

Abstract
Stress is a growing public health concern and can lead to significant disabilities. The neural response to stressors is thought to be dependent on the extended amygdala. The basolateral amygdala (BLA) is responsible for associations of sensory stimuli with emotional valence and is thought to be involved in stress-induced responses. Previous behavioral and electrophysiological experiments demonstrate that, in response to stress, changes occur in glutamatergic neurotransmission within the BLA and, in particular in transmission at AMPA receptors. Given the established role of AMPA receptors in memory and synaptic plasticity, we tested the hypothesis that stress produces alterations in the distribution of these receptors in a way that might account for stress induced alterations in amygdala circuitry function. We examined the subcellular localization of GluR1 subunits of the AMPA receptor and the electrophysiological characteristics of BLA principal neurons in an animal model of unpredictable stress. Compared to controls, we demonstrated an increase in the ratio of labeled spines to labeled dendritic shafts in the BLA of rats 6 and 14 days post-stress, but not 1 day post-stress. Furthermore, the frequency of mini-EPSCs was increased in stressed animals without a change in general membrane properties, mini-EPSC amplitude, or in paired pulse modulation of glutamate release. Taken together, these data suggest that the shift of GluR1-containing AMPA receptors from dendritic stores into spines may be in part responsible for the persistent behavioral alterations observed following severe stressors.
Introduction
Animals are exposed to myriad stressors and have developed complex response mechanisms designed to reestablish their original homeostatic state. However, severe stressors can induce a variety of maladaptive stress responses resulting in pathologic behavior. For example, pathological levels of stress have been associated with onset or relapse in drug addiction (Belujon and Grace, 2011; Goeders, 2003), depression (Lloyd and Nemeroff, 2011; Palazidou, 2012), and schizophrenia (Falloon et al., 1996; Goel and Bale, 2009), as well as post-traumatic stress disorder (Jovanovic and Ressler, 2010). Characteristic of the latter syndrome is a persistent alteration in how the individual responds to their environment, including increased arousal and avoidance behavior (Jovanovic and Ressler, 2010). To provide better treatment for these conditions we need to understand the neurobiological basis for such stress-induced, long-lasting behavioral changes.
Both clinical studies and animal research have implicated the extended amygdala, including the basolateral amygdala (BLA) in the neural response to stressors (Gale et al., 2004; Gutman et al., 2008; Rainnie and Ressler, 2009; Vermetten et al., 2007). The BLA integrates information from sensory cortex and thalamus as well as the hippocampus (McDonald, 1998; Sah et al., 2003). It then sends glutamatergic afferents to other regions involved in modulating anxiety and fear responses, including the central nucleus of the amygdala (CeA) and the bed nucleus of the stria terminalis (BNST) (Davis et al., 1994; McDonald, 1998). Behavioral research has indicated that the BLA is involved in assigning emotional significance to sensory stimuli (Davis et al., 1994; Nader et al., 2001; Walker and Davis, 2008). Moreover, excitotoxic lesions or pharmacological manipulations of the BLA disrupt the acquisition of fear memories (Fanselow and Kim, 1994; Fanselow and LeDoux, 1999; Onishi and Xavier, 2010), as well as their maintenance and extinction (Flavell and Lee, 2012; Shekhar et al., 2005). The formation of fear memories is associated with long-term potentiation (LTP) in the BLA (Goosens and Maren, 2002; Rogan et al., 1997). Intense stressors have been shown to facilitate memory formation (McGaugh and Roozendaal, 2002; Shors, 2001). Thus, the BLA could become abnormally responsive to benign stimuli, resulting in the association of inappropriate levels of emotionality to neutral stimuli following severe stressors. Imaging studies have demonstrated increased basal activity levels of the amygdala in patients with anxiety disorders (Etkin and Wager, 2007; Rabinak et al., 2011). Additionally, patients with anxiety disorders show increased amygdala responses to fear-inducing stimuli compared to normal controls (Linnman et al., 2011; Rauch and Shin, 1997; Rauch et al., 2006; Shin and Liberzon, 2010). Patients with PTSD have also demonstrated increased amygdala responses to neutral stimuli as measured by fMRI (Brunetti et al., 2010; Hendler et al., 2003), indicating aberrant functioning of the amygdala under non-threatening conditions.
Altered activity patterns can change the distribution of signal transduction proteins in the brain. This has been particularly well studied with respect to the role of glutamatergic systems in LTP. For example, AMPA receptors have been shown to be incorporated into synapses as a mechanism of producing LTP (Makino and Malinow, 2009; Yudowski et al., 2007) and changes in AMPA receptor trafficking are also induced in the amygdala by a fear conditioning paradigm (Nedelescu et al., 2010; Rumpel et al., 2005). Furthermore, various types of stress and stress hormones can also modify the electrophysiological characteristics of BLA neurons via modulations of neurotransmitter receptor systems, including both glutamate and GABA systems (McCool et al., 2010; Rainnie et al., 2004; Shekhar et al., 2005; Vouimba et al., 2004). These experiments suggest that stress could alter the neuronal localization of AMPA receptors in the rat BLA. We have shown that unpredictable shock stress resulted in a long lasting generalized increase in startle (Hazra et al., 2012). Since previous experiments show a modulation of AMPA receptor protein localization as a mechanism for alterations in neuronal function, we hypothesized that stress would produce a lasting alteration in the distribution of the AMPA receptor subunit, GluR1, in the BLA and that this change might produce alterations on excitatory synaptic transmission in BLA principal neurons.
Materials and Methods
Animals
All experimental protocols strictly conformed to National Institutes of Health guidelines for the Care and Use of Laboratory. Animals, and were approved by the Institutional Animal Care and Use Committees of the Atlanta VA and Emory University. For immunohistochemical experiments we used 34 adult, male, Sprague–Dawley rats that were 43 days old at the beginning of the first footshock session (Charles River, Wilmington, MA, USA). For electrophysiological experiments, we used 13 male Sprague–Dawley rats 52 days old (the same age as our 6 day post-stress animals).
For our stress studies we used a repeated unpredictable foot shock protocol (Hazra et al., 2012). Here, rats receiving shock stress were placed in a testing chamber (Lafayette Instruments) and exposed to 16 randomly timed foot shocks during a 30 minute period and then returned to their home cages. The shocks were 0.5mA and lasted for 0.5s, and the half-hour session was broken down into a 5-minute habituation session followed by three eight-minute periods. Rats received eight random shocks in the first and third eight-minute periods. Rats underwent this shock protocol daily for four days. All rats were 43 days old on the first stress day, and age-matched control groups remained in home cages without shocks.
Stressed rats and their age matched controls were sacrificed at either one, six or fourteen days following the final day of the footshock protocol. For the immunohistochemical studies, rats were given an injection of pentobarbital (100 mg/kg i.p.), and within minutes of loss of tail-pinch reflex, transcardially perfused with at least 100 ml of cold, oxygenated Ringer’s solution. This was followed by perfusion with 500 ml of fixative containing 4.0% paraformaldehyde and 0.1% glutaraldehyde, in phosphate buffer (PB, 0.1 M, pH 7.4). Brains were removed from the skull and stored overnight in 4.0% paraformaldehyde overnight at 4°C. The brains were then rinsed in phosphate-buffered saline (PBS) and cut into 60 μm-thick coronal sections on a vibratome. Sections were put in a 1.0% sodium borohydride in phosphate buffered saline (PBS) for 20 min and rinsed with PBS before being processed for immunohistochemistry. In order to improve penetration of immunolabeling, a freeze thaw protocol was used. Sections were transferred to a cryoprotectant solution (PB, 0.05 M, pH 7.4 containing 25% sucrose and 10% glycerol) for 20 min. They were then frozen in a −80 °C freezer for 20 min, returned to a decreasing gradient of cryoprotectant solutions and rinsed in PBS. Sections then underwent immunohistochemical procedures for the immunoperoxidase localization of GluR1.
Immunohistochemical Experiments
Immunohistochemistry and preparation for electron microscopy was done according to standard techniques (Muly et al., 2010). We used an affinity purified rabbit polyclonal antibody directed against the cytoplasmic domain of the GluR1 subunit of the AMPA glutamate receptor (ab1504; Millipore; Temicula, CA) used at 0.3 μg/ml and biotinylated goat-anti-rabbit secondary antisera (Vector, Burlingame, CA, USA) diluted 1:200. Specificity of the antibody has been confirmed by Western blotting (Wenthold et al., 1992). After immunolabeleing the sections were postfixed in 1% osmium tetroxide, dehydrated in ethanol and placed in uranyl acetate to increase contrast for the electron microscope. The sections were embedded in epoxy resin (Durcupan, Sigma-Aldrich, St. Louis, MO) and, following hardening blocks of the BLA were prepared and ultrathin sections collected and stained with lead citrate. The region of the BLA sampled was in the middle to slightly rostral region of the BLA (bregma −2.1 to −3.6) and dorsoventrally in the middle of the nucleus with a bias toward the basal half. The grids were examined with a JEOL 1011 electron microscope (JEOL; Munchen, Germany). The immunoperoxidase labeling for GluR1 penetrated deep into the tissue, so we were able to sample material away from the tissue–resin interface. We randomly selected fields of immunoreactive elements from the blocks, and images were taken at a magnification of ×40,000 using a Gatan 785 camera and examined using Gatan Digital Micrograph software (Gatan, Inc.; Pleasonton CA). Data were collected from two blocks from each stressed animal and one from each control animal, and in total thirty four animals were examined. On each micrograph, DAB-labeled profiles were identified based on their content of patches of DAB reaction product, darker than any adjacent mitochondria, without sharp borders suggesting crystalline deposits and contained within a membrane bound element and not appearing to overlay multiple profiles. These labeled profiles were classified as spines, dendritic shafts, terminals, axons, or glia based on ultrastructural criteria (Peters, 1991). Although the presence of DAB within the elements complicates their identification, whenever possible both the presence and the absence of multiple criteria were used to make each determination (Muly et al., 2003). If strong DAB, ultrastructural flaws, or absence of clear features made identification difficult, the profile was discarded. Profiles were identified as spines based on size (0.3–1.5μm in diameter), presence of spine apparatus, absence of mitochondria or microtubules, and in some cases presence of asymmetric synaptic contacts. Dendritic shafts were identified by their greater size (0.5μm or more in diameter) and the presence of microtubules, mitochondria, and in some cases synaptic contacts. Axon terminals were characterized by the presence of numerous vesicles, mitochondria, and occasionally a presynaptic specialization. Preterminal, unmyelinated axons were identified by their small size (0.1–0.3μm in diameter); regular, round shape; and occasional presence of synaptic vesicles or neurofilaments. Glial profiles were identified based on their characteristic shape, which appears to fill in the space between other, nearby profiles, and a relatively clear cytoplasm, which occasionally contained numerous filaments. The numbers of profiles immunoreactive for GluR1 were tabulated and the distributions compared with an ANOVA. An additional analysis was made between the control group and the 6 day post stress group. Micrographs were examined and spines and dendritic shafts were identified without regard to whether they were immunolabeled. The ratio of spines to shaft profiles was determined for each animal and compared to determine whether a change in the relative number of either element was caused by the stress protocol.
Electrophysiological Experiments
Slices of 350 μm thickness containing the BLA were obtained as has been described previously (Rainnie, 1999) from control animals or animals sacrificed six days after the conclusion of the stress protocol. Briefly, rats were anesthetized with isoflurane (Fisher Scientific, Hanoverpark, IL, USA) and the brains rapidly dissected out and immersed in a cold (4 °C) 95–5% oxygen/carbon dioxide oxygenated “cutting solution” with the following composition (in mM): NaCl (130), NaHCO3 (30), KCl (3.50), KH2PO4 (1.10), MgCl2 (6.0), CaCl2 (1.0), glucose (10), supplemented with kynurenic acid (2.0). Slices containing the BLA were cut using a Leica VTS-1000 Vibratome (Leica Microsystems Inc., Bannockburn, IL, USA). After cutting, slices were maintained at 34°C in oxygenated “cutting solution” for at least 50min before transferring to regular artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl (130), NaHCO3 (30), KCl (3.50), KH2PO4 (1.10), MgCl2 (1.30), CaCl2 (2.50), and glucose (10). Slices were kept in the regular ACSF for at least 30 min before recording.
Individual slices were then transferred to a submersion-type recording chamber mounted on the fixed stage of a Leica DMLFS microscope (Leica Microsystems Inc., Bannockburn, IL, USA), and continuously perfused by gravity-fed oxygenated 32 °C ACSF at a flow rate of 1–2 ml/min. Slices were viewed using differential interference contrast (DIC) optics and infrared (IR) illumination with an IR sensitive CCD camera (Orca ER, Hamamatsu, Tokyo Japan). Whole-cell patch-clamp recordings were obtained using standard techniques (Rainnie et al., 2004), by an experimenter blind to the identity of the animal from which the slices were obtained. Thin-walled borosilicate glass patches electrodes (WPI, Sarasota, FL, USA) which had a resistance of 4–6 MΩ were filled with (in mM): 130K-gluconate, 2 KCl, 10 HEPES, 3 MgCl2, 2 K-ATP, 0.2 NaGTP, and 5 phosphocreatine, adjusted to pH 7.3 with KOH, and having an osmolarity of 280–290 mOsm. Whole-cell access resistances measured in voltage clamp ranged from 5–20 MΩ were monitored throughout each experiment; and neurons showing a 15% change of access resistance were discarded.
Individual BLA projection neurons were visualized in situ using DIC microscopy in combination with a 40x water immersion objective, and displayed in real time on a computer monitor. Projection neurons were identified according to their characteristic size and shape (McDonald et al., 2005), as well as their physiological characteristics, and were normally located between 50 and 120 μM beneath the surface of the slice. Data acquisition and analysis were performed using a MultiClamp700B amplifier in conjunction with pClamp10.0 software and a DigiData 1320A AD/DA interface (Molecular Devices, Burlingame, CA, USA). Whole cell patch clamp recordings were obtained in current clamp and voltage clamp modes and filtered at 2 kHz and digitized at 10–20 kHz. The membrane potential was held at −60 mV for all neurons. The access resistance were monitored throughout experiment and neurons had more than 15% change of access resistance were discarded.
At the start of each experiment, a series of standardized current clamp protocols were performed to further validate the identity of BLA projection neurons (Rainnie et al., 1993). To examine the effects of the stress manipulation on excitatory synaptic transmission in the BLA, 100μM picrotoxin was added to the patch recording solution to block GABAA receptor-mediated currents only in the recorded neuron. Spontaneous mEPSCs were recorded in the presence of TTX (1 mM) and the selective GABAB receptor antagonist, CGP36742 (2 μM). Spontaneous synaptic events were captured continuously for 30s in both control and stressed animals. All events were detected offline and their amplitude and frequency calculated using MiniAnalysis 6.0 (Synaptosoft Inc., Decatur, GA). A change of mEPSCs amplitude is consistent with an increase in AMPA receptor density in postsynaptic structures that already possess the receptor. A change in mEPSC frequency is consistent with either a presynaptic change in release probability or the insertion of AMPA receptors postsynaptic to release sites that did not previously have access to them.
To calculate the paired-pulse ratio (PPR) two EPSCs were evoked with an inter-stimulus-interval of 20, 50, 100 and 200 ms, PPR was calculated as (eEPSC1/eEPSC2), where eEPSC1 and eEPSC2 represent the amplitude of the first and the second eEPSC, respectively. Alterations in the PPR are thought to represent changes in release probability in the presynaptic terminal.
Statistics
For the neuroanatomical studies, data was first checked for normality using the Shapiro-Wilk test. The groups were then compared using a 2-way ANOVA with age and labeled element as factors. Additionally, treatment groups were compared with a 1-way ANOVA with the ratio of labeled spines to labeled dendritic shafts as the factor. Pairwise comparisons were performed using post-hoc Scheffe tests. All data are expressed as the mean ± SEM. For the physiological studies, statistical tests were conducted using Graphpad Prism 4.0. Data of mEPSCs was analyzed using the Mann-Whitney U test. A paired t test was performed to test PPR. A p<0.05 was considered statistically significant for all cases.
Results
In order to test our hypothesis that stress induces a persistent alteration in the distribution of AMPA receptor subunits in the BLA, we examine three time points: 1, 6, and 14 days after the four day stress protocol. We first determined the localization of GluR1 in control animals at three different ages: 47, 52 and 60 days (n= 6, 5 and 5 respectively), which correspond to the ages of animals at the three different time points after completion of the stress protocol, in order to determine whether age might have a confounding effect on measuring AMPA receptor distribution in stressed animals at these time points. We found the vast majority of GluR1 immunoreactivity was in postsynaptic elements, i.e. dendritic shafts and spines (Figure 1A), consistent with previous reports (Farb et al., 1995). While dendritic shafts and spines comprised the bulk of GluR1-labeled profiles, unlabeled spines and shafts were also noted even in regions that were extensively labeled (Figure 1A). In order to quantify our impression, we examined material from the aforementioned 16 control animals and imaged and classified 180 to 275 labeled elements within the neuropil of the BLA from each animal. We determined the percentage of labeled elements in five different components of neuropil for each control animal. These data were analyzed by a two-way ANOVA with factors of age and labeled element. There was a significant main effect of labeled element (F(4,65)=1253.410, p<0.0001), but no significant effect of age (F(2,65)=0.004, p=0.996), nor a significant interaction (F(8,65)=1.710, p=0.113). Accordingly, data for the distribution of GluR1 in control BLA was pooled from the three age groups and is depicted in Figure 2. As suggested by our qualitative observations, the overwhelming majority of GluR1-labeled elements in BLA neuropil comprised the dendritic arbor, with over 96% of the labeling seen in dendritic shafts and spines.
Figure 1.

Electron micrograph of GluR1 immunoreactivity in the BLA from a control rat (A) and a 6-day post stress rat (B). Arrowheads indicate GluR1-immunoreactive spines; arrows indicate GluR1-immunoreactive dendritic shafts; and open arrowheads indicate unlabeled spines. Note that there are non-labeled spines and dendritic shafts intermingled with the labeled ones. Scale bar in (B) is 500nm.
Figure 2.
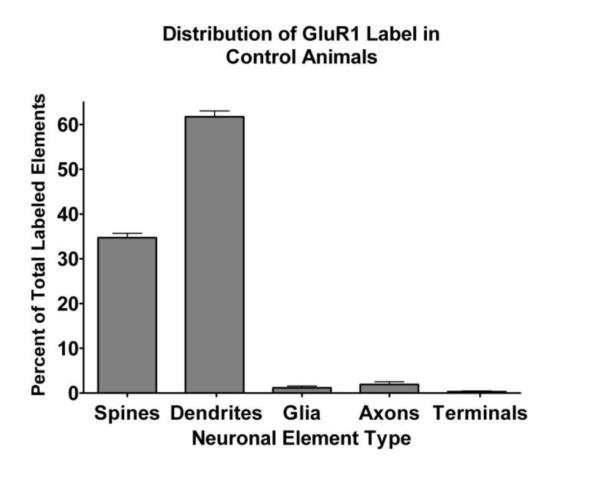
Bar chart of the distribution of GluR1 immunoreactivity in different types of neuronal elements, pooled from control subjects. GluR1 is primarily located in dendritic shafts and spines with very little observed in axonal elements or glia. Bars indicate mean with standard errors (n=16).
Having established the distribution of GluR1 in rat BLA across the relevant time frame, we next examined the effect of stress on this distribution. Here, we studied animals that were sacrificed 1, 6 and 14 days after the completion of the unpredictable shock stress paradigm (n=6 for each group) and compared them to the 16 control animals. For each of the stress animals, we imaged and classified between 300-600 labeled elements per animal within the neuropil of the BLA. Because the vast majority of all GluR1 labeling was observed in dendritic shafts and spines, as in the control group, we focused our analysis on these compartments, calculating the ratio of labeled spines to labeled dendritic shafts (Figure 1B, 3). There was a significant main effect of treatment on the ratio of spine to shaft GluR1 labeling (F(3,30)=15.582, p<0.0001). Post hoc Scheffe tests found no significant difference between control and 1 day post stress (p=0.81) or between day 6 and day 14 post stress (p=0.64), but a significant increase in ratio of spine to shaft labeling for day 6 stress animals compared to control (p<0.0001) and day 1 (p=0.0001), as well as a significant increase in ratio of spine to shaft labeling for day 14 stress animals compared to control (p=0.0055) and day 1 (p=0.0039). These results indicate that exposure to stress causes a relative shift in the distribution of the AMPA receptor subunit GluR1 from dendritic shafts to dendritic spines, which is delayed in onset by at least one day but that then persists until at least 14 days post stress exposure.
This result could be explained either by a stress induced shift in GluR1 localization of BLA neurons from dendritic shafts into spines such that more spines now contained GluR1 or by a stress induced sprouting of dendritic spines which contained GluR1 at the same rate as spines in the control condition. In order to differentiate between these two possibilities, we determined the ratio of spines to shafts in neuropil of control animals without regard to whether those profiles were immunolabeled and compared it with that for the 6-day post stress group. No significant difference was found between these two groups (p=0.890), indicating that a morphological difference between the neurons in the control and stress groups is not responsible for the change in the ratio of labeled spines to labeled dendritic shafts under these conditions.
In order to determine if a change in the ratio of GluR1-labeled spines to dendritic shafts could be related to a loss of GluR1 labeling in interneuron dendrites we conducted a secondary analysis. Unlike spiny principal neurons (McGuire et al., 1991), interneurons receive asymmetric synaptic contacts on their dendritic shafts and soma and not on spines. Thus the presence of asymmetric synaptic contacts onto dendritic shafts has been used as a indicator that these dendrites belong to interneurons. While this measure is inexact, in that there are some asymmetric contacts onto principal neuron dendritic shafts, it is the only means to address this question given the absence of protein markers for interneurons that are found throughout the dendritic arbor of all interneurons. We found that 10.5 +/− 1.1% of GluR1-labeled dendrites in control material received asymmetric synapses. There was no effect of stress treatment on this percentage (F(3,30)=0.077, p=0.97). Thus it seems unlikely that the increase in the ratio of labeled spines to dendritic shafts after stress is due to a reduction in labeling of interneuron dendrites.
Since we observed an increase in the relative amount of GluR1 immunoreactivity in BLA spines following stress, we examined the effect of stress on glutamatergic neurotransmission in this nucleus to determine if the shift of GluR1 to spines was reflected in the electrophysiological properties of these neurons. We first looked to see if the stress manipulation itself changed any of the intrinsic membrane properties of BLA principal neurons. As shown in Table 1, no significant differences were observed in any of the electrophysiological properties examined in tissue from control and stressed animals. Hence, we next examined the effect of stress on the frequency and amplitude of spontaneous miniature excitatory postsynaptic currents (mEPSCs) in BLA principal neurons. Here, mEPSCs were recorded in the presence of intracellular picrotoxin (100 μM), and bath applied CGP36742 (2 μM) and TTX (1mM) to block GABA receptor-mediated inhibitory synaptic currents and action potential dependent synaptic transmission respectively (Figure 4A,B). Stress significantly increased the mean frequency of mEPSCs from 0.98±0.19 Hz in control animals to 2.71±0.69 Hz in stressed animals (control: n=14, stress: n=17; p < 0.05, Mann-Whitney U test, Figure 4C), but had no effect on the amplitude of the mEPSCs (control: 24.82±1.05 pA; stress: 24.03±0.82 pA; p =0.98, Mann-Whitney U test, Figure 4D). Quantitative analysis showed that stress caused a leftward shift of the cumulative probability curve for the frequency of the mEPSCs, suggesting that in stress animals the inter-event intervals were decreased. In contrast, the relative distribution of the amplitudes for these spontaneous events was unchanged (Figures 4E,F). The stress-induced increase of mEPSC frequency together with its lack of effect on mEPSC amplitude initially suggested a presynaptic locus for the stress induced changes, although postsynaptic mechanisms could not be ruled out. In order to test this, we compared the paired-pulse ratio (PPR) of stimulus-evoked EPSCs, a form of synaptic plasticity that also depends on alterations in presynaptic release probability, in control and stressed rats. Interestingly, the PPR at inter-pulse intervals of 20, 50, 100 and 200 ms was not changed in slices from stressed animals compared to controls (n=14, P=0.79), which paradoxically suggested that presynaptic glutamate release probability was not changed in BLA from stressed animals (Figure 5). A possible explanation for this apparent contradiction is that the increased mEPSCs frequency in stressed animals was not due to changes of presynaptic glutamate release, but due to an increase in the number of functional synapses in principal neurons from stressed animals.
TABLE 1.
Intrinsic membrane properties of BLA projection neurons in Control and Stress rats respectively
| Control (n=21) | Stress (n=20) | |
|---|---|---|
| RMP(mV) | −60.4±0.6 | −60.2±0.4 |
| Tau(ms) | 22.1±1.9 | 22.2±1.5 |
| Rm(MΩ) | 68.1±4.8 | 69.0±3.9 |
| Threshold(mV) | −38.3±1.7 | −38.9±1.4 |
Figure 4.

Stress increases the frequency of mEPSCs. Spontaneous mEPSCs of BLA principal neurons were recorded with intracellular application of Picrotoxin (100 μM), bath application of CGP36742 (2 μM) and TTX (1 mM). (A) and (B) Representative traces showing mEPSCs of BLA principal neurons from Control and Stress animals respectively. (C) and (D) Group data indicate unpredictable shock stress increased the frequency but not the amplitude of mEPSCs. (Control: n=14, Stress: n=17; * p < 0.05). (E) and (F) Cumulative data plots showing that unpredictable shock stress decreased the inter-event interval but not the amplitude of mEPSCs.
Figure 5.

(A) Raw data show the effects of unpredictable shock stress on the PPR of eEPSCs. eEPSC amplitude was measured using paired stimuli pulses separated by 50 ms, in slices from control and stress animals respectively. (B) A plot chart showing the group data for the effects of unpredictable shock stress on PPR using paired stimuli pulses separated by 20, 50, 100 and 200 ms. Note: The PPR was not changed (n=14, P=0.79) in slices from stress animals (triangles) compared to control animals (squares). Error bars indicate S.E.M.
Discussion
In this study we found that stress produced a delayed increase in the localization of GluR1 protein to spines compared with dendritic shafts. This shift in localization was not due to a relative change in the composition of the neuropil of the BLA or to differences in animal age at the different time points. Stress induced alteration in receptor distribution was coupled with a stress induced increase in the frequency, but not the amplitude of mEPSCs and with no effect on paired-pulse ratio. Thus, we propose that the observed change in localization pattern of GluR1 represents a shift of receptor from dendritic stores into dendritic spines that prior to the stressful experience had little to no GluR1-containing AMPA receptors. These changes could contribute to the persistent hyper-arousal and increased anxiety observed following stress (Hazra et al., 2012).
A number of studies have suggested that stress produces morphologic alterations in the neurons of the hippocampus, amygdala and BNST. In the BLA, chronic immobilization stress has been shown to increase dendritic arborization in projection neurons (Vyas et al., 2002) and increase spine density (Eiland and McEwen, 2012; Qin et al., 2011; Vyas et al., 2006). Alternately, chronic unpredictable stress, which used a variety of different stressors over a 10-day period, produced atrophy of the dendritic arbor, but only in BLA bipolar cells (Vyas et al., 2002). While our examination of the distribution of GluR1 could be sensitive to morphological changes that alter the relative contribution of spines and dendritic shafts to the neuropil compartment, we found no change in the overall ratio of spines to shafts occurred following our stress protocol. While this finding does not address here whether there was a general hypertrophy or atrophy of the dendritic arbors of BLA principal neurons, it does demonstrate that there was not a significant increase in the spinyness of the BLA dendrites present, and thus that the increased localization of GluR1 in spines was not due to spine sprouting alone. Furthermore, we found no significant effect of stress on membrane input resistance (Rm) in BLA neurons, a marker that is sensitive to the overall degree of dendritic arborization, arguing against significant increase or decrease in overall dendritic arbor in the BLA after stress. One reason for the difference between our study and previous work on spine density and stress may be the stress protocol used. Our paradigm used half-hour sessions of mild footshock for four days, while the aforementioned studies that observed increased spine density after stress used from 10 to 20 days of a minimum of two hours continuous exposure to the stressors (Eiland and McEwen, 2012; Qin et al., 2011; Vyas et al., 2006). It has been shown that the duration of a stressor can affect the temporal development of these plastic changes (Mitra et al., 2005). The relatively brief duration of our stressor exposure appears to have limited any structural changes while still inducing a change in glutamate receptor localization and an increase in anxiety behavior (Hazra et al., 2012). It remains to be determined what the effect of longer or more intense footshock sessions might be on spine density and GluR1 localization.
The BLA plays a key role in the association of external cues with emotional salience, particularly negative salience (Davis et al., 1997; Davis and Whalen, 2001; Walker and Davis, 2008), and glutamatergic afferents and neurotransmission are critical to this process (Davis et al., 1994; McDonald, 1998). Thus, stress-induced alterations in glutamatergic receptors are likely to significantly alter the processing of sensory information in the BLA. Indeed, both restraint (Reznikov et al., 2007) and footshock have been shown to cause glutamate release in the BLA (Hegoburu et al., 2009). In addition, stress hormones themselves can produce a complex enhancement of glutamatergic transmission (Karst et al., 2010; Rainnie et al., 2004). Furthermore, many studies of plasticity and of stress have demonstrated that enhanced glutamatergic signaling causes an increased presence of membrane-bound AMPA receptors (Makino and Malinow, 2009; Yudowski et al., 2007). The data presented here indicates that this new, membrane-bound AMPA receptor is directed to dendritic spines that previously had little or no GluR1-containing AMPA receptor.
Alterations of neuronal activity in response to stress are a form of plasticity, and it has been theorized that these changes underlie the symptoms of chronic stress disorders, such as heightened anxiety (Sigurdsson et al., 2007). Plasticity is a major feature of glutamatergic synapses, and long term potentiation (LTP) has been demonstrated at BLA synapses (Li et al., 2011a; Rogan and LeDoux, 1995; Rogan et al., 1997; Yu et al., 2008). Additionally, previous studies have demonstrated glutamate receptors in the amygdala are involved in fear conditioning (Walker and Davis, 2002), and that LTP in the amygdala is associated with an increased cell surface presence of AMPA receptors (Yu et al., 2008). Our results demonstrate that significant stress results in an altered glutamatergic system in the BLA and that this involves AMPA glutamate receptors. Furthermore, the stress paradigm we are using has been shown to facilitate the induction of LTP in the amygdala (Li et al., 2011b). Consistent with our results, withdrawal from chronic ethanol inhalation produced anxiety behavior and an increased frequency of mEPSCs in rat BLA (Lack et al., 2007). The increase in AMPA function does not seem to be mediated by increased AMPA receptor subunit peptide levels, as measured by Western blot analysis (Christian et al., 2012; McCool et al., 2010). The insertion of AMPA receptor subunits into synapses during LTP has further been demonstrated to occur from pre-existing stores of protein (Makino and Malinow, 2009). These studies are consistent with results reported here which suggest a shift of AMPA receptor from the shafts of dendrites into spines where glutamatergic inputs are typically found. It is important to recognize that AMPA receptor proteins can undergo plastic changes, including insertion into synapses, in relatively shorter time frames than we investigated using fear memory paradigms (Nedelescu et al., 2010; Rumpel et al., 2005). We believe our experiments speak to a different phenomenon: that stress produces a long lasting shift in AMPA receptors that are then available in the spine, either in the synapse or close by, to participate in glutamatergic transmission. Interestingly, GABAergic neurotransmission is reduced following stress, which leads to hyperexcitability of BLA neurons (Duvarci and Pare, 2007; Rainnie et al., 2004; Rodriguez Manzanares et al., 2005). Thus, stress produces a variety of changes in the BLA which together promote enhanced excitability of BLA neurons and contribute to the expression of pathological stress behavior as seen by an enhanced startle response following the unpredictable footshock paradigm (Hazra et al., 2012).
An intriguing implication of our findings is that it is possible that our model of stress could be activating putative silent synapses in the BLA. Silent synapses are glutamatergic synapses that either contain no AMPA receptors or inactivated AMPA receptors. Silent synapses were originally defined as not producing an EPSC at resting membrane potential (Merrill and Wall, 1972). It was later discovered in the hippocampus that events that produce LTP also have the ability to unsilence these synapses. Evidence accrued that functional postsynaptic AMPA receptors were responsible for this change (Kerchner and Nicoll, 2008). Relevant to this point, we found that in control animals, GluR1 was found in both dendritic shafts and spines, but that there were clear examples of unlabeled spines in the midst of labeled elements. Thus, a possible explanation for our anatomical and physiological findings is that stress promotes the insertion of AMPA receptors containing the GluR1 subunit into silent synapses, which allows them to become active synapses. This unmasking of previously silent synapses could facilitate induction of LTP observed following stress (Li et al., 2011b) and perhaps might underlie the generalization of anxiety responses to previously neutral stimuli seen following stress. Alternately, it is also possible that the unlabeled spines contain functional AMPA receptors composed of GluR2 and GluR3 subunits. It will be important to determine how the localization of other AMPA receptor subunits is modulated be stress.
Figure 3.
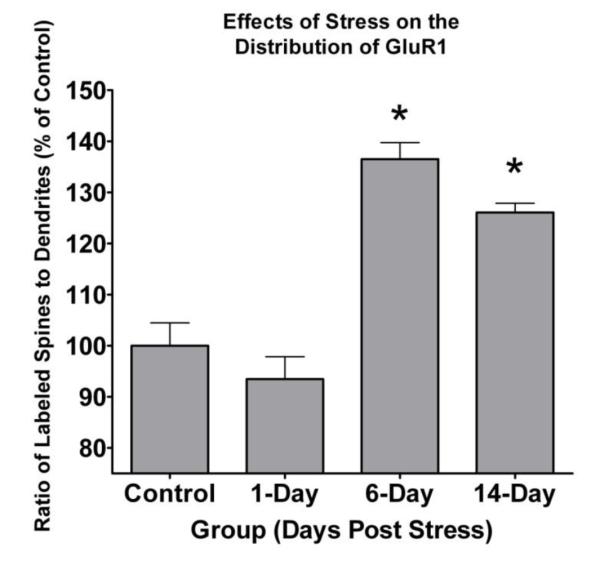
GluR1 immunoreactive spine to dendritic shaft ratio is increased following exposure to stress. The proportion of GluR1 labeled spines to GluR1 labeled dendritic shafts was calculated in control subjects and in 3 groups of subjects exposed to 4 daily 30 minute footshock sessions (1-, 6-, and 14-day post shock groups). There was a significant effect of time post stress on the spine to dendrite ratio (F(3,30)=15.582, p<0.0001). The labeled spine to dendrite ratio is increased in the 6- (p<0.0001) and 14-day (p=0.0055) post stress groups as compared to the control group, but not in the 1-day (p=0.81) post stress group. Asterisks indicate a significance level of p<0.05.
Acknowledgments
The authors would like to thank Marcelia Maddox for technical assistance on the project.
This work was supported by a Merit Award from the Office of Research and Development, Department of Veterans Affairs to ECM; by NIMH grant MH069852 to DGR; and by an NIH/NCRR base grant [Grant P51RR000165] to Yerkes National Primate Research Center.
References
- Belujon P, Grace AA. Hippocampus, amygdala, and stress: interacting systems that affect susceptibility to addiction. Annals of the New York Academy of Sciences. 2011;1216:114–121. doi: 10.1111/j.1749-6632.2010.05896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti M, Sepede G, Mingoia G, Catani C, Ferretti A, Merla A, Del Gratta C, Romani GL, Babiloni C. Elevated response of human amygdala to neutral stimuli in mild post traumatic stress disorder: neural correlates of generalized emotional response. Neuroscience. 2010;168(3):670–679. doi: 10.1016/j.neuroscience.2010.04.024. [DOI] [PubMed] [Google Scholar]
- Christian DT, Alexander NJ, Diaz MR, Robinson S, McCool BA. Chronic intermittent ethanol and withdrawal differentially modulate basolateral amygdala AMPA-type glutamate receptor function and trafficking. Neuropharmacology. 2012;62(7):2430–2439. doi: 10.1016/j.neuropharm.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17(5):208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Davis M, Walker DL, Lee Y. Roles of the amygdala and bed nucleus of the stria terminalis in fear and anxiety measured with the acoustic startle reflex. Possible relevance to PTSD. Annals of the New York Academy of Sciences. 1997;821:305–331. doi: 10.1111/j.1749-6632.1997.tb48289.x. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6(1):13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Pare D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci. 2007;27(16):4482–4491. doi: 10.1523/JNEUROSCI.0680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiland L, McEwen BS. Early life stress followed by subsequent adult chronic stress potentiates anxiety and blunts hippocampal structural remodeling. Hippocampus. 2012;22(1):82–91. doi: 10.1002/hipo.20862. [DOI] [PubMed] [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. The American journal of psychiatry. 2007;164(10):1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falloon IR, Kydd RR, Coverdale JH, Laidlaw TM. Early detection and intervention for initial episodes of schizophrenia. Schizophr Bull. 1996;22(2):271–282. doi: 10.1093/schbul/22.2.271. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Kim JJ. Acquisition of contextual Pavlovian fear conditioning is blocked by application of an NMDA receptor antagonist D,L-2-amino-5-phosphonovaleric acid to the basolateral amygdala. Behav Neurosci. 1994;108(1):210–212. doi: 10.1037//0735-7044.108.1.210. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, LeDoux JE. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23(2):229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- Farb CR, Aoki C, Ledoux JE. Differential localization of NMDA and AMPA receptor subunits in the lateral and basal nuclei of the amygdala: a light and electron microscopic study. The Journal of comparative neurology. 1995;362(1):86–108. doi: 10.1002/cne.903620106. [DOI] [PubMed] [Google Scholar]
- Flavell CR, Lee JL. Post-training unilateral amygdala lesions selectively impair contextual fear memories. Learn Mem. 2012;19(6):256–263. doi: 10.1101/lm.025403.111. [DOI] [PubMed] [Google Scholar]
- Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, Wiltgen B, Fanselow MS. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci. 2004;24(15):3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeders NE. The impact of stress on addiction. Eur Neuropsychopharmacol. 2003;13(6):435–441. doi: 10.1016/j.euroneuro.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Goel N, Bale TL. Examining the intersection of sex and stress in modelling neuropsychiatric disorders. J Neuroendocrinol. 2009;21(4):415–420. doi: 10.1111/j.1365-2826.2009.01843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Long-term potentiation as a substrate for memory: evidence from studies of amygdaloid plasticity and Pavlovian fear conditioning. Hippocampus. 2002;12(5):592–599. doi: 10.1002/hipo.10099. [DOI] [PubMed] [Google Scholar]
- Gutman AR, Yang Y, Ressler KJ, Davis M. The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J Neurosci. 2008;28(48):12682–12690. doi: 10.1523/JNEUROSCI.2305-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra R, Guo JD, Dabrowska J, Rainnie DG. Differential distribution of serotonin receptor subtypes in BNST(ALG) neurons: Modulation by unpredictable shock stress. Neuroscience. 2012 doi: 10.1016/j.neuroscience.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegoburu C, Sevelinges Y, Thevenet M, Gervais R, Parrot S, Mouly AM. Differential dynamics of amino acid release in the amygdala and olfactory cortex during odor fear acquisition as revealed with simultaneous high temporal resolution microdialysis. Learn Mem. 2009;16(11):687–697. doi: 10.1101/lm.1584209. [DOI] [PubMed] [Google Scholar]
- Hendler T, Rotshtein P, Yeshurun Y, Weizmann T, Kahn I, Ben-Bashat D, Malach R, Bleich A. Sensing the invisible: differential sensitivity of visual cortex and amygdala to traumatic context. Neuroimage. 2003;19(3):587–600. doi: 10.1016/s1053-8119(03)00141-1. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. The American journal of psychiatry. 2010;167(6):648–662. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst H, Berger S, Erdmann G, Schutz G, Joels M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107(32):14449–14454. doi: 10.1073/pnas.0914381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nature reviews. 2008;9(11):813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack AK, Diaz MR, Chappell A, DuBois DW, McCool BA. Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol. 2007;98(6):3185–3196. doi: 10.1152/jn.00189.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Dabrowska J, Hazra R, Rainnie DG. Synergistic activation of dopamine D1 and TrkB receptors mediate gain control of synaptic plasticity in the basolateral amygdala. PLoS One. 2011a;6(10):e26065. doi: 10.1371/journal.pone.0026065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Hazra R, Stair S, Rainnie D. Repeated shock stress facilitates basolateral amygdala synaptic plasticity through a decrease cAMP-specific phosphodiesterase type IV (PDE4) expression. Society for Neuroscience. 2011b doi: 10.1007/s00429-017-1575-z. Abstracts:191.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnman C, Zeffiro TA, Pitman RK, Milad MR. An fMRI study of unconditioned responses in post-traumatic stress disorder. Biol Mood Anxiety Disord. 2011;1(1):8. doi: 10.1186/2045-5380-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RB, Nemeroff CB. The role of corticotropin-releasing hormone in the pathophysiology of depression: therapeutic implications. Curr Top Med Chem. 2011;11(6):609–617. doi: 10.2174/1568026611109060609. [DOI] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64(3):381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, Christian DT, Diaz MR, Lack AK. Glutamate plasticity in the drunken amygdala: the making of an anxious synapse. Int Rev Neurobiol. 2010;91:205–233. doi: 10.1016/S0074-7742(10)91007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55(3):257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F, Mania I, Rainnie DG. Evidence for a perisomatic innervation of parvalbumin-containing interneurons by individual pyramidal cells in the basolateral amygdala. Brain research. 2005;1035(1):32–40. doi: 10.1016/j.brainres.2004.11.052. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12(2):205–210. doi: 10.1016/s0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- McGuire BA, Gilbert CD, Rivlin PK, Wiesel TN. Targets of horizontal connections in macaque primary visual cortex. The Journal of comparative neurology. 1991;305(3):370–392. doi: 10.1002/cne.903050303. [DOI] [PubMed] [Google Scholar]
- Merrill EG, Wall PD. Factors forming the edge of a receptive field: the presence of relatively ineffective afferent terminals. J Physiol. 1972;226(3):825–846. doi: 10.1113/jphysiol.1972.sp010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2005;102(26):9371–9376. doi: 10.1073/pnas.0504011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muly EC, Maddox M, Khan ZU. Distribution of D1 and D5 dopamine receptors in the primate nucleus accumbens. Neuroscience. 2010;169(4):1557–1566. doi: 10.1016/j.neuroscience.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Muly EC, Maddox M, Smith Y. Distribution of mGluR1alpha and mGluR5 immunolabeling in primate prefrontal cortex. The Journal of comparative neurology. 2003;467(4):521–535. doi: 10.1002/cne.10937. [DOI] [PubMed] [Google Scholar]
- Nader K, Majidishad P, Amorapanth P, LeDoux JE. Damage to the lateral and central, but not other, amygdaloid nuclei prevents the acquisition of auditory fear conditioning. Learn Mem. 2001;8(3):156–163. doi: 10.1101/lm.38101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelescu H, Kelso CM, Lazaro-Munoz G, Purpura M, Cain CK, Ledoux JE, Aoki C. Endogenous GluR1-containing AMPA receptors translocate to asymmetric synapses in the lateral amygdala during the early phase of fear memory formation: an electron microscopic immunocytochemical study. The Journal of comparative neurology. 2010;518(23):4723–4739. doi: 10.1002/cne.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi BK, Xavier GF. Contextual, but not auditory, fear conditioning is disrupted by neurotoxic selective lesion of the basal nucleus of amygdala in rats. Neurobiol Learn Mem. 2010;93(2):165–174. doi: 10.1016/j.nlm.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Palazidou E. The neurobiology of depression. Br Med Bull. 2012;101:127–145. doi: 10.1093/bmb/lds004. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay S, Webster H. The Fine Structure of the Nervous System. Oxford Press; New York: 1991. [Google Scholar]
- Qin M, Xia Z, Huang T, Smith CB. Effects of chronic immobilization stress on anxiety-like behavior and basolateral amygdala morphology in Fmr1 knockout mice. Neuroscience. 2011;194:282–290. doi: 10.1016/j.neuroscience.2011.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinak CA, Angstadt M, Welsh RC, Kenndy AE, Lyubkin M, Martis B, Phan KL. Altered amygdala resting-state functional connectivity in post-traumatic stress disorder. Front Psychiatry. 2011;2:62. doi: 10.3389/fpsyt.2011.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG. Serotonergic modulation of neurotransmission in the rat basolateral amygdala. J Neurophysiol. 1999;82(1):69–85. doi: 10.1152/jn.1999.82.1.69. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69(4):1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24(14):3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Ressler KJ. Physiology of the Amygdala: Implications for PTSD. In: Shiromani PJ, Keane TM, LeDoux JE, editors. Post-Traumatic Stress Disorder: Basic Science and Clinical Practice. Humana Press; New York, New York, USA: 2009. pp. 39–78. [Google Scholar]
- Rauch SL, Shin LM. Functional neuroimaging studies in posttraumatic stress disorder. Annals of the New York Academy of Sciences. 1997;821:83–98. doi: 10.1111/j.1749-6632.1997.tb48271.x. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research--past, present, and future. Biol Psychiatry. 2006;60(4):376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Reznikov LR, Grillo CA, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J. Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci. 2007;25(10):3109–3114. doi: 10.1111/j.1460-9568.2007.05560.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez Manzanares PA, Isoardi NA, Carrer HF, Molina VA. Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J Neurosci. 2005;25(38):8725–8734. doi: 10.1523/JNEUROSCI.2260-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15(1):127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390(6660):604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308(5718):83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83(3):803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Truitt W, Rainnie D, Sajdyk T. Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress. 2005;8(4):209–219. doi: 10.1080/10253890500504557. [DOI] [PubMed] [Google Scholar]
- Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35(1):169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ. Acute stress rapidly and persistently enhances memory formation in the male rat. Neurobiol Learn Mem. 2001;75(1):10–29. doi: 10.1006/nlme.1999.3956. [DOI] [PubMed] [Google Scholar]
- Sigurdsson T, Doyere V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52(1):215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Vermetten E, Schmahl C, Southwick SM, Bremner JD. Positron tomographic emission study of olfactory induced emotional recall in veterans with and without combat-related posttraumatic stress disorder. Psychopharmacol Bull. 2007;40(1):8–30. [PMC free article] [PubMed] [Google Scholar]
- Vouimba RM, Yaniv D, Diamond D, Richter-Levin G. Effects of inescapable stress on LTP in the amygdala versus the dentate gyrus of freely behaving rats. Eur J Neurosci. 2004;19(7):1887–1894. doi: 10.1111/j.1460-9568.2004.03294.x. [DOI] [PubMed] [Google Scholar]
- Vyas A, Jadhav S, Chattarji S. Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience. 2006;143(2):387–393. doi: 10.1016/j.neuroscience.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22(15):6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Davis M. The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacology, biochemistry, and behavior. 2002;71(3):379–392. doi: 10.1016/s0091-3057(01)00698-0. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. Role of the extended amygdala in short-duration versus sustained fear: a tribute to Dr. Lennart Heimer. Brain Struct Funct. 2008;213(1-2):29–42. doi: 10.1007/s00429-008-0183-3. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Yokotani N, Doi K, Wada K. Immunochemical characterization of the non-NMDA glutamate receptor using subunit-specific antibodies. Evidence for a hetero oligomeric structure in rat brain. J Biol Chem. 1992;267(1):501–507. [PubMed] [Google Scholar]
- Yu SY, Wu DC, Liu L, Ge Y, Wang YT. Role of AMPA receptor trafficking in NMDA receptor-dependent synaptic plasticity in the rat lateral amygdala. J Neurochem. 2008;106(2):889–899. doi: 10.1111/j.1471-4159.2008.05461.x. [DOI] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci. 2007;27(41):11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]