (200 ppm) did not attain puberty (vaginal opening) until PND 54 [38]. Even the F1 rats from mothers exposed to a lower dose of CrVI (50 ppm) attained puberty around PND 47 (unpublished data). Therefore, three age groups were chosen to cover the entire phase of follicle development in CrIII-treated rats. On PND 25, female pups from 5 mother rats (n=20; 4 pups/mother) from each group were euthanized under CO2 anesthesia followed by cervical dislocation, and blood and ovaries were collected. After weaning, the remaining pups were maintained separately and fed with regular drinking water and diet. On PND 45, rats from another 5 mother rats (n=20) from each group were euthanized, and blood and ovaries were collected. On PND 65, the remaining rats (n=20) were euthanized, and blood and ovaries were collected. Cycling rats from control and experimental groups were euthanized on the next proestrus phase of the estrus cycle after PND 45 or PND 65.
Histological evaluation of follicular atresia
Ovaries were collected and fixed in Bouin’s solution for 24 h and transferred to 70% ethanol. After fixation, the tissues were dehydrated, embedded, serially sectioned (5 μm), mounted on glass slides, and stained with hematoxylin and eosin. Large and small atretic follicles were counted in serial sections of the ovary. Every 12th section was used to count atretic follicle numbers [44]. The criteria to identify atretic follicles were according to Osman [45] and Borgeest et al. [46] as indicated: degenerative changes in the GC wall, which shows cell shrinkage, pyknosis, and karyorrhexis (rupture of nucleus with disintegration of chromatin into granules), and/or degenerative changes in the oocyte such as the breakdown of the nuclear membrane with oocyte fragmentation. Antral follicles were considered atretic if they contained disorganized GCs with at least 20 apoptotic bodies in the GC layer(s).
TUNEL assay on paraffin-embedded tissue sections and primary cultures of granulosa and theca cells
Paraffin-embedded tissue sections were deparaffinized in xylene and dehydrated in a graded ethanol series: 90, 80, 70% for 3–6 min, followed by a washing in double-distilled water. Nuclei of tissue sections were stripped of proteins by incubating with 20 μg/ml proteinase K (in 200 ml of 10 mM Tris–HCl, pH 7.4) for 30 min at 21–37 °C and washed in phosphate-buffered saline (PBS). Endogenous peroxidase was inactivated by incubating sections with 3% H2O2 in methanol for 10 min at room temperature, blocked with 3% BSA in PBS for 30 min, and washed in PBS. For the positive control, sections were incubated with DNase I (diluted in 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 1 mg/ml BSA) for 10 min at 15–25 °C to induce DNA strand breaks. Slides were rinsed twice in PBS followed by addition of 50 μl of TUNEL reaction mixture to the sections and incubation for 60 min at 37 °C in a humidified chamber in the dark. The sections were rinsed and incubated with the TUNEL detection antibody (Roche) labeled with peroxidase. The peroxidase was visualized with diaminobenzidine. Slides were mounted and analyzed by light microscopy. The intensity of staining for each protein was quantified in 10 randomly selected high-power fields from each experimental group, using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA) as described previously [47] according to the manufacturer’s instructions.
Measurement of oxidative damage in plasma and the ovary
Oxidative damage was measured by estimating the levels of H2O2 and LPO in plasma and the ovary. H2O2 production was measured spectrophotometrically using a commercial kit (Cayman Chemical). Briefly, H2O2 was detected using 10-acetyl-3,7-dihydroxyphenoxazine in the presence of horseradish peroxidase, which reacts with H2O2 at a 1:1 stoichiometry to produce highly fluorescent resorufin that can be quantified at 590 nm. LPO levels were measured by a direct method, using a colorimetric measurement kit as per the manufacturer’s instructions (Cayman Chemical). The kit measures hydroperoxides directly by utilizing the redox reactions with ferrous ions. Briefly, LPO was extracted into chloroform and directly used in the assay thereby eliminating any interference caused by H2O2 and/or endogenous ferric ions in the sample. Hydroxyperoxides are highly unstable and react with ferrous ions to produce ferric ions. The resulting ferric ions were detected using thiocyanate ion as the chromogen and quantified at 500 nm.
Measurement of vitamin C and antioxidant enzymes
Vitamin C and AOX activities were estimated both in the plasma and in the ovaries of F1 offspring exposed to Cr through mother’s milk. Ovaries were dissected out and homogenized in 50 mM Tris–HCl buffer, pH 7.5, and centrifuged at 10,000g for 15 min. The supernatants were used for the assay.
Vitamin C (ascorbic acid)
A commercial kit (Cat. No. 700420; Cayman Chemical) was used to measure the vitamin C levels according to the manufacturer’s protocol. The kit utilizes the condensation reaction of dehydroascorbic acid with o-phenylenediamine giving 3-(dihydroxyethyl)-furo [3,4–b]-quinoxaline-1-one, which can be analyzed by fluorescence using an excitation wavelength between 340 and 350 nm and an emission wavelength between 420 and 430 nm.
Superoxide dismutase
A commercial kit (Cayman Chemical) was used to measure the SOD activity according to the manufacturer’s protocol. The kit utilizes the interaction between hypoxanthine and xanthine oxidase to produce radicals, which are detected using tetrazolium salt and can be read at 460 nm. The kit detects all three types of SOD metalloenzymes: copper/zinc, manganese, and iron. One unit of SOD is defined as the amount of enzyme required to dismutate 50% of the radicals.
Catalase
The catalase activity was measured using a commercial kit (Cayman Chemical). In this assay, catalase reacts with methanol in the presence of H2O2 to produce formaldehyde, which is then measured by adding 4-amino-3-hydrazino-5-mercapto-1,2,4-triazole to form a purple reaction product that can be detected spectrophotometrically at 540 nm.
Glutathione peroxidase
A commercial kit (Cayman Chemical) was used to measure the GPx levels according to the manufacturer’s protocol. The kit measures the GPx activity indirectly by a coupled reaction with GR. Oxidized glutathione (GSSG), produced upon reduction of hydroperoxide by GPx, is recycled to its reduced state by GR and NADPH. The oxidation of NADPH to NADP+ is accompanied by a decrease in absorbance at 340 nm.
Glutathione S-transferase
A commercial kit (Cayman Chemical) was used to measure the GST levels according to the manufacturer’s protocol. The kit measures the total GST activity by measuring the conjugation of 1-chloro-2,4-dinitrobenzene with reduced glutathione. The conjugation is accompanied by an increase in absorbance and it is directly proportional to the GST activity and can be measured at 340 nm.
Glutathione reductase
A commercial kit (Cayman Chemical) was used to measure the GR levels according to the manufacturer’s protocol. GR activity was measured as the rate of NADPH oxidation. The oxidation of NADPH to NADP+ is accompanied by a decrease in absorbance at 340 nm and is directly proportional to the GR activity.
In vitro studies
Isolation and primary culture of granulosa cells from immature rat ovaries
Granulosa cells were isolated from a total of 160 ovaries collected from 80 immature rats. Female Sprague–Dawley rats of postnatal day 23–26 were harvested as described [48,49], with few modifications in the procedure. Briefly, ovaries were cleared from the surrounding fat under a stereomicroscope and punctured with 25-gauge needles. Cells were collected in phenol red-free Dulbecco’s modified Eagle’s medium (DMEM)–F12 containing 0.2% BSA, 10 mM Hepes, and 6.8 mM EGTA; incubated for 15 min at 37 °C; and centrifuged for 5 min at 250g. The pellets were suspended in a solution containing 0.5 M sucrose, 0.2% BSA, and 1.8 mM EGTA in DMEM–F12 and incubated for 5 min. After incubation, the suspension was diluted with 3 vol DMEM–F12, centrifuged at 250g, and treated sequentially with trypsin (20 μg/ml) for 1 min, 300 μg/ml soybean trypsin inhibitor for 5 min, and DNase I (100 μg/ml) for 5 min at 37 °C. The cells were washed with medium and suspended in DMEM–F12. Cells obtained from the 160 ovaries were cultured in 36 tissue culture dishes in DMEM–F12 supplemented with 20 mM Hepes (pH 7.4), 4 mM glutamine, 100 IU penicillin/ml, and 100 μg/ml streptomycin and subjected to treatments with potassium dichromate, a form of CrVI, with or without pretreatment with vitamin C as described under in vitro experimental design for CrVI treatment.
Isolation and primary culture of theca cells from immature rat ovaries
TCs were isolated as described [50]. Briefly, freshly collected ovaries were placed in medium 199 containing 25 mM Hepes (pH 7.4), 2 mM L-glutamine, 1 mg/ml BSA, 100 U/ml penicillin, and 100 μg/ml streptomycin. The ovaries were then freed from adhering fat and actively punctured with a 27-gauge needle under a stereomicroscope to release GC and blood cells. The remaining ovarian tissue was washed three times with DMEM-F12 to release any remaining GC. The tissue was then minced and incubated for 30 min at 37 °C in the same medium supplemented with 0.65 mg/ml collagenase type 1 plus 10 μg/ml deoxyribonuclease. The dispersion of TCs was enhanced by aspirating and releasing the ovarian tissue suspension repeatedly with a 10-ml pipette. The TCs released by enzymatic digestion were centrifuged at 250g for 5 min and washed in medium three times to eliminate remaining collagenase. Cells were resuspended in McCoy’s 5A medium containing 2 mM L-glutamine, 1 mg/ml BSA, 100 U/ml penicillin, and 100 μg/ml streptomycin and subjected to unit gravity sedimentation for 5 min to eliminate small fragments of undispersed ovarian tissue. Cell viability was assessed by trypan blue exclusion. TCs were seeded in 60-mm plates (3 × 106 viable cells) and were maintained overnight in McCoy’s 5A medium containing 2 mM L-glutamine, 1 mg/ml BSA, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere of 95% oxygen and 5% CO2 at 37 °C.
SIGC culture
The spontaneously immortalized rat granulosa cell line SIGC [41] was cultured in DMEM-F12 (Sigma-Aldrich) with 5% fetal bovine serum (Hyclone), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml amphotericin-B in a humidified atmosphere with 95% air, 5% CO2 at 37 °C.
In vitro experimental design for CrVI treatment
At 70% confluency, primary cultures of ovarian GCs and TCs or the SIGCs were serum-starved for 24 h with or without vitamin C in the medium and divided into six treatment groups, as follows: (1) control, cells were treated with medium; (2) CrVI−12 h, cells were treated with 10 μM potassium dichromate for 12 h; (3) CrVI−24 h, cells were treated with 10 μM potassium dichromate for 24 h; (4) vitamin C, cells were treated with 1 mM vitamin C for 24 h; (5) vitamin C+CrVI−12 h, cells were pretreated with 1 mM vitamin C for 24 h and treated with 10 μM potassium dichromate for 12 h; (6) vitamin C+CrVI−24h, cells were pretreated with 1 mM vitamin C for 24 h and treated with 10 μM potassium dichromate for 24 h. After the treatment, cells were harvested using 0.1% trypsin-EDTA and mixed with 75 μl of RNAprotect cell reagent (Qiagen). Total RNA was isolated using an RNA isolation kit (Qiagen), quantified, and used for real-time PCR All treatments were performed in triplicate on the same day and each experiment was repeated three times on different days to test the reproducibility of the results.
Measurement of steroid hormones and FSH by ELISA
Steroid hormones E2, P4, and T and peptide hormone FSH were estimated using ELISA kits (DRG International) according to the manufacturer’s instructions. The intra- and interassay coefficients of variation ranged from 5.0 to 8.5%.
Protein extraction and immunoblotting
After the CrVI treatment with or without vitamin C pretreatment, protein from GCs was isolated and immunoblotting/Western blotting was performed as described previously [38]. Briefly, cells were harvested using 1% trypsin–EDTA and pelleted. Cell lysates were sonicated in sonication buffer, which consisted of 20 mM Tris–HCl, 0.5 mM EDTA, 100 μM diethyldithiocarbamate, 1% Tween, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail tablets Complete EDTA-free (1 tablet/50 ml) and PhosStop (1 tablet/10 ml). Sonication was performed using a Microson ultrasonic cell disruptor (Microsonix, Farmingdale, NY, USA). Protein concentration was determined using the Bradford method and a Bio-Rad protein assay kit. Protein samples (75 μg) were resolved using 7.5,10, or 12.5% SDS–PAGE. Chemiluminescent substrate was applied according to the manufacturer’s instructions (Pierce Biotechnology). The blots were exposed to Blue X-Ray film and densitometry of autoradiograms was performed using an Alpha Imager (Alpha Innotech, San Leandro, CA, USA).
Real-time reverse transcription (RT)-PCR
Total RNA was isolated from GCs, SIGCs, and TCs using an RNeasy Mini Kit (Cat. No. 74104; Qiagen) according to the manufacturer’s instruction. The purity and concentration of RNA were determined spectrophotometrically by measuring the absorbance at 260/280 nm, and a purity of 1.8–2.0 was considered satisfactory for the real-time RT-PCR analysis. The first-strand cDNA was synthesized using 1 μg total RNA from the QuantiTect RT kit according to the manufacturer’s instructions. cDNA (2 μl) was mixed with 10 μl master mix (2 × reaction buffer, dNTP mix, Hot Gold Star DNA polymerase, MgCl2, and SybrGreen dye) and sense and antisense oligonucleotide primers for the respective genes and the β-actin gene for internal control, with the total reaction volume made up to 20 μl with RNase-free water. The reaction cycles were as follows: PCR enzyme initial activation at 95 °C for 15 min, initial denaturation at 94 °C for 15 s, annealing at 56 °C for 30 s, and elongation at 72 °C for 30 s. All reactions were run in triplicate. The PCR amplification of all transcripts was performed on an MX3000P real-time PCR machine (Stratagene, Santa Clara, CA, USA). The fold differences were calculated by normalizing the relative expression of the gene of interest with β-actin and the results expressed as fold changes.
Statistical analyses
All numerical data were subjected to one-way ANOVA to detect the effects of treatment and dose interactions. Tukey-Kramer HSD test was used to adjust for multiple pair-wise comparisons of means. Least-squares regression analysis was used to determine the effects of treatment, dose, and treatment × dose interactions in in vivo studies. We took into account the nested structure in the design. Mixed models analysis was used to account for any correlation between the results of pups from the same mother. Mixed models were used to model both fixed effects (in this case treatment) and random effects (in this case mothers and pups) [51]. For in vitro studies, least-squares regression analysis was used to determine effects of treatment (control, CrVI, CrVI+vitamin C), time (CrVI 12 h, CrVI 24 h), and treatment × time interactions. Each value is the mean ± SEM from three plates per treatment. Similar results were obtained in three different experiments performed on 3 different days/time. Statistical analyses were performed using general linear models of Statistical Analysis System (SAS, Cary, NC, USA) and P < 0.05 was considered significant.
Results
Supplementary Table 2 depicts an overall summary of the effects of Cr on the levels of ROS and activity of AOX enzymes in vivo, and Supplementary Table 3 depicts an overall summary of AOX activities and mRNA expression of cytosolic and mitochondrial AOX genes in GCs, SIGCs, and TCs in vitro. Mitigative or inhibitory effects of vitamin C against Cr toxicity varied depending on the dose of CrVI and/or the age of rats.
In vivo studies
Cr increased follicular atresia
Atretic follicle numbers were counted in serial sections of the ovaries of F1 female offspring on PNDs 25, 45, and 65 (Fig. 1). CrIII accelerated atresia of follicles compared to control in a dose-dependent manner. An age-dependent change in the magnitude of follicle atresia/apoptosis was also observed, i.e., an overall percentage of follicular atresia was higher in PND 25 rats compared to PND 45 and PND 65 rats. Interestingly, supplementation of vitamin C inhibited the follicular atresia in F1 rats from the CrVI 50 ppm group and mitigated follicular atresia in the CrVI 100 ppm and CrVI 200 ppm groups.
Fig. 1.
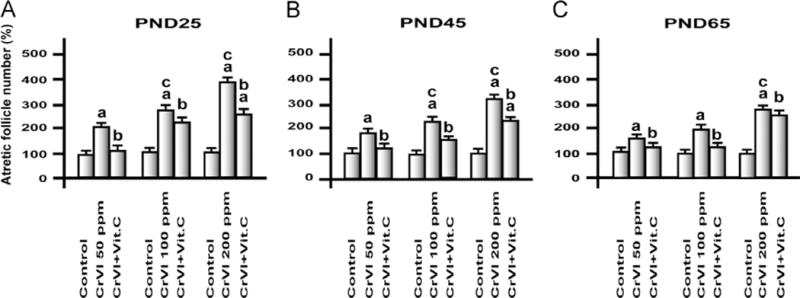
Effects of lactational exposure to CrIII on follicular atresia of F1 offspring. Experimental details are described under Materials and methods. Lactating mother rats received 50, 100, or 200 ppm CrVI in drinking water, with or without vitamin C supplementation through gavage. Suckling F1 female offspring received CrIII through mother’s milk during PNDs 1–21. After PND 21 pups from both control and treatment groups were continued on regular diet and water and were euthanized on PND 25. Ovaries were fixed, embedded, and serially sectioned, and every 12th section was used for counting atretic follicles, and the remaining unstained sections were used for TUNEL assay. aControl vs CrVI or CrVI+vitamin C; bCrVI vs CrVI+vitamin C; cCrVI 50 ppm vs CrVI 100 ppm or CrVI 200 ppm. Each value represents the mean±SEM of 20 rats, P<0.05.
Cr increased apoptosis of follicular cells
Ovaries from F1 rats that were lactationally exposed to CrIII showed a significant increase in percentage of atretic follicles with degenerated oocytes and disorganized and disintegrated GCs with pyknotic nuclei on PND 25 (Fig. 2). Larger follicles were more vulnerable to the toxic effects of CrIII. Vitamin C inhibited CrIII-induced follicular atresia in the 50 and 100 ppm groups, and it mitigated CrIII effects in the 200 ppm group. Quantitative evaluation of the TUNEL assays revealed approximately 48, 64, and 80% apoptotic cells in the 50, 100, and 200 ppm groups, respectively. Control ovaries contained less than 7% positively labeled cells per ovarian section. Vitamin C inhibited apoptosis of GC and oocytes exposed to CrIII in the 50 and 100 ppm groups, whereas it mitigated the effects of CrIII in the 200 ppm group.
Fig. 2.
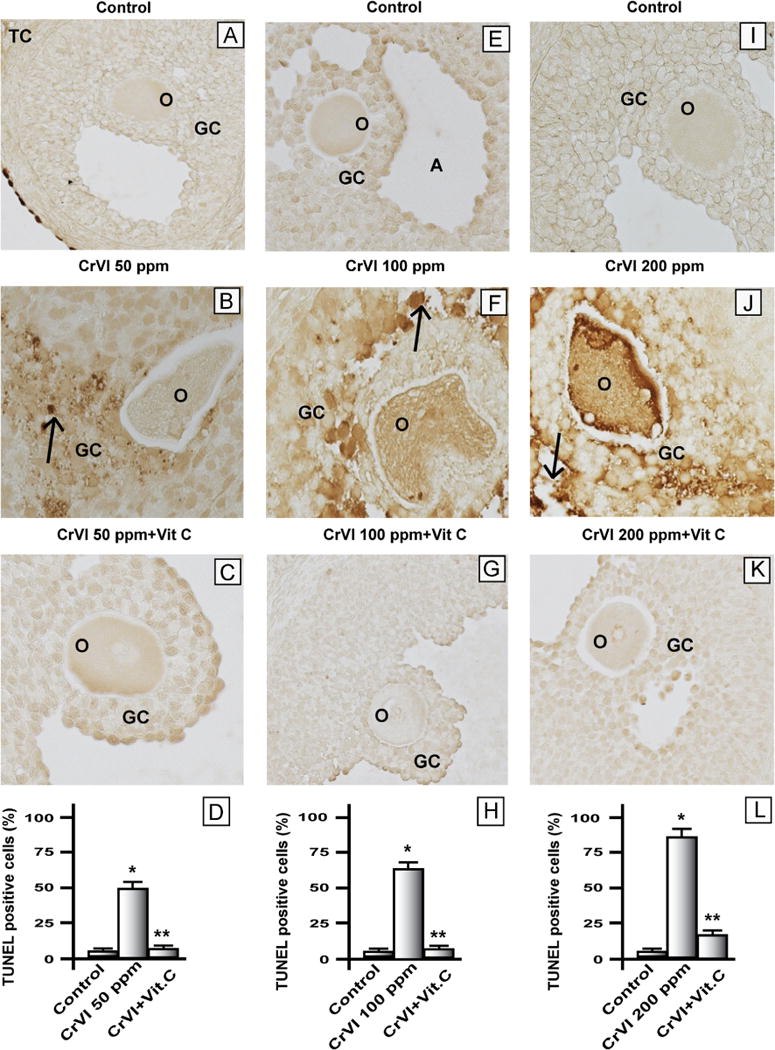
Effect of lactational exposure to CrIII on apoptosis of granulosa cells in F1 offspring. Lactating mother rats received 50, 100, or 200 ppm CrVI in drinking water, with or without vitamin C supplementation through gavage. Suckling F1 female offspring received Cr through their mother’s milk from PND 1 to 21. On PND 25 the F1 offspring were euthanized and ovaries collected, fixed in 4% paraformaldehyde, embedded, and sectioned. Apoptotic, TUNEL-positive cells were stained brown. A representative image for each treatment group is shown. (A–C) Representative images of ovaries: (A) control PND 25 rats that received no treatment, (B) CrVI 50 ppm, and (C) CrVI 50 ppm+vitamin C supplementation. (D) Histogram showing percentage of TUNEL-positive cells from (A), (B), and (C). (E–G) Representative images of ovaries: (E) control PND 25 rats that received no treatment, (F) CrVI 100 ppm, and (G) CrVI 100 ppm+vitamin C supplementation. (H) Histogram showing percentage of TUNEL-positive cells from (E), (F), and (G). (I–K) Representative images of ovaries: (I) control PND 25 rats that received no treatment, (J) CrVI 200 ppm, and (K) CrVI 200 ppm+vitamin C supplementation. (L) Histogram showing percentage of TUNEL-positive cells from (I), (J), and (K). O, oocyte; GC, granulosa cell; TC, theca cell; A, antral cavity. Arrows indicate apoptotic cells. *Control vs CrVI, **CrVI vs CrVI+vitamin C, P<0.05.
Cr decreased steroid and increased FSH hormone levels
The effects of lactational exposure to CrIII on the levels of E2, T, P4, and FSH were analyzed (Table 1). CrIII significantly (P<0.05) decreased E2, T, and P4 in a dose-dependent manner irrespective of age. Vitamin C inhibited or mitigated the effects of CrIII on E2, T, and P4. Overall, vitamin C mitigated the effects of CrIII in the 200 ppm group, whereas it inhibited the CrIII effects in the 50 and 100 ppm groups. CrIII increased FSH levels in F1 rats of all age groups irrespective of the dose of CrVI to the mothers. Vitamin C inhibited the effects of CrIII on FSH in all age groups.
Table 1.
Effect of lactational exposure to CrIII on circulating levels of estradiol, testosterone, progesterone and FSH in F1 rats on postnatal days 25, 45 and 65.
| Age | Treatment | Hormone profile
|
|||
|---|---|---|---|---|---|
| Estradiol (pg/ml) | Testosterone (pg/ml) | Progesterone (ng/ml) | FSH (ng/ml) | ||
| PND 25 | Control | 30.2 ± 2.0 | 923 ± 69 | 5.3 ± 3.1 | 14.2 ± 1.0 |
| Cr 50ppm | 21.3 ± 1.3a | 615 ± 45a | 1.2 ± 2.0a | 29.5 ± 2.1a | |
| Cr 50ppm + Vit. C | 28.4 ± 1.8b | 856 ± 69b | 4.5 ± 2.0b | 15.4 ± 0.7b | |
| Control | 30.2 ± 2.0 | 923 ± 69 | 5.3 ± 3.1 | 14.2 ± 1.0 | |
| Cr 100ppm | 15.1 ± 0.7a | 548 ± 41a | 1.5 ± 2.0a | 32.4 ± 2.3a | |
| Cr 100 ppm + Vit. C | 27.9 ± 2.2b | 825 ± 48b | 4.0 ± 2.0b | 21.2 ± 1.6b | |
| Control | 30.2 ± 2.0 | 923 ± 69 | 5.3 ± 3.1 | 14.2 ± 1.0 | |
| Cr 200 ppm | 12.5 ± 0.8a | 359 ± 29a | 0.7 ± 0.05a | 35.3 ± 2.3a | |
| Cr 200 ppm + Vit. C | 22.4 ± 1.5ab | 656 ± 48ab | 2.0 ± 0.09ab | 21.4 ± 1.6b | |
| PND 45 | Control | 40.3 ± 2.0 | 950 ± 86 | 6.9 ± 0.4 | 14.2 ± 1.2 |
| Cr 50 ppm | 21.3 ± 1.4a | 610 ± 32a | 3.0 ± 0.2a | 27.4 ± 2.0a | |
| Cr 50 ppm + vitamin C | 41.2 ± 3.0b | 913 ± 82b | 8.5 ± 0.7b | 16.3 ± 1.0b | |
| Control | 40.3 ± 2.0 | 950 ± 86 | 6.9 ± 0.4 | 14.2 ± 1.2 | |
| Cr 100 ppm | 12.4 ± 1.0a | 620 ± 35a | 2.8 ± 0.1a | 30.2 ± 2.0a | |
| Cr 100 ppm + Vit. C | 41.5 ± 3.1b | 847 ± 69b | 7.2 ± 0.5b | 13.5 ± 1.0b | |
| Control | 40.3 ± 2.0 | 950 ± 86 | 6.9 ± 0.4 | 14.2 ± 1.2 | |
| Cr 200 ppm | 12.2 ± 0.9a | 390 ± 38a | 1. 0 ± 0.05a | 35.3 ± 2.1a | |
| Cr 200 ppm + Vit. C | 28.5 ± 1.9ab | 725 ± 40ab | 7.3 ± 0.4b | 15.5 ± 1.1b | |
| PND 65 | Control | 42.2 ± 3.4 | 950 ± 52 | 9.8 ± 0.6 | 13.8 ± 1.0 |
| Cr 50 ppm | 28.4 ± 2.1a | 717 ± 42a | 5.4 ± 0.3a | 25.4 ± 1.6a | |
| Cr 50 ppm + vitamin C | 41.5 ± 2.5b | 850 ± 53b | 9.2 ± 0.5b | 14.3 ± 0.9b | |
| Control | 42.2 ± 3.4 | 950 ± 52 | 9.8 ± 0.6 | 13.8 ± 1.0 | |
| Cr 100 ppm | 28.2 ± 2.1a | 719 ± 67a | 3.5 ± 2.0a | 32.4 ± 3.0a | |
| Cr 100 ppm + Vit. C | 43.4 ± 2.0b | 870 ± 69b | 8.2 ± 2.0b | 19.5 ± 1.4b | |
| Control | 42.2 ± 3.4 | 950 ± 52 | 9.8 ± 0.6 | 13.8 ± 1.0 | |
| Cr 200 ppm | 18.3 ± 1.4a | 585 ± 46a | 3.4 ± 0.1a | 35.1 ± 2.2a | |
| Cr 200 ppm + Vit. C | 39.7 ± 2.4b | 909 ± 78b | 6.3 ± 0.4ab | 17.6 ± 1.2b | |
p<0.05. Each value represents mean ± SEM of 20 female rats (from 5 mothers). See materials and methods for experimental details. PND – Postnatal day.
Control vs Cr or Cr+ vitamin C.
Crvs Cr+ vitamin C.
Chromium downregulated FSH receptor (FSHR) mRNA and protein
FSH regulates GC proliferation and inhibits apoptosis [49]. CrIII upregulated plasma FSH through a negative-feedback loop. Therefore, FSHR mRNA in the whole ovary extract and protein in GC were measured. CrIII downregulated FSHR mRNA expression in the ovary (Fig. 3A) and vitamin C mitigated the effect of CrIII in the 50 and 100 ppm groups, but not the 200 ppm group. CrIII downregulated FSHR protein in GC (Figs. 3B and 3C), and vitamin C mitigated CrIII effects in vitro.
Fig. 3.
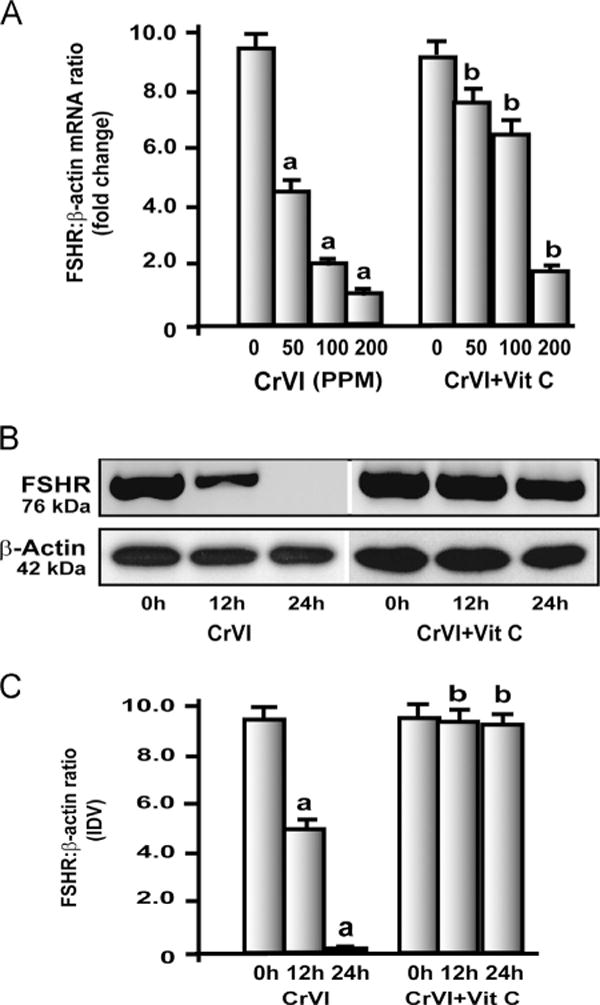
Effects of CrIII on FSH-receptor (FSHR) mRNA expression in the ovary. Experimental details are described under Materials and methods. (A) FSHR mRNA was quantified by real-time PCR in the ovary of rats that were exposed to CrIII through mother’s milk between PND 1 and 21. (B, C) Effects of CrVI treatment on FSHR protein expression in primary cultures of granulosa cells (GC). In vitro experiments were carried out in triplicates in the six groups as follows: control, CrVI 12 h, CrVI 24 h, vitamin C, CrVI 12 h+vitamin C, and CrVI 24h+vitamin C. Cells were treated with 10 μM potassium dichromate with or without pretreatment with 1 mM vitamin C. Expression of FSHR protein was quantified by Western blot analysis. (B) Representative immunoblots of FSHR and β-actin proteins and (C) histogram showing FSHR:β-actin ratio. CrVI treatment decreased FSHR in GCs in a time-dependent manner, and vitamin C inhibited the effects of CrVI treatment. Each value is the mean±SEM of three different plates per treatment, P<0.05; aCrVI treatment, 0 h vs 12 or 24 h; bCrVI+vitamin C (pretreatment), 0 h vs 12 or 24 h.
Chromium increased oxidative stress
Cr is well known to induce oxidative stress [29]. To understand the mechanism behind CrIII-induced follicular atresia, oxidative stress markers LPO and H2O2 were measured both in plasma and in the ovary (Fig. 4). CrIII significantly (P<0.05) increased LPO and H2O2 levels in plasma and ovary in a dose-dependent manner, with a maximum increase induced in F1 rats from the 200 ppm group. Vitamin C significantly inhibited CrIII-induced increases in LPO and H2O2 in the 50 and 100 ppm groups, whereas it mitigated these effects in the 200 ppm group.
Fig. 4.
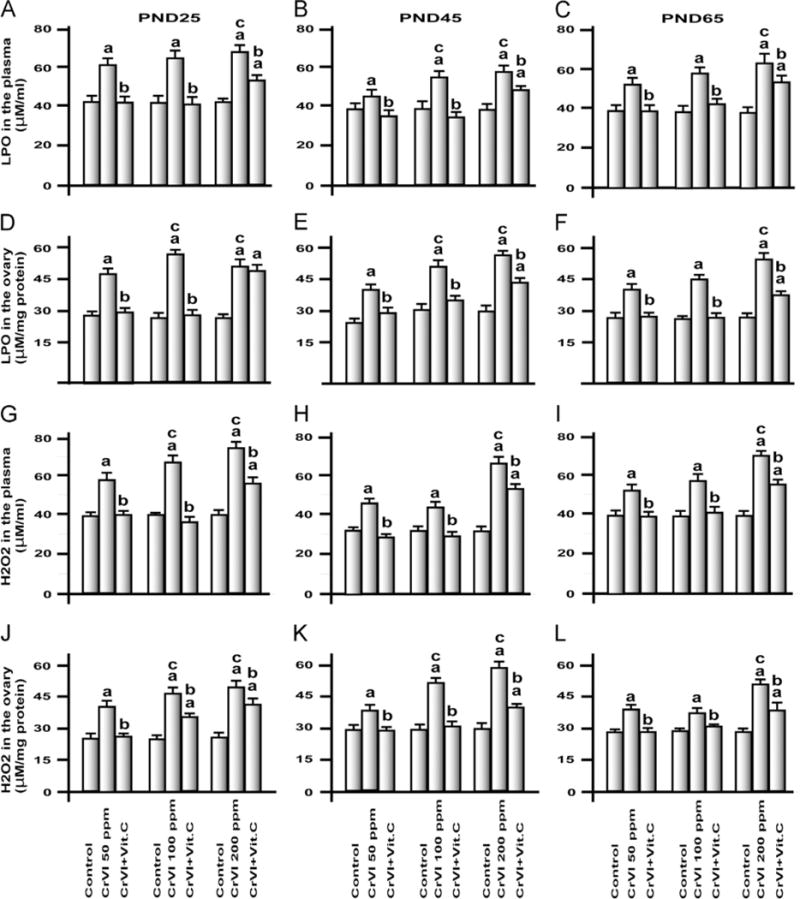
Effects of lactational exposure to CrIII on H2O2 and LPO in the plasma and ovary of F1 offspring. Experimental details are described under Materials and methods. H2O2 and LPO were measured in plasma and ovary of rats that were exposed to CrIII through mother’s milk between PND 1 and 21. LPO in plasma of (A) PND 25, (B) PND 45, and (C) PND 65 rats. LPO in the ovary of (D) PND 25, (E) PND 45, and (F) PND 65 rats. H2O2 in plasma of (G) PND 25, (H) PND 45, and (I) PND 65 rats. H2O2 in the ovary of (J) PND 25, (K) PND 45, and (L) PND 65 rats. CrIII treatment increased LPO and H2O2 in plasma and ovary, and vitamin C supplementation to CrVI-treated mothers mitigated or inhibited the adverse effects of CrIII on the ovary of F1 rats. aControl vs CrVI or CrVI+vitamin C; bCrVI vs CrVI+vitamin C; cCrVI 50 ppm vs CrVI 100 ppm or CrVI 200 ppm. Each value represents the mean±SEM of 20 rats, P<0.05.
Chromium decreased the antioxidants vitamin C, GPx, and GR and increased GST in plasma and ovary
Vitamin C was measured in the plasma and the ovary to estimate the increase in vitamin C levels due to supplementation of vitamin C, as well as to understand the effects of CrIII on vitamin C levels. Oxidative stress changes the ratio between ROS and antioxidants, because the latter are utilized by the cells to quench/scavenge ROS [30]. To understand the effect of CrIII on AOXs in the ovary, the activities of AOXs were measured (Fig. 5). CrIII decreased vitamin C in plasma and ovary, and supplementation of vitamin C resulted in a significant increase in both plasma and ovarian vitamin C (Figs. 5A and B). CrIII invariably increased GST in plasma and ovary of F1 rats from the 50, 100, and 200 ppm groups and vitamin C had no effect on GST (Figs. 5C and D). In PND 25 rats, CrIII significantly (P<0.05) decreased GPx at all doses, whereas in PND 45 and PND 65 rats it increased GPx in the 50 ppm group and decreased it in the 100 and 200 ppm groups (Figs. 5E and F). GR was decreased by CrIII in plasma and ovary in all three groups, irrespective of age (Figs. 5G and H). Interestingly, vitamin C mitigated or inhibited the effects of CrIII on GPx and GR, but not on GST.
Fig. 5.
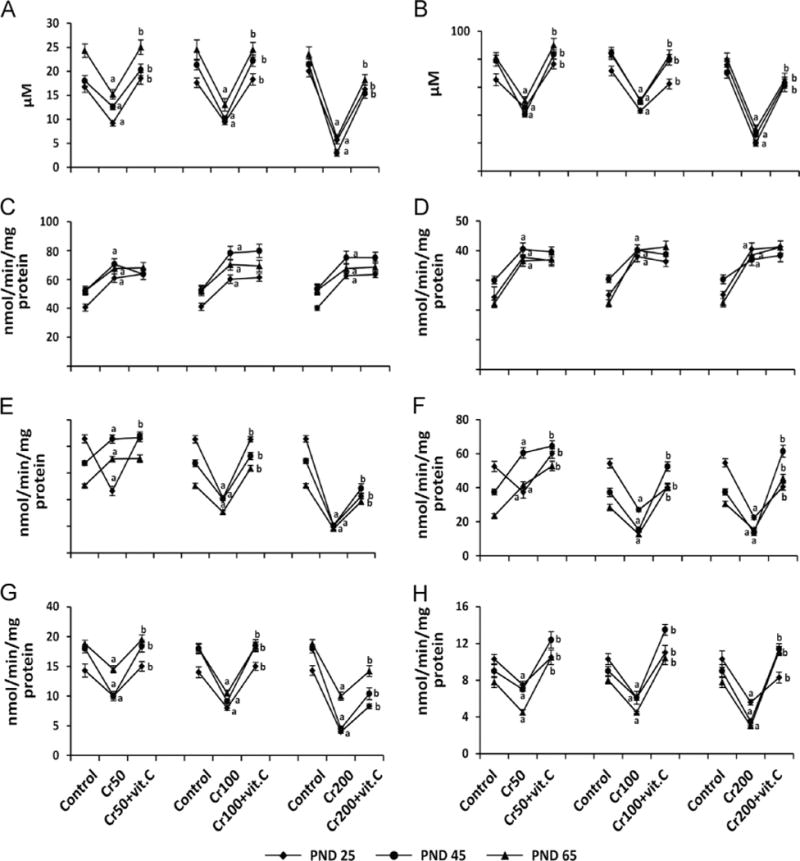
Effects of lactational exposure to CrIII on vitamin C, GST, GPx, and GR in plasma and ovary of F1 offspring. Experimental details are described under Materials and methods. Vitamin C, GPx, GST, and GR were measured in plasma and the whole ovary extracts from rats that were exposed to CrIII through mother’s milk between PND 1 and 21. Vitamin C in (A) plasma and (B) ovary; GST in (C) plasma and (D) ovary; GPx in (E) plasma and (F) ovary; and GR in (G) plasma and (H) ovary of PND 25, PND 45, and PND 65 rats exposed to CrIII lactationally through mother’s milk. Cr treatment decreased vitamin C, GPx, and GR and increased GST in plasma and ovary of F1 offspring in a dose-dependent manner, and vitamin C mitigated or inhibited the effects of CrIII, except on GST. aControl vs CrVI; bCrVI vs CrVI+vitamin C. Each value represents the mean±SEM of 20 rats, P<0.05.
Chromium decreased the antioxidants SOD and catalase in plasma and ovary
CrIII significantly (P<0.05) decreased SOD and catalase in plasma and ovary of PND 25, 45, and 65 rats in a dose-dependent manner, whereas the mitigative effect of vitamin C varied depending on the dose of CrVI to the mothers and the age of F1 rats (Fig. 6). In PND 25 rats, vitamin C inhibited the effects of CrIII on SOD in plasma and ovary in the 50 ppm group and mitigated its effects in the 100 and 200 ppm groups (Figs. 6A and B). In PND 45 and 65 rats, vitamin C inhibited the effects of CrIII on SOD in the 50 and 100 ppm groups and mitigated the effects of CrIII in the 200 ppm group (Figs. 6A and B). Mitigative or inhibitory effects of vitamin C on CrIII-induced changes in catalase were the same as those of SOD (Figs. 6C and D).
Fig. 6.
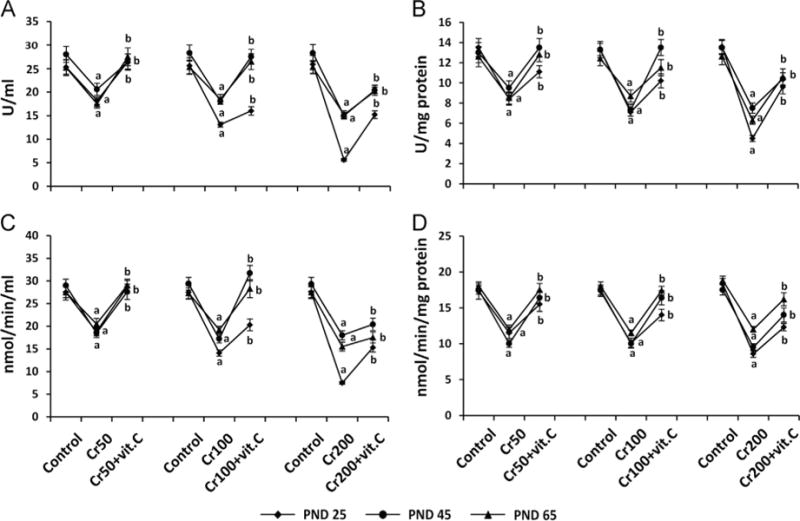
Effects of lactational exposure to CrIII on SOD and catalase in the plasma and the ovary of F1 offspring. Experimental details are described under Materials and methods. SOD and catalase were measured in plasma and whole ovary extracts from rats that were exposed to CrVI through mother’s milk between PND 1 and 21. SOD in (A) plasma and (B) ovary and catalase in (C) plasma and (D) ovary of PND 25, PND 45, and PND 65 rats exposed to CrIII lactationally through mother’s milk. CrIII treatment decreased SOD and catalase in plasma and ovary of F1 offspring in a dose-dependent manner, and vitamin C mitigated or inhibited the effects of CrIII. aControl vs CrVI; bCrVI vs CrVI+vitamin C. Each value represents the mean±SEM of 20 rats, P<0.05.
In vitro studies
Exposure to CrVI increased ROS in granulosa and theca cells in vitro
To understand the cell-specific response of GCs and TCs to CrVI treatment and intervention of vitamin C, primary cultures of GCs and TCs from immature rats were treated with CrVI with or without vitamin C pretreatment. Immortalized granulosa cells, SIGCs, were also used to compare the responses with primary cultures of GC and to validate SIGCs as a model to understand heavy metal toxicity (Fig. 7). Data indicate that CrVI treatment increased LPO and H2O2 levels in GC and TC in vitro by 12 and 24 h, whereas in SIGCs, LPO and H2O2 were increased only at 24 h after CrVI treatment. Vitamin C mitigated or inhibited the increase in LPO and H2O2 due to CrVI treatment in GCs, SIGCs, and TCs.
Fig. 7.
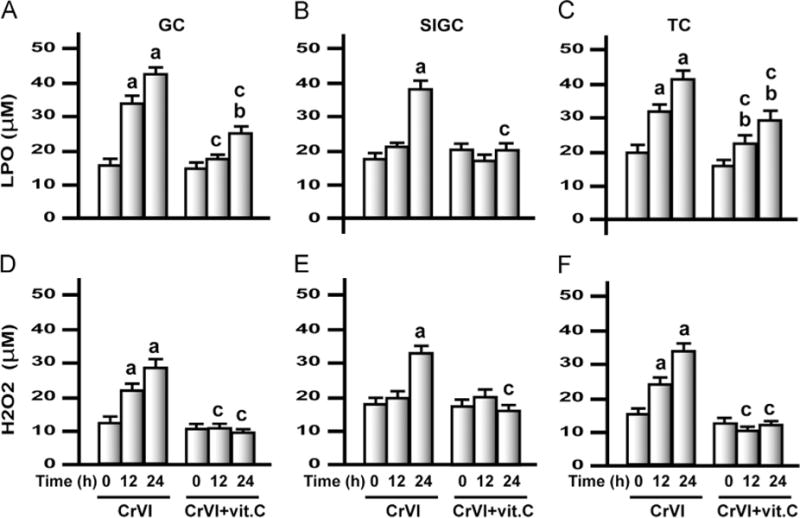
Effects of exposure to CrVI on LPO and H2O2 in primary cultures of granulosa cells (GCs), the spontaneously immortalized rat granulosa cell line (SIGCs), and theca cells (TCs). In vitro experiments were carried out in triplicates in six groups as follows: control, CrVI 12 h, CrVI 24 h, vitamin C, CrVI 12 h+vitamin C, and CrVI 24 h+vitamin C. Cells were treated with 10 μM potassium dichromate with or without pretreatment with 1 mM vitamin C. H2O2 and LPO were measured in the culture medium. Histograms depict LPO in (A) GCs, (B) SIGCs, and (C) TCs and H2O2 in (D) GCs, (E) SIGCs, and (F) TCs. CrVI increased LPO and H2O2 in GCs, SIGCs, and TCs in a time-dependent manner, and vitamin C pretreatment mitigated or inhibited the effects of CrVI treatment. Each value is the mean±SEM of three different plates per treatment, P<0.05. aCrVI treatment, 0 h vs 12 or 24 h; bCrVI+vitamin C, 0 h vs 12 or 24 h; cCrVI (12 or 24 h) vs CrVI+vitamin C (12 or 24 h).
Exposure to CrVI decreased vitamin C and antioxidant levels in granulosa and theca cells in vitro
The effects of CrVI treatment on the intracellular level of vitamin C and the activity of AOXs were measured in culture medium. CrVI treatment significantly (P<0.05) decreased vitamin C, GPx, GST, GR, SOD, and catalase of GCs, SIGCs, and TCs after 12 and 24 h of treatment in a time-dependent manner (Fig. 8). Vitamin C pretreatment elevated its own concentration in the cells and mitigated the effects of CrVI treatment on all the enzymes from GCs, SIGCs, and TCs and inhibited the effects of CrVI on catalase in SIGCs alone.
Fig. 8.
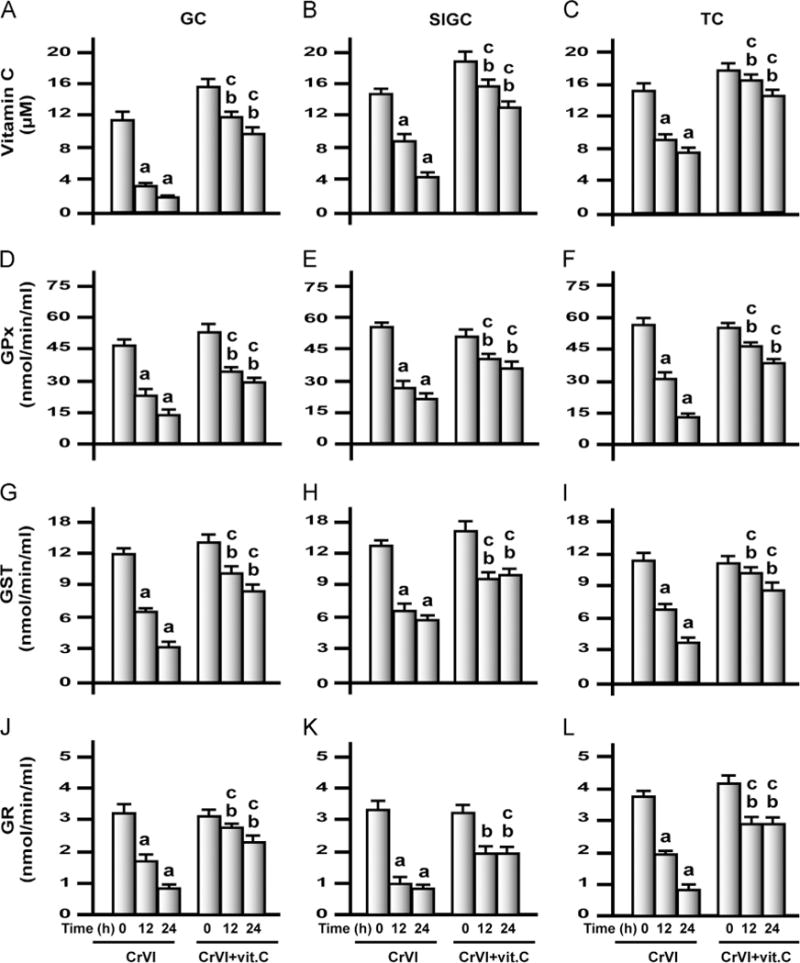
Effects of exposure to CrVI on vitamin C, GPx, GST, and GR in GCs, SIGCs, and TCs. GCs, TCs, and SIGCs were cultured and treated with CrVI with or without vitamin C pretreatment, as described in the legend to Fig. 7. Histograms depict vitamin C in (A) GCs, (B) SIGCs, and (C) TCs; GPx in (D) GCs, (E) SIGCs, and (F) TCs; GST in (G) GCs, (H) SIGCs, and (I) TCs; and GR in (J) GCs, (K) SIGCs, and (L) TCs. CrVI treatment decreased vitamin C, GPx, GST, and GR in GCs, SIGCs, and TCs in a time-dependent manner, and vitamin C pretreatment mitigated or inhibited the effects of CrVI treatment. Each value is the mean±SEM of three different plates per treatment, P<0.05. aCrVI treatment, 0 h vs 12 or 24 h; bCrVI+vitamin C, 0 h vs 12 or 24 h; cCrVI (12 or 24 h) vs CrVI+vitamin C (12 or 24 h).
Exposure to CrVI decreased mRNA expression of antioxidant enzymes in granulosa and theca cells in vitro
GSH depletion within the mitochondria is an important trigger for the apoptotic pathway [52]. GSTs catalyze GSH conjugation to electrophilic compounds through thioether linkages [53]. GSSG, the product of GSH oxidation, which is toxic to cells, is reduced by GR to restore GSH levels. As a consequence, the ratio of GSH/GSSG in cells is normally maintained at about 100:1 [54]. From our earlier study, it is evident that mitochondria play a crucial role in CrVI-induced apoptosis [38]. Mitochondrial levels of H2O2 were elevated under depletion of PRDX3 [35]. Even though GPx is present in the mitochondria, identification of mitochondria-specific AOXs such as PRDX3 and TXN has drawn more attention to understanding the mechanism of apoptosis mediated through mitochondrial ROS. Therefore, we studied the effects of CrVI exposure on the mRNA expression of important cytosolic and mitochondrial AOXs in GCs, SIGCs, and TCs (Figs. 9–12), and the overall trend of modulation is summarized in Supplementary Table 3.
Fig. 9.
Effects of exposure to CrVI on SOD and catalase in GCs, SIGCs, and TCs. GCs, SIGCs, and TCs were cultured and treated with CrVI with or without vitamin C pretreatment, as described in the legend to Fig. 7. Histograms depict SOD in (A) GCs, (B) SIGCs, and (C) TCs and catalase in (D) GCs, (E) SIGCs, and (F) TCs. CrVI treatment decreased SOD and catalase in GCs, SIGCs, and TCs in a time-dependent manner, and vitamin C pretreatment mitigated or inhibited the effects of CrVI. Each value is the mean±SEM of three different plates per treatment, P<0.05. aCrVI treatment, 0 h vs 12 or 24 h; bCrVI+vitamin C (pretreatment), 0 h vs 12 or 24 h; cCrVI (12 or 24 h) vs CrVI+vitamin C (12 or 24 h).
Fig. 12.
Effects of exposure to CrVI on mRNA expression of TXN1, TXN2, TXNRD2, and PRDX3 in GCs, SIGCs, and TCs. GCs, SIGCs, and TCs were cultured and treated with CrVI with or without vitamin C pretreatment, as described in the legend to Fig. 7. Histograms depict expression of TXN1 mRNA in (A) GCs, (B) SIGCs, and (C) TCs; TXN2 mRNA in (D) GCs, (E) SIGCs, and (F) TCs; TXNRD2 mRNA in (G) GCs, (H) SIGCs, and (I) TCs; and PRDX3 mRNA in (J) GCs, (K) SIGCs, and (L) TCs. CrVI treatment decreased mRNA expression of TXN1, TXN2, TXNRD2, and PRDX3 and vitamin C mitigated or inhibited the effects of exposure to CrVI in GCs and SIGCs. In TCs, vitamin C failed to mitigate effects CrVI treatment on TXN1 and TXN2. Each value is the mean±SEM of three different samples per treatment, P<0.05. aCrVI treatment, 0 h vs 12 or 24 h; bCrVI+vitamin C (pretreatment), 0 h vs 12 or 24 h; cCrVI (12 or 24 h) vs CrVI + vitamin C (12 or 24 h).
Treatment of GCs, SIGCs, and TCs with CrVI resulted in the downregulation of mRNA expression of all 12 AOX enzymes (SOD1, SOD2, catalase, GLRX1, GSTM1, GSTM2, GSTA, GR, TXN1, TXN2, TXNRD2, and PRDX3) by 12 and 24 h in a time-dependent manner (Figs. 9–12). Obviously, the reduction in mRNA levels of all the enzymes was higher after 24 h of treatment compared to 12 h. We observed cell-specific changes in the intervention effects of vitamin C against CrVI treatment. In GCs, vitamin C mitigated CrVI effects on SOD1, SOD2, GLRX1, GSTM1, GSTA, GSHR, TXN1, TXN2, TXNRD2, and PRDX3 after 12 and 24 h. Vitamin C also inhibited the CrVI effects on GSTM2 after 12 h but did not mitigate the effect of CrVI on catalase. In SIGCs, vitamin C mitigated CrVI effects on all the enzymes just as in GCs, with the exception of catalase after 12 h of treatment. In TCs, vitamin C mitigated CrVI effects on SOD1, SOD2, catalase, GLRX1, GSTA, GR, TXNRD2, and PRDX3 by 12 and 24 h; inhibited the effects on SOD2 and GSTA4 after 12 h; but failed to mitigate CrVI effects on GSTM1, GSTM2, TXN1, and TXN2 after either 12 or 24 h.
Discussion
Our previous studies indicated that lactational exposure to CrIII during the neonatal/postnatal period (PND 1–21) resulted in a marked delay in pubertal onset due to an arrest in follicle development at the secondary follicle stage and failure in the development of antral follicles [55]. Exposure to CrVI induced granulosa cell apoptosis through the p53 pathway activated by ERK1/2 and by shifting the subcellular translocation of mitochondrial apoptotic machinery of the intrinsic apoptotic pathway [38]. Treatment with CrVI also resulted in the downregulation of cyclins and cyclin-dependent kinases (CDK) and upregulated CDK inhibitors in GCs to manifest an arrest of cell cycle progression in G1–S/G2–M phase transitions of the cell cycle [49], which could be an additional mechanism for the delay/arrest in follicle development. However, the key regulator(s) for many of the above-mentioned adverse and toxic effects of Cr has not yet been identified. Our data clearly indicated that lactational exposure to CrIII accelerated follicular atresia in PND 25, 45, and 65 rats in a dose-dependent and age-dependent manner. This was accompanied by an increase in oxidative stress and depletion of AOXs in the plasma and the ovary. To understand the effects of CrVI on GCs and TCs, we studied key AOXs localized in the cytosol and mitochondria that contribute to AOX defense and scavenging of free radicals in the ovarian microenvironment. Our data provide clear evidence for the effects of CrVI treatment that increased ROS, with a concurrent decrease in mRNA expression of AOXs in both GCs and TCs.
Long-term (90 days) exposure to high-dose CrVI (520 mg/L) in drinking water has been reported to induce anemia and oxidative stress in plasma and intestine in rodents [22,23]. Similarly, exposure of rats to methimazole, an anti-thyroid drug, during gestation and lactation induced anemia in the mother rats and suckling pups, resulting in increased oxidative stress and depletion of antioxidants [56]. Direct exposure of rats to CrVI increased oxidative stress in erythrocytes [57]. Exposure to sodium dichromate dihydrate (0–1000 mg/L) for 3 months caused a microcytic hypochromic anemia in rats and mice [58]. In support of the above studies, the current in vivo study suggests that lactational exposure to CrIII resulted in increased oxidative stress and depletion of antioxidants in plasma and ovary of suckling offspring. Taken together, one or more of the above mechanisms might have contributed to the CrIII-induced follicular atresia and GC apoptosis in F1 offspring.
CrVI is rapidly transported into the cells via plasma membrane anion transporters and reduced/detoxified to CrIII [17,18]. Whereas CrVI enters cells by facilitated transport, CrIII enters cells by passive diffusion or phagocytosis of precipitates, resulting in much lower uptake [59,60]. However, Sayato et al. [61] demonstrated the comparative metabolic fate of labeled chromium chloride (CrIII) and sodium chromate (CrVI) and interaction of these compounds in rat liver and blood after their oral and intravenous administration. Gastrointestinal absorption of both compounds was below 1% of the oral dose with similar rate of absorption. Studies in mice and yeast clearly indicate that CrIII causes more damage to the DNA than CrVI [62]. In the cellular reduction of CrVI, a spectrum of ROS ( , H2O2, and •OH) is produced. Once inside the cells or in body fluids, most CrVI will be converted to CrIII. Obviously, the predominant form of chromium in the mother’s milk in this study was likely to be CrIII. CrIII induces oxidative stress to a lesser extent compared to CrVI [60]. Although CrIII was thought to be relatively nontoxic, spin trapping studies by Ozawa and Hanaki [63] have demonstrated that CrIII can be reduced to CrII by biological reductants such as L-cysteine and NADH. The newly formed CrII reacts with hydrogen peroxide to generate hydroxyl radical, resulting in the tissue-damaging effects and systemic toxicity. This could also be attributed to CrIII-induced oxidative stress and follicular atresia in suckling offspring in the current study, apart from the anticipated anemic effects of CrIII or CrII in the mothers and/or suckling pups.
Our data indicated that lactational exposure to CrIII increased lipid peroxidation and elevated H2O2 concentration in plasma and ovary in a dose-dependent manner, suggesting that CrIII may have induced follicular atresia through oxidative stress. This is supported by similar studies from other groups [46,64]. Free radicals have a dual role in the reproductive tract, as they are among the key signaling molecules modulating various reproductive functions [65]. Therefore, it may not be enough to consider an increase in ROS alone as a contributing factor to follicular atresia. To further understand the effects of CrIII on AOXs, we have estimated GPx, GR, GST, SOD, and catalase. Our data indicated that lactational exposure to CrIII decreased GR, GPx, and catalase in plasma and ovary, in a dose-dependent manner, which is either mitigated or inhibited by vitamin C. GPx present in the mitochondria plays a crucial role in metabolizing H2O2. Even though catalase also removes H2O2, it is not present in mitochondria [66]. GPx and catalase convert H2O2 into water and molecular O2[67]. CrIII increased H2O2 levels both in the plasma and in the ovary while decreasing GPx and catalase. It is suggested that the ovary of the F1 rats exposed to CrIII may have utilized the available GPx and catalase to remove elevated H2O2 and therefore resulted in depletion of GPx and catalase. Interestingly, CrIII increased GST in plasma and ovary in all the age groups. GSTs not only catalyze the conjugation of reduced glutathione via sulfhydryl groups but also bind to toxins and function as transport proteins. We suggest that increased GSTs (total) resulting from CrIII toxicity may facilitate the excretion of CrIII, to protect the ovary. In addition, CrIII decreased total SOD in plasma and ovary of PND 25, 45, and 65 rats. On the other hand, results from the in vitro experiments reveal that treatment with CrVI also decreased mRNA levels of SOD1, SOD2, and catalase in GCs, SIGCs, and TCs. It has been established through knockout studies that SOD1, and not SOD2, is important to maintain normal fertility in mice [31]. However, this study suggests that SOD1 may be important to maintain normal physiology of the ovary, whereas SOD2 (localized in mitochondria) plays a defensive role to protect the ovary from mitochondrial ROS induced by Cr. A recent novel finding demonstrated that ovarian expression of genes associated with ovarian thiol redox balance and cellular detoxification of electrophilic compounds, such as PRDX3, TXN2, GRX1, and GSTM2, were downregulated during aging in the mouse ovary [36]. Mitochondria-specific AOXs, such as PRDX3, TXN2, and TXNRD2, provide a major line of defense against mitochondrial ROS [36]. PRDX3 depletion in cells leads to increased H2O2 levels in mitochondria [68]. This study indicated that treatment with CrVI decreased GLRX1 mRNA levels in GCs, SIGCs, and TCs in a time-dependent manner. GLRX is important for the survival of the oocytes. GLRX binds to apoptosis signal-regulating kinase and prevents the onset of apoptosis [69]. Degenerating oocytes showed a significant decrease or absence of GLRX expression [70]. Moreover, cells overexpressing GLRX are more resistant to cadmium-induced apoptosis [71]. Thus the Cr-induced decrease in GLRX expression may predispose GCs and TCs to undergo apoptosis.
Lactational exposure to CrIII increased H2O2 levels in the ovary. Intracellular H2O2 is removed mostly by catalase, GPx, and PRDX. The PRDX concentration in mitochondria is 30 times higher than GPx1 [68]; and GPx1 clears only 15% of mitochondrial H2O2. PRDX3 is more abundant in mitochondria and is a major regulator of mitochondrial H2O2 concentration and apoptosis [68]. PRDX3 functions as a suppressor of mitochondria-mediated apoptosis by eliminating H2O2 in mitochondria [68]. Moreover, an age-related decrease in the expression of PRDX3 and TXN2 is indicative of increased susceptibility of mitochondria in aging ovaries to oxidative damage [36]. Therefore, we also studied the effects of CrVI treatment on the mitochondrial AOXs in addition to cytosolic AOX components. The mRNA of the mitochondrial AOXs TXN2 and PRDX3 was downregulated by CrVI treatment in GCs, SIGCs, and TCs, suggesting that cellular thiol redox homeostasis is disturbed by CrVI exposure, leading to increased vulnerability of the ovary to oxidative stress and follicular atresia. GSTM1, GSTM2, and GSTA4 play key roles in the cellular detoxification of electrophilic compounds through GSH conjugation and in protection against lipid peroxidation. The observed decrease in GSTM1, GSTM2, and GSTA4 in GCs and TCs suggests a decrease in cellular resistance to oxidative stress caused by CrVI treatment. Current data showed a discrepancy between in vivo and in vitro analyses of GSTs. Whereas CrVI exposure increased total GST activity in vivo, GSTM1, GSTM2, and GSTA4 were downregulated by CrVI treatment in GCs and TCs in vitro. There are several isoforms of GST that have different roles to play in oxidative damage [72]. It is suggested that although GSTM1, GSTM2, and GSTA4 were decreased by CrVI treatment in GCs and TCs, other isoforms may have increased under in vivo conditions. Further studies are needed on all other different isoforms of GST to understand the reason for this discrepancy. Interestingly, mRNA expression of AOXs showed a cell-specific response to vitamin C in mitigating the effect of CrVI treatment. Vitamin C did not rescue TC from the effect of CrVI treatment on GSTM1, GSTM2, TXN1, and TXN2. Even though AOXs such as vitamin C and GSH are present in the ovary at very high concentrations, it is possible that different AOX systems may operate in different ovarian cell types to defend against oxidative damage. Overall, this study clearly indicates that Cr accelerated follicular atresia and apoptosis of GCs through the downregulation of cytosolic and mitochondrial AOXs and disruption of the cellular thiol-redox balance leading to increased oxidative damage to mitochondria.
Understanding the mechanisms through which various toxicants affect/disrupt ovarian steroidogenesis and folliculogenesis at the molecular and cellular levels requires readily available cells for in vitro studies [73]. Because of several limitations in obtaining granulosa cells, particularly from human, and to reduce the use of experimental animals, the use of granulosa cell lines for in vitro model systems has become an attractive alternative. Our recent study validated SIGCs and compared their responses to CrVI treatment with primary cultures of GCs [49], suggesting SIGCs as a good model system to study metal toxicity and mitigative effects of vitamin C. This study also indicates that the cellular responses of SIGCs to CrVI treatment in altering the AOX to ROS balance and predisposing cells to apoptotic death are comparable to those of GCs; thus SIGCs could stand as a functionally relevant granulosa cell line model to understand the mechanism of metal toxicity on the ovary.
E2 plays important roles in preventing oxidative stress. E2 depletion leads to apoptosis and DNA damage, as well as elevated oxidative stress, in the lymphocytes of rats, which are reversed by supplementing E2[74]. E2 supplementation reversed the age-related depletion of AOXs and increase in free radicals in rat liver [75]. Therefore, this study suggests that decreased levels of E2, T, and P4 due to increased follicular atresia may predispose the ovary to decreased AOX gene transcription and activity, resulting in oxidative stress. On the other hand, decreased AOX levels may also have indirectly contributed to the decrease in steroidogenic machinery and diminished steroidogenesis. However, more studies are needed to confirm the role of hormones on AOXs, and that of AOXs on the stepwise synthesis of steroids by ovarian cells. Moreover, CrIII increased FSH levels in F1 rats, through a negative-feedback mechanism. Interestingly, FSHR mRNA was downregulated in the ovary of F1 rats. This is also confirmed by the data from the in vitro studies, in that CrVI treatment downregulated FSHR protein in GCs, which was inhibited by vitamin C. This suggests that increased apoptosis of granulosa cells of the ovary and accelerated follicular atresia, despite the elevated FSH, could be partly due to the downregulation of FSHR by CrIII.
Our recent studies have documented a protective role for vitamin C against Cr-induced reproductive toxicity at the cellular and molecular levels [38,49,55]. In this study, vitamin C played a protective role against the adverse effects of CrVI treatment on transcription of mitochondrial AOX enzymes in GCs and TCs. However, the actual mechanism by which vitamin C ameliorates Cr-induced ovarian toxicity is not clearly understood. Vitamin C is transported into the cells via the sodium-dependent vitamin C transporters 1 and 2, which lead to accumulation of vitamin C within cells against a concentration gradient [76]. Interestingly, vitamin C has been reported to mitigate or inhibit ROS by either decreasing oxidative stress or increasing AOX enzymes or doing both in various tissues against a variety of toxicants such as cisplatin-induced testicular toxicity [77], endosulfan-induced systemic toxicity [78], imidaclopid- [79] or ethanol- [80] induced hepatotoxicity, arsenic-induced liver and testicular toxicity [81], and Cr-induced ovarian toxicity [38,49]. Vitamin C enters mitochondria via facilitative glucose transporters 1 and 10 and confers mitochondrial protection against oxidative injury [82]. Based on the previous reports from our studies and others, and on the current data, we suggest that the protective effects of vitamin C against Cr-induced ovarian toxicity may be attained through one or more of the following mechanisms: (i) reduction in free radicals; (ii) increase in cytosolic AOXs; (iii) increased intramitochondrial transport of vitamin C to scavenge mitochondrial ROS, thus preventing changes in mitochondrial membrane potential and leakage of cytochrome c; and (iv) vitamin C-induced increase in mitochondrial AOXs. In conclusion, these data suggest that CrIII induced follicular atresia and apoptosis of GCs by increasing intraovarian ROS levels along with a depletion of cytoplasmic and mitochondrial AOXs. Supplementation of vitamin C to CrVI-treated rats, or pretreatment with vitamin C of CrVI-treated cells, mitigated or inhibited the adverse effects of Cr on follicular atresia in F1 offspring or GC/TC apoptosis by decreasing ROS and increasing AOXs.
Supplementary Material
Fig. 10.
Effects of exposure to CrVI on mRNA levels of SOD1, SOD2, and catalase and GLRX1 in GCs, SIGCs, and TCs. GCs and TCs were isolated from control immature rats (PND 23–26), as described under Materials and methods. GCs, SIGCs, and TCs were cultured and treated with CrVI with or without vitamin C pretreatment, as described in the legend to Fig. 7. Histograms depict expression of SOD1 mRNA in (A) GCs, (B) SIGCs, and (C) TCs; SOD2 mRNA in (D) GCs, (E) SIGCs, and (F) TCs; catalase mRNA in (G) GCs, (H) SIGCs, and (I) TCs; and GLRX1 mRNA in (J) GCs, (K) SIGCs, and (L) TCs. CrVI treatment decreased mRNA expression of SOD1, SOD2, catalase, and GLRX1, and vitamin C pretreatment mitigated or inhibited the effects of CrVI treatment. Each value is the mean±SEM of three different samples per treatment, P<0.05. aCrVI treatment, 0 h vs 12 or 24 h; bCrVI+vitamin C (pretreatment), 0 h vs 12 or 24 h; cCrVI (12 or 24 h) vs CrVI+vitamin C (12 or 24 h).
Fig. 11.
Effects of exposure to CrVI on mRNA expression of GSTM1, GSTM2, GSTA4, and GSHR in GCs, SIGCs, and TCs. GCs and TCs were isolated from control immature rats (PND 23–26), as described under Materials and methods. GCs, SIGCs, and TCs were cultured and treated with CrVI with or without vitamin C pretreatment, as described in the legend to Fig. 7. Histograms depict expression of GSTM1 mRNA in (A) GCs, (B) SIGCs, and (C) TCs; GSTM2 mRNA in (D) GCs, (E) SIGCs, and (F) TCs; GSTA4 mRNA in (G) GCs, (H) SIGCs, and (I) TCs; and GSHR mRNA in (J) GCs, (K) SIGCs, and (L) TCs. CrVI treatment decreased mRNA expression of GSTM1, GSTM2, GSTA4, and GSHR, and vitamin C pretreatment mitigated or inhibited the effects of CrVI in GCs and SIGCs. In TCs, vitamin C failed to mitigate the effects of CrVI on GSTM1 and GSTM2. Each value is the mean±SEM of three different samples per treatment, P<0.05. aCrVI treatment, 0 h vs 12 or 24 h; bCrVI+vitamin C (pretreatment), 0 h vs 12 or 24 h; cCrVI (12 or 24 h) vs CrVI+vitamin C (12 or 24 h).
Acknowledgments
This work was supported by National Institutes of Health/National Institute of Environmental Health Sciences Grants ES016605-A2 and ES020561-01 to S.K.B. We acknowledge Dr. Rola Barhoumi, Department of Integrative Biosciences, Texas A&M University, for the assistance with statistical analysis. We acknowledge Dr. Gerald Bratton and Dr. Robert Taylor for their help in measuring Cr from the breast milk, drinking water, and bedding of the animal cages. We acknowledge Dr. Vincent Gresham and Ms. Andrea Moss for their help in collecting milk from rats to confirm Cr in the breast milk.
Abbreviations
- AOX
antioxidant enzyme
- BAD
B cell lymphoma 2-associated agonist of cell death
- BAX
B cell lymphoma 2-associated X protein
- BSA
bovine serum albumin
- CDK
cyclin-dependent kinase
- DMEM
Dulbecco’s modified Eagle medium
- E2
estradiol
- ERK
extracellular-signal-regulated kinase
- FSH
follicle-stimulating hormone
- GC
granulosa cell
- GLRX
glutaredoxin
- GPx
glutathione peroxidase
- GSH
glutathione
- GSSG
oxidized glutathione
- GST
glutathione S-transferase
- Hepes
(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- LPO
lipid hydroperoxide
- GR
glutathione reductase
- P4
progesterone
- PND
postnatal day
- PRDX3
peroxiredoxin 3
- ROS
reactive oxygen species
- SIGC
spontaneously immortalized rat granulosa cell
- SOD
superoxide dismutase
- T
testosterone
- TC
theca cell
- TXN
thioredoxin
- TXNRD
thioredoxin reductase
Appendix A. Supplementary Information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed. 2013.02.006.
References
- 1.Costa M, Klein CB. Toxicity and carcinogenicity of chromium compounds in humans. Crit Rev Toxicol. 2006;36:155–163. doi: 10.1080/10408440500534032. [DOI] [PubMed] [Google Scholar]
- 2.OSHA. Occupational exposure to hexavalent chromium; final rule. Washington, DC: Department of Labor, Occupational Safety and Health Administration; 2006. [Google Scholar]
- 3.Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol. 2008;21:28–44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Honeycutt ME. Hexavalent chromium in Texas drinking water. Toxicol Sci. 2010;119:423–424. doi: 10.1093/toxsci/kfq347. [DOI] [PubMed] [Google Scholar]
- 5.Layton L. Probable carcinogen hexavalent chromium found in drinking water of 31 U.S. cities. Washington DC: The Washington Post; 2010. [Google Scholar]
- 6.Luippold RS, Mundt KA, Austin RP, Liebig E, Panko J, Crump C, Crump K, Proctor D. Lung cancer mortality among chromate production workers. Occup Environ Med. 2003;60:451–457. doi: 10.1136/oem.60.6.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonde JP. The risk of male subfecundity attributable to welding of metals: studies of semen quality, infertility, fertility, adverse pregnancy outcome and childhood malignancy. Int J Androl. 1993;1:1–29. doi: 10.1111/j.1365-2605.1993.tb01367.x. [DOI] [PubMed] [Google Scholar]
- 8.Shmitova LA. [Content of hexavalent chromium in the biological substrates of pregnant women and puerperae engaged in the manufacture of chromium compounds] Gig Tr Prof Zabol. 1980;2:33–35. [PubMed] [Google Scholar]
- 9.Jendryczko A, Drozdz M, Magner K. [Preliminary studies of chromium concentration in the myometrium in the third trimester of pregnancy, in chorionic tissue in the first trimester and in the blood of pregnant women] Ginekol Pol. 1984;55:691–694. [PubMed] [Google Scholar]
- 10.Greene LE, Riederer AM, Marcus M, Lkhasuren O. Associations of fertility and pregnancy outcomes with leather tannery work in Mongolia: a pilot study. Int J Occup Environ Health. 2010;16:60–68. doi: 10.1179/107735210800546100. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Cai WW, Lee DJ. Occupational hazards and pregnancy outcomes. Am J Ind Med. 1992;21:397–408. doi: 10.1002/ajim.4700210312. [DOI] [PubMed] [Google Scholar]
- 12.Barceloux DG. Chromium. J Toxicol Clin Toxicol. 1999;37:173–194. doi: 10.1081/clt-100102418. [DOI] [PubMed] [Google Scholar]
- 13.Campbell MA, Li L-H, Wu KL, Dunn A, Roth L, Beaumont J. Reproductive and cancer hazard assessment section office of environmental health hazard assessment. California Environmental Protection Agency; 2009. Evidence on the developmental and reproductive toxicity of chromium (hexavalent compounds) pp. 3–97. [Google Scholar]
- 14.Andrews RE, Shah KM, Wilkinson JM, Gartland A. Effects of cobalt and chromium ions at clinically equivalent concentrations after metal-on-metal hip replacement on human osteoblasts and osteoclasts: implications for skeletal health. Bone. 2011;49:717–723. doi: 10.1016/j.bone.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Marouani N, Tebourbi O, Mokni M, Yacoubi MT, Sakly M, Benkhalifa M, Ben Rhouma K. Embryotoxicity and fetotoxicity following intraperitoneal administrations of hexavalent chromium to pregnant rats. Zygote. 2011;19:229–235. doi: 10.1017/S0967199410000274. [DOI] [PubMed] [Google Scholar]
- 16.Apostoli P, Catalani S. Metal ions affecting reproduction and development. Met Ions Life Sci. 2011;8:263–303. [PubMed] [Google Scholar]
- 17.DeFlora S, Serra D, Basso C, Zanacchi P. Mechanistic aspects of chromium carcinogenicity. Arch Toxicol. 1989;13:28–39. doi: 10.1007/978-3-642-74117-3_3. [DOI] [PubMed] [Google Scholar]
- 18.Alexander J, Aaseth J. Uptake of chromate in human red blood cells and isolated rat liver cells: the role of the anion carrier. Analyst. 1995;120:931–933. doi: 10.1039/an9952000931. [DOI] [PubMed] [Google Scholar]
- 19.Wetterhahn KE, Hamilton JW. Molecular basis of hexavalent chromium carcinogenicity: effect on gene expression. Sci Total Environ. 1989;86:113–129. doi: 10.1016/0048-9697(89)90199-x. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Q, Kluz T, Salnikow K, Costa M. Comparison of the cytotoxicity, cellular uptake, and DNA–protein crosslinks induced by potassium chromate in lymphoblast cell lines derived from three different individuals. Biol Trace Elem Res. 2002;86:11–22. doi: 10.1385/BTER:86:1:11. [DOI] [PubMed] [Google Scholar]
- 21.Zhitkovich A. Importance of chromium–DNA adducts in mutagenicity and toxicity of chromium(VI) Chem Res Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]
- 22.Thompson CM, Proctor DM, Suh M, Haws LC, Hebert CD, Mann JF, Shertzer HG, Hixon JG, Harris MA. Comparison of the effects of hexavalent chromium in the alimentary canal of F344 rats and B6C3F1 mice following exposure in drinking water: implications for carcinogenic modes of action. Toxicol Sci. 2012;125:79–90. doi: 10.1093/toxsci/kfr280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson CM, Proctor DM, Haws LC, Hebert CD, Grimes SD, Shertzer HG, Kopec AK, Hixon JG, Zacharewski TR, Harris MA. Investigation of the mode of action underlying the tumorigenic response induced in B6C3F1 mice exposed orally to hexavalent chromium. Toxicol Sci. 2011;123:58–70. doi: 10.1093/toxsci/kfr164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Chen T, LaCerte C, Wise SS, Holmes A, Martino J, Wise JP, Jr, Thompson WD, Wise JP., Sr Comparative cytotoxicity and genotoxicity of particulate and soluble hexavalent chromium in human and sperm whale (Physeter macrocephalus) skin cells. Comp Biochem Physiol Toxicol Pharmacol. 2012;155:143–150. doi: 10.1016/j.cbpc.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proctor DM, Thompson CM, Suh M, Harris MA. A response to “A quantitative assessment of the carcinogenicity of hexavalent chromium by the oral route and its relevance to human exposure.”. Environ Res. 2011;111:468–470. doi: 10.1016/j.envres.2011.01.019. discussion 471–462. [DOI] [PubMed] [Google Scholar]
- 26.Thompson CM, Haws LC, Harris MA, Gatto NM, Proctor DM. Application of the U.S. EPA mode of action framework for purposes of guiding future research: a case study involving the oral carcinogenicity of hexavalent chromium. Toxicol Sci. 2011;119:20–40. doi: 10.1093/toxsci/kfq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wise SS, Holmes AL, Qin Q, Xie H, Katsifis SP, Thompson WD, Wise JP., Sr Comparative genotoxicity and cytotoxicity of four hexavalent chromium compounds in human bronchial cells. Chem Res Toxicol. 2010;23:365–372. doi: 10.1021/tx900363j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Chen T, Wise SS, Holmes A, Shaffiey F, Wise JP, Jr, Thompson WD, Kraus S, Wise JP., Sr Cytotoxicity and genotoxicity of hexavalent chromium in human and North Atlantic right whale (Eubalaena glacialis) lung cells. Comp Biochem Physiol Toxicol Pharmacol. 2009;150:487–494. doi: 10.1016/j.cbpc.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 30.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Matzuk MM, Dionne L, Guo Q, Kumar TR, Lebovitz RM. Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology. 1998;139:4008–4011. doi: 10.1210/endo.139.9.6289. [DOI] [PubMed] [Google Scholar]
- 32.Matos L, Stevenson D, Gomes F, Silva-Carvalho JL, Almeida H. Superoxide dismutase expression in human cumulus oophorus cells. Mol Hum Reprod. 2009;15:411–419. doi: 10.1093/molehr/gap034. [DOI] [PubMed] [Google Scholar]
- 33.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 34.Pedrajas JR, Miranda-Vizuete A, Javanmardy N, Gustafsson JA, Spyrou G. Mitochondria of Saccharomyces cerevisiae contain one-conserved cysteine type peroxiredoxin with thioredoxin peroxidase activity. J Biol Chem. 2000;275:16296–16301. doi: 10.1074/jbc.275.21.16296. [DOI] [PubMed] [Google Scholar]
- 35.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 36.Lim J, Luderer U. Oxidative damage increases and antioxidant gene expression decreases with aging in the mouse ovary. Biol Reprod. 2011;84:775–782. doi: 10.1095/biolreprod.110.088583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee YC, Huang HY, Chang CJ, Cheng CH, Chen YT. Mitochondrial GLUT10 facilitates dehydroascorbic acid import and protects cells against oxidative stress: mechanistic insight into arterial tortuosity syndrome. Hum Mol Genet. 2010;19:3721–3733. doi: 10.1093/hmg/ddq286. [DOI] [PubMed] [Google Scholar]
- 38.Banu SK, Stanley JA, Lee J, Stephen SD, Arosh JA, Hoyer PB, Burghardt RC. Hexavalent chromium-induced apoptosis of granulosa cells involves selective sub-cellular translocation of Bcl-2 members, ERK1/2 and p53. Toxicol Appl Pharmacol. 2011;251:253–266. doi: 10.1016/j.taap.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yacobi K, Wojtowicz A, Tsafriri A, Gross A. Gonadotropins enhance caspase-3 and −7 activity and apoptosis in the theca-interstitial cells of rat preovulatory follicles in culture. Endocrinology. 2004;145:1943–1951. doi: 10.1210/en.2003-1395. [DOI] [PubMed] [Google Scholar]
- 40.Tatemoto H, Sakurai N, Muto N. Protection of porcine oocytes against apoptotic cell death caused by oxidative stress during in vitro maturation: role of cumulus cells. Biol Reprod. 2000;63:805–810. doi: 10.1095/biolreprod63.3.805. [DOI] [PubMed] [Google Scholar]
- 41.Stein LS, Stoica G, Tilley R, Burghardt RC. Rat ovarian granulosa cell culture: a model system for the study of cell–cell communication during multistep transformation. Cancer Res. 1991;51:696–706. [PubMed] [Google Scholar]
- 42.Li Chen T, Wise SS, Kraus S, Shaffiey F, Levine KM, Thompson WD, Romano T, O’Hara T, Wise JP., Sr Particulate hexavalent chromium is cytotoxic and genotoxic to the North Atlantic right whale (Eubalaena glacialis) lung and skin fibroblasts. Environ Mol Mutagen. 2009;50:387–393. doi: 10.1002/em.20471. [DOI] [PubMed] [Google Scholar]
- 43.Goodale BC, Walter R, Pelsue SR, Thompson WD, Wise SS, Winn RN, Mitani H, Wise JP., Sr The cytotoxicity and genotoxicity of hexavalent chromium in medaka (Oryzias latipes) cells. Aquat Toxicol. 2008;87:60–67. doi: 10.1016/j.aquatox.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 44.Devine PJ, Perreault SD, Luderer U. Roles of reactive oxygen species and antioxidants in ovarian toxicity. Biol Reprod. 2012;86:1–10. doi: 10.1095/biolreprod.111.095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osman P. Rate and course of atresia during follicular development in the adult cyclic rat. J Reprod Fertil. 1985;73:261–270. doi: 10.1530/jrf.0.0730261. [DOI] [PubMed] [Google Scholar]
- 46.Borgeest C, Symonds D, Mayer LP, Hoyer PB, Flaws JA. Methoxychlor may cause ovarian follicular atresia and proliferation of the ovarian epithelium in the mouse. Toxicol Sci. 2002;68:473–478. doi: 10.1093/toxsci/68.2.473. [DOI] [PubMed] [Google Scholar]
- 47.Banu SK, Lee J, Speights VOJ, Starzinski-Powitz A, Arosh JA. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 induces apoptosis of human endometriotic cells through suppression of ERK1/2, AKT, NFkappaB, and beta-catenin pathways and activation of intrinsic apoptotic mechanisms. Mol Endocrinol. 2009;23:1291–1305. doi: 10.1210/me.2009-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kayampilly PP, Menon KM. Follicle-stimulating hormone increases tuberin phosphorylation and mammalian target of rapamycin signaling through an extracellular signal-regulated kinase-dependent pathway in rat granulosa cells. Endocrinology. 2007;148:3950–3957. doi: 10.1210/en.2007-0202. [DOI] [PubMed] [Google Scholar]
- 49.Stanley JA, Lee J, Nithy TK, Arosh JA, Burghardt RC, Banu SK. Chromium-VI arrests cell cycle and decreases granulosa cell proliferation by down-regulating cyclin-dependent kinases (CDK) and cyclins and up-regulating CDK-inhibitors. Reprod Toxicol. 2011;32:112–123. doi: 10.1016/j.reprotox.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palaniappan M, Menon KM. Human chorionic gonadotropin stimulates theca-interstitial cell proliferation and cell cycle regulatory proteins by a cAMP-dependent activation of AKT/mTORC1 signaling pathway. Mol Endocrinol. 2010;24:1782–1793. doi: 10.1210/me.2010-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kratzer DD, Littell RC. Appropriate statistical methods to compare dose responses of methionine sources. Poult Sci. 2006;85:947–954. doi: 10.1093/ps/85.5.947. [DOI] [PubMed] [Google Scholar]
- 52.Biroccio A, Benassi B, Fiorentino F, Zupi G. Glutathione depletion induced by c-Myc downregulation triggers apoptosis on treatment with alkylating agents. Neoplasia. 2004;6:195–206. doi: 10.1593/neo.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raza H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J. 2011;278:4243–4251. doi: 10.1111/j.1742-4658.2011.08358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 55.Banu SK, Samuel JB, Arosh JA, Burghardt RC, Aruldhas MM. Lactational exposure to hexavalent chromium delays puberty by impairing ovarian development, steroidogenesis and pituitary hormone synthesis in developing Wistar rats. Toxicol Appl Pharmacol. 2008;232:180–189. doi: 10.1016/j.taap.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 56.Amara IB, Hakim A, Troudi A, Soudani N, Makni FA, Zeghal KM, Zeghal N. Protective effects of selenium on methimazole-induced anemia and oxidative stress in adult rats and their offspring. Hum Exp Toxicol. 2011;30:1549–1560. doi: 10.1177/0960327110392403. [DOI] [PubMed] [Google Scholar]
- 57.Soudani N, Ben Amara I, Troudi A, Hakim A, Bouaziz H, Ayadi Makni F, Zeghal KM, Zeghal N. Oxidative damage induced by chromium (VI) in rat erythrocytes: protective effect of selenium. J Physiol Biochem. 2011;67:577–588. doi: 10.1007/s13105-011-0104-4. [DOI] [PubMed] [Google Scholar]
- 58.NTP. National Toxicology Program Technical Report Series. Bethesda: NIH NIEHS, NTP; 2008. Toxicology and carcinogenesis studies of sodium dichromate dihydrate (CAS No. 7789-12-0) in F344/N rats and B6C3F1 mice (drinking water studies) pp. 1–192. [PubMed] [Google Scholar]
- 59.ATSDR. Toxicological profile of chromium. Atlanta: U.S. Department of Health and Human Services; 2012. pp. 1–2. [Google Scholar]
- 60.Bucher J. Toxicity Report Series. Bethesda: NIH NIEHS, NTP; 2007. NTP toxicity studies of sodium dichromate dihydrate (CAS No. 7789-12-0) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice; pp. 1–G4. [PubMed] [Google Scholar]
- 61.Sayato Y, Nakamuro K, Matsui S, Ando M. Metabolic fate of chromium compounds. I. Comparative behavior of chromium in rat administered with Na251CrO4and 51CrCl3. J Pharmacobio-dyn. 1980;3:17–23. doi: 10.1248/bpb1978.3.17. [DOI] [PubMed] [Google Scholar]
- 62.Kirpnick-Sobol Z, Reliene R, Schiestl RH. Carcinogenic Cr(VI) and the nutritional supplement Cr(III) induce DNA deletions in yeast and mice. Cancer Res. 2006;66:3480–3484. doi: 10.1158/0008-5472.CAN-05-3944. [DOI] [PubMed] [Google Scholar]
- 63.Ozawa T, Hanaki A. Spin-trapping studies on the reactions of Cr(III) with hydrogen peroxide in the presence of biological reductants: is Cr(III) nontoxic? Biochem Int. 1990;22:343–352. [PubMed] [Google Scholar]
- 64.Rao MV, Chawla SL, Sharma SR. Protective role of vitamin E on nickel and/or chromium induced oxidative stress in the mouse ovary. Food Chem Toxicol. 2009;47:1368–1371. doi: 10.1016/j.fct.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 65.Agarwal A, Gupta S, Sikka S. The role of free radicals and antioxidants in reproduction. Curr Opin Obstet Gynecol. 2006;18:325–332. doi: 10.1097/01.gco.0000193003.58158.4e. [DOI] [PubMed] [Google Scholar]
- 66.Scibior D, Czeczot H. Catalase: structure, properties, functions. Postepy Hig Med Dosw. 2006;60:170–180. [PubMed] [Google Scholar]
- 67.Heck DE, Shakarjian M, Kim HD, Laskin JD, Vetrano AM. Mechanisms of oxidant generation by catalase. Ann N Y Acad Sci. 2010;1203:120–125. doi: 10.1111/j.1749-6632.2010.05603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 69.Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee YJ. Role of glutaredoxin in metabolic oxidative stress: glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 70.Gonzalez-Fernandez R, Gaytan F, Martinez-Galisteo E, Porras P, Padilla CA, Criado JES, Barcena JA. Expression of glutaredoxin (thioltransferase) in the rat ovary during the oestrous cycle and postnatal development. J Mol Endocrinol. 2005;34:625–635. doi: 10.1677/jme.1.01715. [DOI] [PubMed] [Google Scholar]
- 71.Chrestensen CA, Starke DW, Mieyal JJ. Acute cadmium exposure inactivates thioltransferase (glutaredoxin), inhibits intracellular reduction of protein–glutathionyl-mixed disulfides, and initiates apoptosis. J Biol Chem. 2000;275:26556. doi: 10.1074/jbc.M004097200. [DOI] [PubMed] [Google Scholar]
- 72.Fiander H, Schneider H. Compounds that induce isoforms of glutathione S-transferase with properties of a critical enzyme in defense against oxidative stress. Biochem Biophys Res Commun. 1999;262:591–595. doi: 10.1006/bbrc.1999.1262. [DOI] [PubMed] [Google Scholar]
- 73.Havelock Rainey JC, Carr WEBR. Ovarian granulosa cell lines. Mol Cell Endocrinol. 2004;228:67–78. doi: 10.1016/j.mce.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 74.Tang XL, Liu XJ, Tian Q, Zhang W. Dynamic oxidative stress and DNA damage induced by oestrogen deficiency and protective effects of puerarin and 17β-oestradiol in ovariectomized rats. Basic Clin Pharmacol Toxicol. 2012;111:87–91. doi: 10.1111/j.1742-7843.2012.00864.x. [DOI] [PubMed] [Google Scholar]
- 75.Kumar P, Kale RK, Baquer NZ. Estradiol modulates membrane-linked ATPases, antioxidant enzymes, membrane fluidity, lipid peroxidation, and lipofuscin in aged rat liver. J Aging Res. 2011;2011:580245. doi: 10.4061/2011/580245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harrison FE, May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol Med. 2009;46:719–730. doi: 10.1016/j.freeradbiomed.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahmed EA, Omar HMSKE, Ragb SM, Nasser AY. The antioxidant activity of vitamin C, DPPD and L-cysteine against cisplatin-induced testicular oxidative damage in rats. Food Chem Toxicol. 2011;49:1115–1121. doi: 10.1016/j.fct.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 78.Ozdem S, Nacitarhan C, Gulay MS, Hatipoglu FS, Ozdem SS. The effect of ascorbic acid supplementation on endosulfan toxicity in rabbits. Toxicol Ind Health. 2011;27:437–446. doi: 10.1177/0748233710388450. [DOI] [PubMed] [Google Scholar]
- 79.El-Gendy KS, Aly NM, Mahmoud FH, Kenawy A, El-Sebae AK. The role of vitamin C as antioxidant in protection of oxidative stress induced by imidacloprid. Food Chem Toxicol. 2010;48:215–221. doi: 10.1016/j.fct.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 80.Abhilash PA, Harikrishnan R, Indira M. Ascorbic acid supplementation causes faster restoration of reduced glutathione content in the regression of alcohol-induced hepatotoxicity in male guinea pigs. Redox Rep. 2012;17:72–79. doi: 10.1179/1351000212Y.0000000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Banerjee P, Bhattacharyya SS, Bhattacharjee N, Pathak S, Boujedaini N, Belon P, Khuda-Bukhsh AR. Ascorbic acid combats arsenic-induced oxidative stress in mice liver. Ecotoxicol Environ Saf. 2009;72:639–649. doi: 10.1016/j.ecoenv.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 82.Sagun KC, Carcamo JM, Golde DW. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005;19:1657–1567. doi: 10.1096/fj.05-4107com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.