

Abstract
α-Ketoglutarate dehydrogenase (KGDH) is reversibly inhibited when rat heart mitochondria are exposed to hydrogen peroxide (H2O2). H2O2-induced inhibition occurs through the formation of a mixed disulfide between a protein sulfhydryl and glutathione. Upon consumption of H2O2, glutaredoxin can rapidly remove glutathione, resulting in regeneration of enzyme activity. KGDH is a key regulatory site within the Krebs cycle. Glutathionylation of the enzyme may therefore represent an important means to control mitochondrial function in response to oxidative stress. We have previously provided indirect evidence that glutathionylation occurs on lipoic acid, a cofactor covalently bound to the E2 subunit of KGDH. However, lipoic acid contains two vicinal sulfhydryls and rapid disulfide exchange might be predicted to preclude stable glutathionylation. The current study sought conclusive identification of the site and chemistry of KGDH glutathionylation and factors that control the degree and rate of enzyme inhibition. We present evidence that, upon reaction of free lipoic acid with oxidized glutathione in solution, disulfide exchange occurs rapidly, producing oxidized lipoic acid and reduced glutathione. This prevents the stable formation of a glutathione–lipoic acid adduct. Nevertheless, 1:1 lipoic acid–glutathione adducts are formed on KGDH because the second sulfhydryl on lipoic acid is unable to participate in disulfide exchange in the enzyme’s native conformation. The maximum degree of KGDH inhibition that can be achieved by treatment of mitochondria with H2O2 is 50%. Results indicate that this is not due to glutathionylation of a subpopulation of the enzyme but, rather, the unique susceptibility of lipoic acid on a subset of E2 subunits within each enzyme complex. Calcium enhances the rate of glutathionylation by increasing the half-life of reduced lipoic acid during enzyme catalysis. This does not, however, alter the maximal level of inhibition, providing further evidence that specific lipoic acid residues within the E2 complex are susceptible to glutathionylation. These findings offer chemical information necessary for the identification of mechanisms and physiological implications of KGDH glutathionylation.
Keywords: Mitochondria, Redox regulation, α-Ketoglutarate dehydrogenase, Glutathionylation, Lipoic acid, Free radicals
The mitochondrial enzyme α-ketoglutarate dehydrogenase (KGDH)1 is a large multienzyme complex that consists of multiple copies of three subunits: E1 (α-ketoglutarate dehydrogenase), E2 (dihydrolipoyl succinyltransferase), and E3 (dihydrolipoyl dehydrogenase). Together, these subunits catalyze the exergonic \conversion of α-ketoglutarate, CoASH, and NAD+ to CO2, succinyl-CoA, and NADH. Enzyme activity is stimulated by Ca2+ and inhibited by the catalytic products succinyl-CoA and NADH to reflect metabolic demand [1–9]. As a key regulatory site within the Krebs cycle, KGDH plays a critical role in cellular energy and biosynthetic homeostasis [10–14]. It is therefore noteworthy that loss of KGDH activity has been observed during numerous degenerative diseases [14–22]. Many of these diseases are associated with oxidative stress. In vitro, KGDH has been shown to be a sensitive target of oxidative damage [22–25]. Oxidative inactivation of KGDH is therefore thought to contribute to the cellular and tissue dysfunction characteristic of these pathophysiological conditions.
In sharp contrast to oxidative damage, KGDH can also undergo reversible inhibition in response to oxidative stress. Treatment of isolated rat cardiac mitochondria with H2O2 results in a loss of KGDH activity and in the rate of ADP-dependent mitochondrial respiration. Upon consumption of H2O2, KGDH and respiratory activities are restored [26]. H2O2-induced inhibition of KGDH is due to formation of a mixed disulfide between a protein sulfhydryl and glutathione (glutathionylation) [27]. In vitro, glutathionylation protects catalytic sulfhydryls on KGDH from oxidative damage [28]. Thus, the reversible nature of this modification could provide for redox regulation as a means to protect the enzyme from oxidative damage. If so, why then would KGDH undergo irreversible oxidative inactivation during certain pathophysiological processes? To adequately address this question, the mechanisms by which KGDH is glutathionylated and the physiological significance of this form of enzyme regulation must be established.
The transient nature of glutathionylation coupled with difficulties in detection has limited mechanistic investigations. Additionally, in disrupted mitochondria or in purified form, KGDH is resistant to glutathionylation-dependent inhibition that can be reversed by glutaredoxin [27]. Previous studies that took advantage of chemical derivatization and sulfhydryl reactivity indirectly identified lipoic acid, a required cofactor covalently linked to the E2 subunit, as the likely site of glutathionylation [28]. This result is, however, perplexing because of the chemical properties of lipoic acid. Lipoic acid has two vicinal sulfhydryls. Therefore, rapid disulfide exchange upon glutathionylation would be predicted to render lipoic acid resistant to stable modification. It is also not clear why H2O2-induced inhibition of KGDH does not exceed 50% [26–28]. This study sought conclusive identification of the site and chemistry of KGDH glutathionylation and factors that control the degree and rate of enzyme inhibition. Chemical, biochemical, and immunochemical approaches were utilized. The results offer important insights on the properties of KGDH that make lipoic acid susceptible to stable glutathionylation and establish a reliable means for exploring the mechanism by which this process occurs.
Materials and methods
Reactions between lipoamide, glutathione, and iodoacetamide
Lipoamide (1.0 mM in 20 mM K2PO4 at pH 8.0) was reduced upon incubation (30 min) with 3.0 mM Tris(2-carboxyethyl) phosphine (TCEP). Agarose-immobilized TCEP (Pierce) and reduced lipoamide were then separated by centrifugation. Reduced lipoamide was subsequently incubated for 30 min at room temperature with GSH or GSSG (5.0 mM), iodoacetamide (2.0 mM), or iodoacetamide (2.0 mM) followed by an additional 30 min with GSSG (5.0 mM). Reaction products were resolved by reverse-phase chromatography (Agilent; Zorbax 300SB-C18 4.6×250 mM column, 1.0 ml/min flow rate, linear gradient of 0–50% acetonitrile, 0.05% trifluoroacetic acid (TFA) relative to H2O, 0.05% TFA in 0–20 min) and the elution profile was monitored at 210 nm using an HPLC system (Shimadzu LC-20A high-precision binary gradient system) equipped with a UV/Vis diode array detector. Products were identified using a ThermoScientific LTQ-XL linear ion trap mass spectrometer equipped with an Eksigent splitless nanoflow HPLC system and Proxeon microspray ion source.
Isolation of mitochondria from rat heart
Male Sprague–Dawley rats (250–300 g) obtained from Harlan Laboratories were euthanized by decapitation. Hearts were immediately excised and perfused with 10 ml ice-cold homogenization buffer (210 mM mannitol, 70 mM sucrose, 1 mM EDTA, 10 mM Mops, pH 7.4) to remove blood. Hearts (0.9–1.1 g) were then minced and homogenized in 20 ml of homogenization buffer using a Polytron homogenizer (low setting, 3 s). The homogenate was centrifuged at 550 g for 5 min (4 °C), and the supernatant was filtered through cheese cloth. The mitochondrial pellet was obtained upon centrifugation of the supernatant at 10,000 g for 10 min (4 °C). After two rinses with ice-cold homogenization buffer, the mitochondria were resuspended into homogenization buffer to a final concentration of 25.0 mg/ml. Protein determinations were made using the bicinchoninic acid method (Pierce), using bovine serum albumin as a standard.
Incubation of mitochondria with H2O2
Mitochondria were diluted to either 0.5 or 1.0 mg/ml in buffer composed of 210 mM mannitol, 70 mM sucrose, 10 mM Mops, and 5.0 mM K2HPO4 at pH 7.4. Respiration was initiated upon the addition of 5.0 mM α-ketoglutarate and allowed to proceed for 2.0 min. H2O2 (25 to 100 μM as indicated) was then added (t=0 in figures). Incubations were performed at room temperature.
Derivatization of sulfenic acids with dimedone
Mitochondria were incubated as described above with the addition of 5.0 mM dimedone [29]. At indicated times, mitochondria were solubilized by the addition of 0.05% Triton X-100 and KGDH activity was measured. Western blot analysis was performed on mitochondrial protein eluted through PD-10 columns (GE Healthcare) to remove excess dimedone and prevent nonspecific dimedone reactions and under reducing conditions to determine the level of native lipoic acid and dimedone-modified KGDH.
Assay for α-ketoglutarate dehydrogenase activity
Mitochondria were diluted to 0.05 mg/ml in 25 mM Mops, 0.05% Triton X-100, pH 7.4. Solubilization of mitochondria with Triton X-100 at a final concentration of 0.05% prevents further H2O2-induced inhibition or reactivation of KGDH [27]. KGDH activity was measured spectrophotometrically (Agilent, 8452A) as the rate of NAD+ reduction to NADH (340 nm, ε=6,200 M−1 cm−1) upon addition of 5.0 mM MgCl2, 2.5 mM α-ketoglutarate, 0.1 mM CoASH, 0.2 mM thiamine pyrophosphate, and 1.0 mM NAD+. The presence of 0.05% Triton X-100 inhibits complex I of the respiratory chain, preventing consumption of NADH. Where indicated, glutaredoxin (1.0 U/ml; CalBiochem) and GSH (1.0 mM) were added to deglutathionylate KGDH before enzyme analysis.
Western blot analysis
Samples were suspended in 27 mM Tris–HCl, 35.2 mM Tris base, 0.5% SDS, 2.5% sucrose, 0.12 mM EDTA, 0.55 mM SERVA Blue G250, and 0.05 mM phenol red at pH 8.0 and incubated for 10 min at 45 °C. Protein was then resolved on a 10% SDS–PAGE gel (Invitrogen Nupage) and electrotransferred onto polyvinylidene difluoride (PVDF) membrane (Bio-Rad). Membrane-immobilized proteins were analyzed utilizing polyclonal rabbit antibodies specific to the E2 subunit of KGDH (anti-E2) or lipoic acid [24] or mouse monoclonal anti-glutathione antibody (Virogen) as specified. Anti-E2 was raised to NH2-(GC)EMRARHKDAFLKKHNLKCOOH, a peptide corresponding to residues 262–278 of the E2 subunit of KGDH from rat heart mitochondria. The peptide was conjugated to keyhole limpet hemocyanin, the immunogen introduced into rabbits, and polyclonal anti-E2 obtained according to standard protocols (Biosynthesis). The antibody was specific to the E2 subunit of KGDH, exhibiting no binding to the E2 subunit of pyruvate dehydrogenase (PDH). Primary antibody binding was visualized utilizing the appropriate peroxidase-conjugated secondary antibody and chemiluminescent substrate (Supersignal West Pico from Pierce).
Native gel electrophoresis
Mitochondrial samples (1.0 mg/ml) were suspended in Native-PAGE sample buffer (Invitrogen) with 0.5% digitonin and incubated on ice for 15 min. Samples were then centrifuged at 16,000 g for 30 min at 4 °C. The supernatant was removed and G250 sample additive was added (final concentration of 0.125%). Protein was then resolved on a 3–12% NativePAGE gel (Invitrogen) at 4 °C. Protein was transferred to a PVDF membrane and fixed with 8% acetic acid before Western blot analysis.
Immunoprecipitation of KGDH
After treatment with H2O2, mitochondria (1.0 mg/ml) were solubilized with 0.05% Triton X-100 to prevent further glutathionylation or deglutathionylation of KGDH [26,27]. Samples were then centrifuged for 10 min at 16,000 g at 4 °C to pellet the membrane fraction. The supernatant was subjected to size-exclusion chromatography (PD-10 column; GE Healthcare) to remove free glutathione. Equal volumes of mitochondrial extracts were then incubated with anti-lipoic acid antibody overnight at 4 °C. Agarose-immobilized antibody was subsequently washed five times with phosphate-buffered saline (PBS) using spin columns (Pierce). Mitochondrial proteins that bound to anti-lipoic acid antibody were eluted with SDS loading buffer in the presence or absence of 100 mM iodoacetamide followed by Western blot analyses. Polyclonal anti-lipoic acid antibodies were first conjugated to biotin and then incubated with streptavidin agarose beads before immunoprecipitation of mitochondrial extracts. Because of the strong binding affinity between biotin and avidin, this procedure minimizes background from denatured antibodies in the blotting procedure. Briefly, anti-lipoic acid antiserum was diluted to approximately 2.5 mg/ml in PBS to a final volume of 1.0 ml. A 10 mM solution of sulfosuccinimidyl-6-[biotin-amido] hexanoate (Pierce) was prepared in water. Biotinylation reagent was added at 20-fold molar excess as recommended by the manufacturer (Pierce). The reaction was incubated at room temperature for 45 min. Excess reagent was removed by size-exclusion chromatography. Biotinylated anti-lipoic acid antibody was then agarose-immobilized upon incubation with streptavidin-conjugated agarose beads for 30 min at room temperature.
Quantification of GSH and GSSG
The levels of GSH and GSSG in mitochondria and cardiac tissue were quantified using reverse-phase HPLC and electrochemical detection [30]. GSH and GSSG were extracted from mitochondria or heart homogenate by treatment with 5% metaphosphoric acid. Proteins were precipitated upon incubation on ice (20 min) and then pelleted by centrifugation (10 min at 16,000 g). The supernatant was filtered (0.45-μm syringe filters) before analysis of GSH and GSSG by HPLC and electrochemical detection (Shimadzu HPLC system, ESA Coularray electrochemical detector 5600A set at 750 mV). GSH and GSSG were eluted through a C18 column (Phenomenex Luna C18(2), 100 Å, 3 μm, 150×4.6 mm) at 0.5 ml/min using an isocratic mobile phase consisting of 25 mM NaH2PO4, 0.5 mM 1-octane sulfonic acid, 4% acetonitrile, pH 2.7. GSH and GSSG concentrations were calculated employing GSH and GSSG standard curves constructed from peak areas.
Preparation and perfusion of isolated rat heart
After euthanasia of rats by decapitation, hearts were rapidly excised and placed in modified Krebs–Henseleit buffer (120 mM NaCl/4.8 mM KCl/2.0 mM CaCl2/1.25 mM MgCl2/1.25 mM KH2 PO4/25 mM NaHCO3/15 mM glutamine at 37 °C). As previously described [31], extraneous tissue was rapidly removed, the aorta was cannulated, and the heart was perfused in retrograde fashion according to Langendorff [32] with modified Krebs–Henseleit buffer (37 °C) saturated with 95% O2/5% CO2. Hearts were placed in a water-jacketed chamber (37 °C) and perfusion pressure was maintained at 60 mm Hg (1 mm Hg=133 Pa). Elapsed time between isolation of the heart and perfusion was approximately 1 min. Hearts were equilibrated in the perfusion buffer for 10 min followed by perfusion with or without 100 μM H2O2 for 15 min. Hearts were then removed from the apparatus and rapidly frozen in liquid nitrogen, pulverized, resuspended in ice-cold 25 mM Mops, 1.0 mM EDTA, pH 7.4, and sonicated with a microtip ultrasonicator. For KGDH activity assays, pulverized tissue was resuspended in 25 mM Mops, 0.05% Triton X-100, pH 7.4. Homogenates were then centrifuged at 16,000 g for 10 min and aliquots of the supernatant (1 to 2 mg/ml protein) were used for measurement of KGDH activity, Western blot analysis, immunoprecipitation, and GSH and GSSG content. Protein concentrations were determined as described above.
Results
Reactions of glutathione with lipoamide
To define conditions required for stable glutathionylation of lipoic acid and to develop methods for direct detection on protein, a series of chemical reactions was performed. Oxidized lipoamide (model for protein-bound lipoic acid) was incubated with immobilized TCEP to reduce the vicinal sulfhydryls (Fig. 1A). Reduced lipoamide was then incubated with either reduced (GSH) or oxidized glutathione (GSSG). Reaction products were resolved by reverse-phase HPLC and identified using mass spectrometry. No reaction was observed between lipoamide and GSH (data not shown). In contrast, incubation of reduced lipoamide with GSSG resulted in a stoichiometric reduction of GSSG to GSH (data not shown) and oxidation of lipoamide (Fig. 1B). The product profile suggests the formation of a 1:1 lipoamide–SG product followed by rapid disulfide exchange with the second sulfhydryl on lipoamide, resulting in the net oxidation of lipoamide and reduction of GSSG. A lipoamide–SG product was formed by first alkylating one sulfhydryl on lipoamide with the sulfhydryl reactive compound iodoacetamide followed by incubation with GSSG (Fig. 1C). Therefore, lipoamide can be stably glutathionylated if one sulfhydryl is sequestered.
Fig. 1.

Reactions between reduced lipoamide and oxidized glutathione. Reactions were allowed to proceed for 30 min at room temperature in 25 mM Na2PO4, pH 8.0. Products were resolved by reverse-phase chromatography and analyzed by mass spectrometry, as shown. (A) Oxidized and reduced lipoamide (1.0 mM each) standards. (B) Products formed upon incubation of reduced lipoamide (1.0 mM) with GSSG (5.0 mM). (C) Products formed upon incubation of reduced lipoamide (1.0 mM) and iodoacetamide (2.0 mM) followed by GSSG (5.0 mM). Numbered peaks and mass spectra represent: (1) reduced lipoamide, (2) oxidized lipoamide, and (3) singly alkylated and glutathionylated lipoamide.
Stabilization of lipoic acid glutathionylation on KGDH
The chemical nature of lipoic acid indicates that for this to be the site of glutathionylation on KGDH, one of the vicinal sulfhydryls must be rendered nonreactive to prevent or diminish disulfide exchange and allow for stable inhibition (Fig. 1). Sequestration within the native conformation of the enzyme could provide such stability. Treatment with iodoacetamide during protein denaturation would be expected to modify the nonglutathionylated lipoic acid sulfhydryl, blocking disulfide exchange and preserving the glutathionylated sulfhydryl for analysis. In contrast, in the absence of iodoacetamide, one would predict that glutathionylated KGDH would not be observed because disulfide exchange would occur upon protein denaturation resulting in the removal of glutathione. To test this possibility and confirm lipoic acid as the site of glutathionylation, isolated rat heart mitochondria respiring with α-ketoglutarate were treated with H2O2. Treatment of mitochondria with 25 μM H2O2 led to 50% inhibition of KGDH at 5.0 min and recovery of enzyme activity at 10.0 min (Fig. 2A). At specified times, mitochondria were solubilized and protein was denatured in the absence or presence of iodoacetamide. Nonreducing SDS Western blot analysis revealed no glutathionylated protein when iodoacetamide was omitted (Fig. 2B). Inclusion of iodoacetamide resulted in the detection of glutathionylated protein that comigrated with the E2 subunit of KGDH (Fig. 2B). Additionally, no glutathionylation was observed before H2O2 treatment or after enzyme reactivation (Fig. 2A and B). Our results support the conclusion that, in the enzyme’s native conformation, one sulfhydryl of lipoic acid is glutathionylated and the other is not available for disulfide exchange.
Fig. 2.
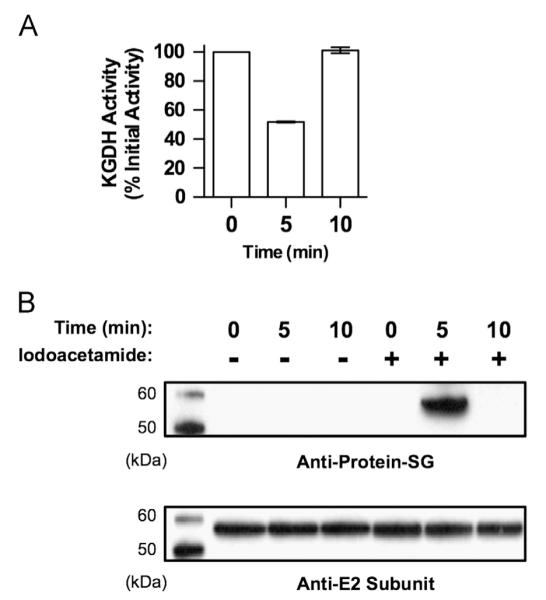
Stabilization of the lipoic acid–glutathione adduct on KGDH. Isolated rat heart mitochondria (0.5 mg/ml) were incubated in the presence of α-ketoglutarate (5.0 mM) as a respiratory substrate. After a 2.0-min preincubation, H2O2 (25 μM) was added. (A) Mitochondria were then solubilized at 0, 5, and 10 min in 25 mM Mops, pH 7.4, containing 0.05% Triton X-100 to prevent further inhibition or reactivation of KGDH. Enzyme activity was then measured spectrophotometrically. Values are represented as means±standard deviation (n=5). (B) At the indicated times, mitochondria were dissolved in SDS-loading buffer in the presence or absence of 100 mM iodoacetamide to alkylate free sulfhydryl groups and prevent disulfide exchange. Western blot analysis was then performed using monoclonal anti-glutathione or polyclonal anti-KGDH E2 subunit antibodies as indicated. The blots shown are representative of five independent experiments.
If the native conformation of KGDH does in fact impart stability to glutathionylated lipoic acid, one would expect to detect glutathionylated KGDH using blue native gel electrophoresis followed by Western blot analysis. After maximal inhibition of KGDH upon treatment of respiring mitochondria with H2O2, a reduction in anti-lipoic acid antibody binding was observed, indicating modification of lipoic acid (Fig. 3). Loss of anti-lipoic acid antibody binding was accompanied by the appearance of glutathionylated protein (Fig. 3). Upon recovery of KGDH activity, glutathionylation was no longer detected and native lipoic acid was restored to previous levels. As judged by mass spectrometry, KGDH subunits were the only proteins detected in the glutathione immunoreactive band (not shown). Thus, the tertiary structure of KGDH prevents disulfide exchange between the unmodified and the glutathionylated sulfhydryls of lipoic acid. It is important to note that PDH, another enzyme that contains lipoic acid and can be visualized by antibody to lipoic acid, does not undergo glutathionylation (Fig. 3).
Fig. 3.
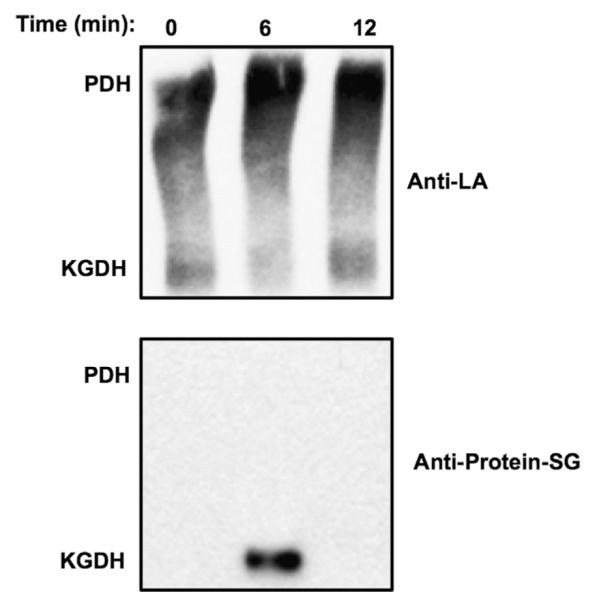
The stability of glutathionylation on native KGDH. Isolated rat heart mitochondria (1.0 mg/ml) were incubated for 2.0 min in the presence of α-ketoglutarate (5.0 mM). H2O2 (100 μM) was then added and, at the indicated times, mitochondria were dissolved in NativePAGE loading buffer. After native gel electrophoresis, Western blot analysis was performed using anti-lipoic acid (Anti-LA) or anti-glutathione (Anti-Protein–SG) antibodies as indicated. The blots shown are representative of five independent experiments. Binding of anti-lipoic acid indicates the presence of pyruvate dehydrogenase (PDH) and KGDH, as indicated.
Effects of Ca2+ on the redox status of lipoic acid and the extent and rate of KGDH glutathionylation and inhibition
Glutathionylation of KGDH requires actively respiring mitochondria and probably reduced lipoic acid on the enzyme’s E2 subunit [27,28]. We sought to define effectors that alter the redox status of lipoic acid and thus the rate and/or extent of H2O2-induced KGDH glutathionylation and inhibition. Mitochondria were incubated in the presence or absence of α-ketoglutarate and/or Ca2+, a known activator of KGDH [6–9]. Mitochondrial protein was then denatured in the presence of iodoacetamide, which reacts with reduced but not oxidized lipoic acid. Alkylation prevents binding of the anti-lipoic acid antibody [28]. Diminished antibody binding therefore indicates increased levels of reduced lipoic acid. In mitochondria respiring on α-ketoglutarate, the presence of 10 μM Ca2+ (a concentration that maximally stimulates ADP-dependent respiration) increased the fraction of reduced lipoic acid residues on KGDH (Fig. 4). In keeping with these effects on the redox status of lipoic acid, Ca2+ was found to significantly increase the rate of KGDH glutathionylation and inhibition upon incubation of respiring mitochondria with H2O2 (Figs. 5A and 5B). Ca2+ did not, however, alter the maximum degree of inhibition. Thus, despite a Ca2+-induced increase in the half-life of reduced lipoic acid on KGDH, not all catalytically active lipoic acid residues are susceptible to glutathionylation.
Fig. 4.
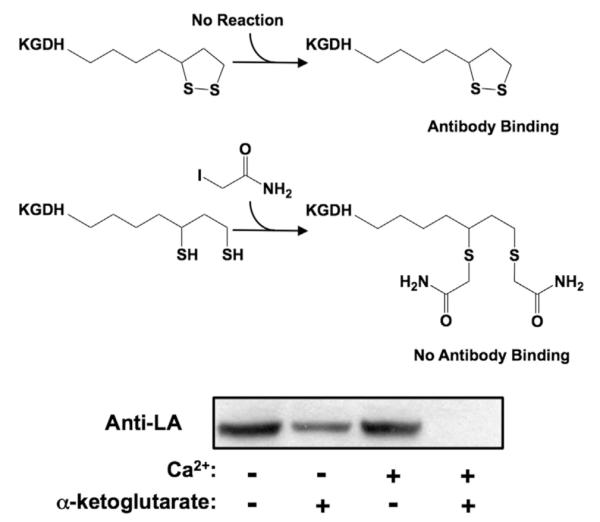
Effects of Ca2+ and α-ketoglutarate on the redox status of lipoic acid on KGDH. Isolated rat heart mitochondria (0.5 mg/ml) were incubated in the presence or absence of α-ketoglutarate (5.0 mM) and/or Ca2+ (10 μM) for 2.0 min. Mitochondria were then dissolved in SDS-loading buffer in the presence of 100 mM iodoacetamide to alkylate reduced lipoic acid groups. Protein (5.0 μg/lane) was resolved by gel electrophoresis, transferred to PVDF membrane, and subjected to Western blot analysis using polyclonal anti-lipoic acid antibodies. The blots shown are representative of three independent experiments.
Fig. 5.

Effects of Ca2+ on the rate and extent of reversible inhibition and glutathionylation of KGDH in response to H2O2. Isolated rat heart mitochondria (0.5 mg/ml) were incubated for 2.0 min with α-ketoglutarate (5.0 mM) as a respiratory substrate in the presence or absence of Ca2+ (10 μM). H2O2 (25 μM) was then added and, at indicated times, (A) mitochondria were solubilized in 25 mM Mops, pH 7.4, containing 0.05% Triton X-100, and KGDH activity was measured spectrophotometrically. Values are represented as means7standard deviation (n=3). (B) Other mitochondria were dissolved in SDS-loading buffer in the presence 100 mM iodoacetamide. Protein (5.0 μg/lane) was resolved by non-reducing gel electrophoresis, transferred to PVDF membrane, and subjected to Western blot analysis using monoclonal anti-glutathione antibodies. The blots shown are representative of three independent experiments.
Differential susceptibilities of lipoic acid residues on KGDH complexes to glutathionylation
The threshold of H2O2-induced glutathionylation and inhibition of KGDH is 50%, irrespective of H2O2 concentration. This may indicate that a subpopulation the enzyme is resistant to glutathionylation. Indeed, two populations of KGDH have been described, one associated with the electron transport chain and a second within the mitochondrial matrix [33–36]. Alternatively, the KGDH complex contains multiple E2 subunits, each with one covalently linked lipoic acid cofactor. Thus, a subset of E2 subunits may be resistant to glutathionylation within each KGDH complex, resulting in a mosaic of modified and unmodified lipoic acid residues. In this scenario, antibody specific to native lipoic acid would be expected to immunoprecipitate KGDH complexes that contain both native and glutathionylated lipoic acid residues. Isolated rat heart mitochondria respiring with α-ketoglutarate as a substrate were incubated with H2O2 to induce maximum inhibition (~50%). Immunoprecipitation using anti-lipoic acid antibodies depleted E2 subunits from the mitochondrial extracts independent of H2O2-induced inhibition (Fig. 6A). Furthermore, glutathionylated E2 subunits were detected in the immunoprecipitates of mitochondria treated with H2O2 with a reduction in the level of native lipoic acid (Fig. 6B). In the absence of iodoacetamide, increased anti-lipoic acid and loss of anti-glutathione antibody binding were observed, verifying lipoic acid as the site of KGDH glutathionylation (Fig. 6B). This is due to disulfide exchange upon denaturation of KGDH and regeneration of native lipoic acid. As a whole, these results offer evidence that each KGDH complex undergoes glutathionylation. However, not all lipoic acid residues within a given KGDH complex are susceptible to glutathionylation.
Fig. 6.
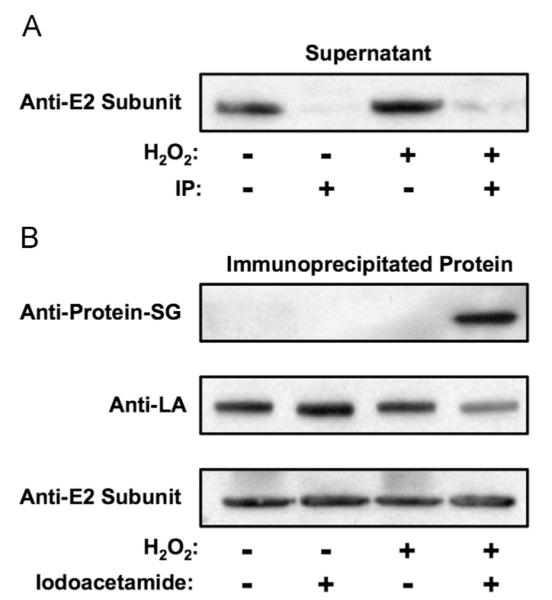
Relative susceptibilities of lipoic acid residues on KGDH complexes to glutathionylation. Isolated rat heart mitochondria (1.0 mg/ml) were incubated for 2.0 min in the presence of α-ketoglutarate (5.0 mM). H2O2 (0 or 25 μM) was then added. After 5.0 min of incubation (maximum 50% inhibition of KGDH in the presence of H2O2), mitochondria were solubilized by the addition of Triton X-100 to a final concentration of 0.05%. Immunoprecipitations were then performed using anti-lipoic acid antibody as detailed under Materials and methods. Western blot analysis was performed (A) on the supernatant fraction to determine the efficiency of immunoprecipitation and (B) on immunoprecipitated protein using anti-glutathione, anti-lipoic acid, or anti-KGDH E2 subunit antibodies as indicated. The blots shown are representative of three independent experiments.
Potential mechanism of KGDH glutathionylation
Glutathionylation of protein sulfhydryls can occur through either disulfide exchange with GSSG or the formation of a sulfenic acid that reacts with a reduced sulfhydryl. To distinguish between these possibilities, we investigated the effects of 25 versus 50 μM H2O2 and sought evidence for oxidation of KGDH lipoic acid sulfhydryls to sulfenic acid. The rate of sulfenic acid formation would be expected to increase with H2O2 concentration. We found that an increase in H2O2 concentration simply prolonged the duration of enzyme inhibition, without affecting the rate and extent of KGDH inhibition and glutathionylation (Fig. 7A and B). Independent of the H2O2 concentration, GSH is rapidly converted to GSSG and the concentration of GSH in the mitochondria remains depressed until the H2O2 has been consumed (Fig. 7C). GSSG content rises rapidly, followed by a decline to a nearly constant level 2 min after H2O2 addition (Fig. 7C). Upon consumption of H2O2 the concentration of GSSG drops, concurrent with a rise in GSH to levels observed before the addition of H2O2 (Fig. 7C). Absolute changes in the levels of GSH and GSSG are the same, regardless of H2O2 concentrations, and glutathionylation and inhibition of KGDH (Fig. 7A and B) occur when GSSG levels reach steady state. These results indicate that the 50% threshold of KGDH inhibition is not likely to be due to depletion of GSH or GSSG. These results do not, however, rule out the possibility that once GSSG falls to a specific level, this limits the degree of KGDH inhibition. It is important to note that a significant lag exists before KGDH glutathionylation and inhibition (Fig. 7A and B), suggesting the involvement of processes that must be activated before glutathionylation. Treatment of mitochondria with H2O2 in the presence of dimedone, a sulfenic acid-reactive compound [29], would trap sulfenylated lipoic acid in a dimedone-modified form, preventing reactivation by glutaredoxin and decreasing the level of native lipoic acid. In the presence of dimedone, H2O2-induced KGDH inhibition could be relieved upon addition of glutaredoxin (Fig. 8A), and no loss of native lipoic acid (Fig. 8B) or appearance of dimedone-modified KGDH was observed (not shown). Taken together, these data indicate that the likely mechanism of KGDH glutathionylation is disulfide exchange between a lipoic acid sulfhydryl and GSSG.
Fig. 7.

Time course of H2O2 induced alterations in KGDH activity, glutathionylation, and mitochondrial content of GSH and GSSG. Isolated rat heart mitochondria (0.5 mg/ml) were incubated for 2.0 min in the presence of α-ketoglutarate (5.0 mM). H2O2 (25 or 50 μM) was then added and, at indicated times, (A) mitochondria were solubilized in 25 mM Mops, pH 7.4, containing 0.05% Triton X-100, and KGDH activity was measured. Values are represented as means±standard deviation (n=3). (B) Other mitochondria were dissolved in SDS-loading buffer in the presence 100 mM iodoacetamide. Proteins (5.0 μg/lane) were resolved by nonreducing gel electrophoresis, transferred to PVDF membrane, and subjected to Western blot analysis using monoclonal anti-glutathione antibodies. The blots shown are representative of three independent experiments. (C) Other mitochondria were incubated with 5.0% metaphosphoric acid to extract GSH and GSSG for HPLC quantification using electrochemical detection. Values are represented as means7standard deviation (n=3).
Fig. 8.

Assessment of H2O2-induced sulfenic acid formation on KGDH. Isolated rat heart mitochondria (0.5 mg/ml) were incubated in the presence of α-ketoglutarate (5.0 mM) as a respiratory substrate in the presence or absence of dimedone (5.0 mM). After a 2.0-min preincubation, H2O2 (25 μM) was added. (A) Mitochondria were then solubilized at 0 and 5.0 min in 25 mM Mops, pH 7.4, containing 0.05% Triton X-100 to prevent further inhibition or reactivation of KGDH. KGDH activity was measured. Where indicated, samples were treated with glutaredoxin (1.0 U/ml) and GSH (1.0 mM) before enzyme analysis. Values are represented as means±standard deviation (n=3). (B and C) Mitochondria were solubilized by the addition of 0.05% Triton X-100. Solubilized mitochondria were eluted through PD-10 desalting columns to remove excess unreacted dimedone. Proteins were then denatured in reducing SDS-loading buffer. Western blot analysis was then performed using (B) polyclonal anti-lipoic acid or (C) polyclonal anti-E2 subunit antibodies as indicated. The blots shown are representative of three independent experiments.
Inability to reconstitute KGDH inhibition and glutathionylation
Previous studies revealed that glutathionylation-dependent glutaredoxin-reversible inhibition of KGDH could not be reconstituted using solubilized mitochondria or purified preparations of the enzyme exposed to various combinations of GSH, GSSG, H2O2, and substrates, cofactors, and effectors, including Ca2+ [27]. Alamethicin is a compound capable of forming pores in the mitochondrial membrane, enabling permeabilization by small molecules such as GSH and GSSG but not protein. To assess whether KGDH inhibition could be recapitulated in alamethicin-treated mitochondria and surpass the 50% threshold with higher concentrations of GSSG, cardiac mitochondria (0.5 mg/ml) were incubated with alamethicin (0 to 30 μM), GSSG (0 to 5.0 mM), GSH (0 to 5.0 mM), H2O2 (0 to 100 μM), KGDH substrates and cofactors, and in the presence or absence of rotenone to preserve NADH. Under all experimental conditions tested, H2O2 did not cause loss of KGDH activity (not shown). H2O2-induced KGDH glutathionylation and inhibition therefore require intact mitochondria and could be governed by several potential factors that limit the maximum achievable degree of inhibition, including recruitment of required components to the site of modification, enzymatic reactions, and/or a unique microenvironment within the mitochondria.
Glutathionylation of KGDH within the heart
We sought evidence that glutathionylation of KGDH can occur in intact heart tissue. Rat hearts were perfused in retrograde fashion according to Langendorff in the presence or absence of 100 μM H2O2 (15 min) [31,32]. Hearts were then frozen in liquid nitrogen, pulverized, and sonicated as described under Materials and methods to ensure complete rupture of the cells. Heart homogenates were analyzed for H2O2-induced KGDH inhibition and glutathionylation and changes in the levels of GSH and GSSG. H2O2 infusion resulted in a significant drop in KGDH activity that was reversed upon incubation with glutaredoxin and GSH (Fig. 9A). Analysis of heart homogenates (Fig. 9B) and proteins immunopurified using anti-lipoic acid (Fig. 9C) indicated that H2O2-induced loss of KGDH activity was accompanied by glutathionylation of the enzyme. KGDH inhibition and glutathionylation were paralleled by a decrease in the ratio of GSH to GSSG (1:1) reflecting rapid oxidation of GSH to GSSG (Fig. 9D). These results demonstrate that KGDH inhibition and glutathionylation can occur in tissue and illustrate the need to clearly define the nature of the modification to accurately assess and quantify its occurrence in biological samples and the (patho)physiological consequences.
Fig. 9.

Alterations in KGDH activity, KGDH glutathionylation, and GSH and GSSG content in rat hearts perfused with H2O2. Rat hearts were perfused for 15 min in the absence or presence of H2O2 (100 μM). Hearts were then frozen in liquid nitrogen, pulverized, resuspended, and sonicated as detailed under Materials and methods. (A) Heart extract was solubilized in 25 mM Mops, pH 7.4, containing 0.05% Triton X-100, and KGDH activity was measured. Data are depicted as the fraction of enzyme activity recoverable upon treatment with glutaredoxin and GSH. (B) Samples were dissolved in SDS-loading buffer in the presence 100 mM iodoacetamide. Proteins (5.0 μg/lane) were resolved by nonreducing gel electrophoresis, transferred to PVDF membrane, and subjected to Western blot analysis using monoclonal anti-glutathione antibodies. (C) KGDH was immunopurified using anti-lipoic acid antibodies as described under Materials and methods. Immunoprecipitates were then subjected to Western blot analysis using monoclonal anti-glutathione antibodies. (D) GSH and GSSG were extracted with 5% metaphosphoric acid, resolved by reverse-phase HPLC, and quantified using electrochemical detection. Data are presented for three independent experiments for control and H2O2-treated hearts (*p<0.003, n=3).
Discussion
KGDH is a key regulatory enzyme within the Krebs cycle critical for energy and biosynthetic homeostasis. H2O2-mediated glutathionylation may therefore represent an important means to regulate mitochondrial function in response to changes in redox status. A clear understanding of the chemistry and factors that control the degree and rate of KGDH glutathionylation is required to define mechanisms by which this occurs and the physiological consequences. Our findings provide evidence that the enzyme’s cofactor lipoic acid is the primary site of glutathionylation. In previous studies we were unable to detect glutathionylation of KGDH. We used an indirect method (biotin switch assay) that suggested lipoic acid as a site of glutathionylation [28]. As demonstrated in this study, glutathionylated lipoic acid must be stabilized by blocking the second sulfhydryl on lipoic acid to prevent intramolecular disulfide exchange upon protein denaturation and enable detection of glutathionylated lipoic acid on KGDH. In addition, differences in the relative susceptibilities of lipoic acid residues within a given KGDH complex to glutathionylation limits H2O2-induced inhibition to a threshold of ~50%. These results provide a valuable means for measuring glutathionylated KGDH and offer important insight into the chemical nature of this process.
Lipoic acid, which contains two vicinal sulfhydryl groups, seems to be an unlikely site for glutathionylation. Chemical evidence indicates that reaction of one lipoic acid sulfhydryl with glutathione is rapidly followed by disulfide exchange and deglutathionylation (Fig. 1). Stable glutathionylation of KGDH could occur as a result of modification of both lipoic acid sulfhydryls by glutathione or through succinylation of one sulfhydryl as part of the normal catalytic cycle followed by glutathionylation. Neither of these possibilities is likely, given that the use of iodoacetamide to block free sulfhydryl groups is required to stabilize glutathionylated lipoic acid upon denaturation of KGDH. Thus, after glutathionylation, steric hindrance of the second lipoic acid sulfhydryl in the enzyme’s native conformation probably prevents deglutathionylation. Steric hindrance could be induced by a change in the conformation of the protein and/or the stereochemistry of lipoic acid on the protein. Alternatively, during the normal catalytic cycle of KGDH, one sulfhydryl on lipoic acid is succinylated, followed by transfer of the succinyl group to CoASH. Thus, there exists heterogeneity in the vicinal sulfhydryls on lipoic acid that may prevent spontaneous disulfide exchange and deglutathionylation.
H2O2-induced inhibition of KGDH does not exceed 50% [26–28], reflecting the inability to glutathionylate all lipoic acids. This may indicate that glutathionylation and deglutathionylation occur simultaneously, reaching an equilibrium reflected in partial glutathionylation. Given that Ca2+ enhances the rate but not the degree of glutathionylation and enzyme inhibition, this is unlikely. Alternatively, a subpopulation of KGDH or E2 subunits within the enzyme complex may be resistant to glutathionylation. Two populations of KGDH have been described, one associated with complex I of the electron transport chain and a second soluble within the mitochondrial matrix [33–36]. However, utilization of antibody specific to native lipoic acid [24,28] results in immunoprecipitation of all KGDH complexes and both native and glutathionylated E2 subunits. Thus, it does not seem that a subpopulation of KGDH is resistant to glutathionylation but rather lipoic acid residues within each KGDH complex are differentially susceptible to glutathionylation.
Variation in reactivity between different lipoic acids within the KGDH complex may be due to reported differences in the geometry of various lipoic acid residues within the E2 core relative to other KGDH subunits. It has been estimated that approximately half of the lipoic acid residues are in a position that enables reactions with multiple E1 and E3 subunits [4,37]. Lipoic acid groups in less favorable positions are less likely to be succinylated by E1 and/or subsequently oxidized by E3. Thus, lipoic acid moieties on each E2 subunit within a given enzyme complex are not geometrically equivalent, which could influence the relative susceptibility to glutathionylation. It is also possible that glutathionylation of a fraction of the lipoic acids alters the susceptibility of the remaining unmodified lipoic acid residues. It is important to note, however, that lipoic acid residues that are not glutathionylated can participate in catalysis given that the threshold of inhibition does not exceed 50%. Moreover, the maximum 50% inhibition is not attributable to loss of glutathione, in particular GSSG.
Demonstrated and proposed mechanisms of protein glutathionylation include: (1) disulfide exchange due to bimolecular collisions between low pKa protein sulfhydryls and GSSG; (2) priming of protein or GSH sulfhydryls by free radical species or prooxidants to enhance reactivity; and (3) enzyme catalyzed [38–42]. Our results strongly suggest that the mechanism of KGDH glutathionylation is disulfide exchange between a lipoic acid sulfhydryl group and GSSG. Reaction of a lipoic acid sulfenic acid with GSH is unlikely given the lack of dependence of KGDH inhibition and glutathionylation on H2O2 concentration. In addition, mitochondria treated with H2O2 in the presence of dimedone did not exhibit modification of KGDH by dimedone and inhibition remained reversible by glutaredoxin. Nevertheless, KGDH appears resistant to reversible glutathionylation-derived inhibition in permeabilized and solubilized mitochondria or purified preparations exposed to various combinations of GSH, GSSG, H2O2, and substrates, cofactors, and effectors, including Ca2+ [27]. These results suggest that glutathionylation and inhibition of KGDH may be enzymatically driven. However, despite an immediate oxidation of GSH to GSSG upon addition of H2O2 to mitochondria, KGDH glutathionylation and inhibition occur only after an appreciable lag (~1 min). The possibility therefore exists that glutathionylation occurs through nonenzymatic processes requiring a unique microenvironment within the mitochondria and/or the recruitment of other factors to the site of modification. Based on the results of this study, it will be important to investigate conditions that increase the half-life of reduced lipoic acid as a determinant of enzymatic or nonenzymatic glutathionylation.
KGDH is an enzyme that, by virtue of the redox chemistry it catalyzes, is highly susceptible to oxidative damage [22–25]. Therefore, loss of the activity of this enzyme during the progression of diseases associated with oxidative stress [14–22] is perhaps not surprising. KGDH can, however, undergo reversible glutathionylation and inhibition that, in vitro, protects the enzyme from certain forms of oxidative damage [28]. The failure of such protective measures may precipitate oxidative damage. Alternatively, glutathionylation of KGDH may not occur primarily as an antioxidant response, but could represent a means to regulate mitochondrial function. This study identified properties of lipoic acid on KGDH that render the cofactor susceptible to glutathionylation and defined biochemical manipulations that can be effectively employed to detect the modification. This provides the basis for delineating the mechanisms of KGDH glutathionylation and accurately assessing the occurrence of this modification in vivo and the physiological consequences.
Acknowledgments
The project described was supported by Grant R01AG016339 from the National Institute on Aging and from the Oklahoma Center for the Advancement of Science and Technology with additional support from the Oklahoma Medical Research Foundation and the Hille Family Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging or the National Institutes of Health.
Abbreviations
- KGDH
α-ketoglutarate dehydrogenase
- PDH
pyruvate dehydrogenase
- E1
α-ketoglutarate dehydrogenase
- E2
dihydrolipoyl succinyltransferase
- E3
dihydrolipoyl dehydrogenase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- protein–SG
glutathionylated protein
- LA
lipoic acid
- IP
immunoprecipitation
References
- [1].Reed LJ. Multienzyme complexes. Acc. Chem. Res. 1974;7:40–46. [Google Scholar]
- [2].Koike K, Hamada M, Tanaka N, Otsuka KI, Ogasahara K, Koike M. Properties and subunit composition of the pig heart 2-oxoglutarate dehydrogenase. J. Biol. Chem. 1974;249:3836–3842. [PubMed] [Google Scholar]
- [3].Koike M, Koike K. Structure, assembly and function of mammalian alphaketo acid dehydrogenase complexes. Adv. Biophys. 1976:187–227. [PubMed] [Google Scholar]
- [4].Waskiewicz DE, Hammes GG. Elementary steps in the reaction mechanism of the alpha-ketoglutarate dehydrogenase multienzyme complex from Escherichia coli: kinetics of succinylation and desuccinylation. Biochemistry. 1984;23:3136–3143. doi: 10.1021/bi00309a005. [DOI] [PubMed] [Google Scholar]
- [5].Perham RN. Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochemistry. 1991;30:8501–8512. doi: 10.1021/bi00099a001. [DOI] [PubMed] [Google Scholar]
- [6].Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978;176:899–906. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hansford RG, Castro F. Effects of micromolar concentrations of free calcium ions on the reduction of heart mitochondrial NAD(P) by 2-oxoglutarate. Biochem. J. 1981;198:525–533. doi: 10.1042/bj1980525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Panov A, Scarpa A. Independent modulation of the activity of alpha-ketoglutarate dehydrogenase complex by Ca2+ and Mg. Biochemistry. 1996;35:427–432. doi: 10.1021/bi952101t. [DOI] [PubMed] [Google Scholar]
- [10].Randle PJ, England PJ, Denton RM. Control of the tricarboxylate cycle and its interactions with glycolysis during acetate utilization in rat heart. Biochem. J. 1970;117:677–695. doi: 10.1042/bj1170677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Smith CM, Bryla J, Williamson JR. Regulation of mitochondrial alphaketoglutarate metabolism by product inhibition at alpha-ketoglutarate dehydrogenase. J. Biol. Chem. 1974;249:1497–1505. [PubMed] [Google Scholar]
- [12].Cooney GJ, Taegtmeyer H, Newsholme EA. Tricarboxylic acid cycle flux and enzyme activities in the isolated working rat heart. Biochem. J. 1981;200:701–703. doi: 10.1042/bj2000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moreno-Sanchez R, Hogue BA, Hansford RG. Influence of NAD-linked dehydrogenase activity on flux through oxidative phosphorylation. Biochem. J. 1990;268:421–428. doi: 10.1042/bj2680421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos. Trans. R. Soc. London B Biol. Sci. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- [16].Gibson GE, Park LC, Sheu KF, Blass JP, Calingasan NY. The alpha-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem. Int. 2000;36:97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- [17].Kish SJ. Brain energy metabolizing enzymes in Alzheimer’s disease: alpha-ketoglutarate dehydrogenase complex and cytochrome oxidase. Ann. N. Y. Acad. Sci. 1997;826:218–228. doi: 10.1111/j.1749-6632.1997.tb48473.x. [DOI] [PubMed] [Google Scholar]
- [18].Lucas DT, Szweda LI. Declines in mitochondrial respiration during cardiac reperfusion: age-dependent inactivation of alpha-ketoglutarate dehydrogenase. Proc. Natl. Acad. Sci. USA. 1999;96:6689–6693. doi: 10.1073/pnas.96.12.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lundberg KC, Szweda LI. Preconditioning prevents loss in mitochondrial function and release of cytochrome c during prolonged cardiac ischemia/reperfusion. Arch. Biochem. Biophys. 2006;453:130–134. doi: 10.1016/j.abb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- [20].Sadek HA, Humphries KM, Szweda PA, Szweda LI. Selective inactivation of redox-sensitive mitochondrial enzymes during cardiac reperfusion. Arch. Biochem. Biophys. 2002;406:222–228. doi: 10.1016/s0003-9861(02)00446-0. [DOI] [PubMed] [Google Scholar]
- [21].Schapira AH. Mitochondrial involvement in Parkinson’s disease, Huntington’s disease, hereditary spastic paraplegia and Friedreich’s ataxia. Biochim. Biophys. Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- [22].Gibson GE, Blass JP, Beal MF, Bunik V. The alpha-ketoglutarate–dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol. Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- [23].Bunik VI, Sievers C. Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur. J. Biochem. 2002;269:5004–5015. doi: 10.1046/j.1432-1033.2002.03204.x. [DOI] [PubMed] [Google Scholar]
- [24].Humphries KM, Szweda LI. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry. 1998;37:15835–15841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- [25].Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J. Biol. Chem. 2001;276:23357–23361. doi: 10.1074/jbc.M100320200. [DOI] [PubMed] [Google Scholar]
- [27].Nulton-Persson AC, Starke DW, Mieyal JJ, Szweda LI. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry. 2003;42:4235–4242. doi: 10.1021/bi027370f. [DOI] [PubMed] [Google Scholar]
- [28].Applegate MA, Humphries KM, Szweda LI. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 2008;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- [29].Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J. Biol. Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- [30].Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic. Biol. Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lucas DT, Szweda LI. Cardiac reperfusion injury: aging, lipid peroxidation, and mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA. 1998;95:510–514. doi: 10.1073/pnas.95.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Langendorff O. Untersuchungen am überlebenden Säugethierherzen. Arch. Gesamte Physiol. Menschen Tiere. 61:291–895. 232. [Google Scholar]
- [33].Fukushima T, Decker RV, Anderson WM, Spivey HO. Substrate channeling of NADH and binding of dehydrogenases to complex I. J. Biol. Chem. 1989;264:16483–16488. [PubMed] [Google Scholar]
- [34].Maas E, Bisswanger H. Localization of the alpha-oxoacid dehydrogenase multienzyme complexes within the mitochondrion. FEBS Lett. 1990;277:189–190. doi: 10.1016/0014-5793(90)80840-f. [DOI] [PubMed] [Google Scholar]
- [35].Porpaczy Z, Sumegi B, Alkonyi I. Interaction between NAD-dependent isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase complex, and NADH:ubiquinone oxidoreductase. J. Biol. Chem. 1987;262:9509–9514. [PubMed] [Google Scholar]
- [36].Sumegi B, Srere PA. Complex I binds several mitochondrial NAD-coupled dehydrogenases. J. Biol. Chem. 1984;259:15040–15045. [PubMed] [Google Scholar]
- [37].Pettit FH, Hamilton L, Munk P, Namihira G, Eley MH, Willms CR, Reed LJ. Alpha-keto acid dehydrogenase complexes. XIX. Subunit structure of the Escherichia coli alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 1973;248:5282–5290. [PubMed] [Google Scholar]
- [38].Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- [39].Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- [40].Hurd TR, Filipovska A, Costa NJ, Dahm CC, Murphy MP. Disulphide formation on mitochondrial protein thiols. Biochem. Soc. Trans. 2005;33:1390–1393. doi: 10.1042/BST0331390. [DOI] [PubMed] [Google Scholar]
- [41].Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signaling. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- [42].Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 2010;285:39646–39654. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]