

Abstract
The title compound, C7H8N6O, represents the keto form and adopts a nearly planar structure (r.m.s. deviation of the non-H atoms = 0.072 Å). In the crystal, molecules form spiral chains along the c axis by N—H⋯N hydrogen bonds. The chains are linked to each other by weak C—H⋯O hydrogen bonds, forming a three-dimensional framework.
Related literature
For review on nucleophilic displacement at the 1,2,4,5-tetrazine ring, see: Clavier & Audebert (2010 ▶); Tolshchina et al. (2013 ▶). For the synthesis of 3-hydroxy-1,2,4,5-tetrazines, see: Ishmetova et al. (2009 ▶); Sheremetev et al. (2012a
▶,b
▶). For the structure of 3-hydroxy-1,2,4,5-tetrazine, see: Yeh et al. (1994 ▶). For a review on oxo-hydroxy tautomerism of various azines, see: Stanovnik et al. (2006 ▶). For standard bond lengths, see: Allen et al. (1987 ▶).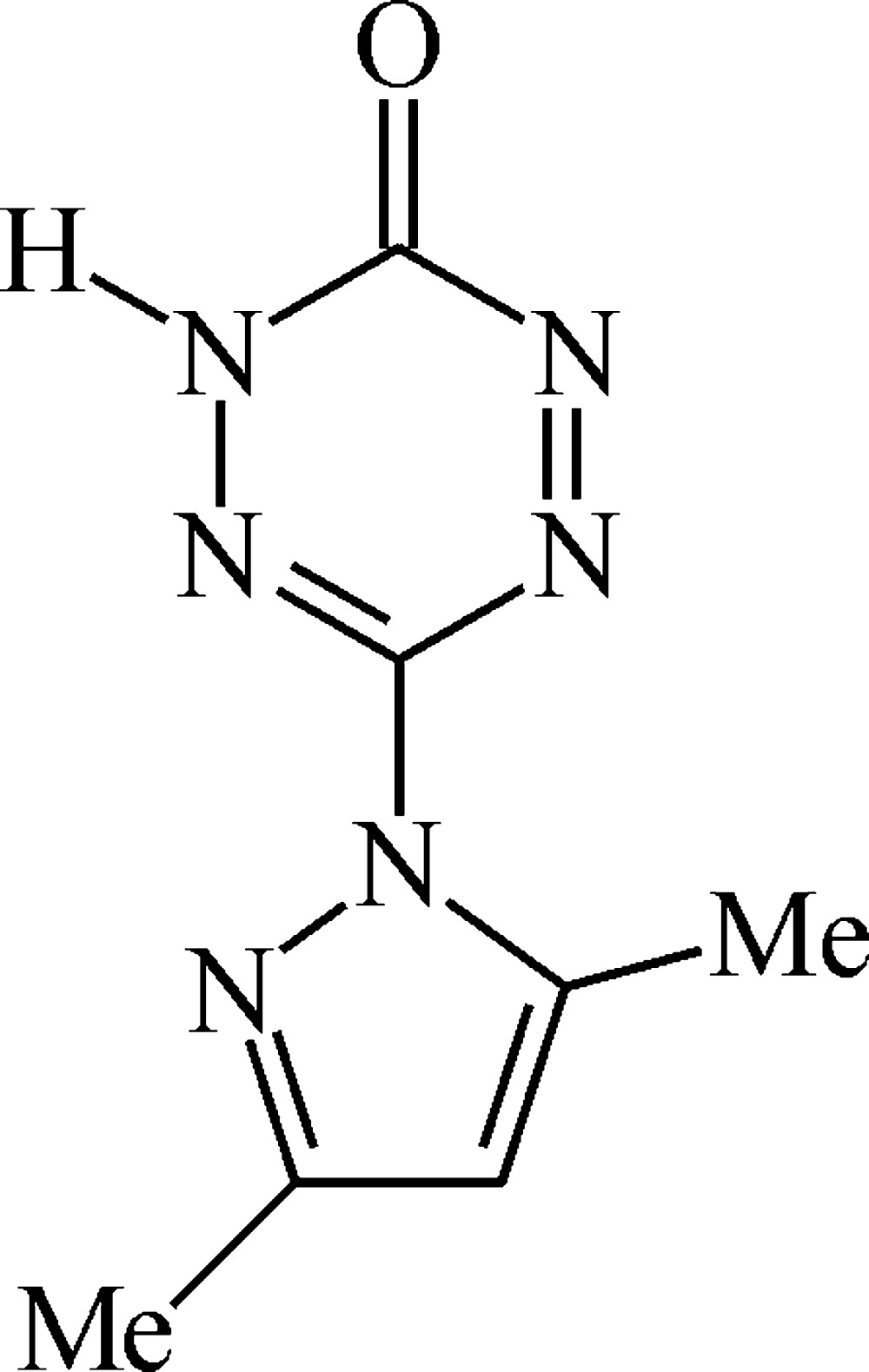
Experimental
Crystal data
C7H8N6O
M r = 192.19
Orthorhombic,

a = 12.1431 (11) Å
b = 12.6551 (12) Å
c = 5.3907 (5) Å
V = 828.40 (13) Å3
Z = 4
Mo Kα radiation
μ = 0.11 mm−1
T = 120 K
0.28 × 0.22 × 0.20 mm
Data collection
Bruker APEXII CCD diffractometer
14157 measured reflections
1353 independent reflections
1228 reflections with I > 2σ(I)
R int = 0.050
Refinement
R[F 2 > 2σ(F 2)] = 0.037
wR(F 2) = 0.094
S = 1.05
1353 reflections
133 parameters
1 restraint
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.40 e Å−3
Δρmin = −0.28 e Å−3
Data collection: APEX (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL, PLATON (Spek, 2009 ▶) and publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S1600536813027360/kq2010sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536813027360/kq2010Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536813027360/kq2010Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N4—H4⋯N1i | 0.89 (3) | 2.00 (3) | 2.863 (2) | 161 (3) |
| C2—H2A⋯O1ii | 0.95 | 2.40 | 3.193 (3) | 141 |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Acknowledgments
This work was supported financially by the Ministry of Education and Science of Russia through the Federal Target Program "Research and Educational Personnel of Innovative Russia at 2009–2013 Years" (grant No. 14.B37.21.0827), and, in part, by a Russian Foundation for Basic Research (grant No. 12–03-31346).
supplementary crystallographic information
1. Comment
Over the last decade, broad studies of nucleophilic displacemet at 1,2,4,5-tetrazine ring have demonstrated that the highly electron-withdrawing aromatic ring exhibit a rich diversity of chemical behavior, and today 3,6-bis(3,5-dimethylpyrazol-1-yl)-1,2,4,5-tetrazine 1 has become common precursor to various tetrazine derivatives (Clavier & Audebert, 2010; Tolshchina et al., 2013). The displacement of the dimethylpyrazolyl moiety in compound 1 by a wider range of N-nucleophiles has been well documented (Sheremetev et al., 2012a, 2012b), while reactions of the tetrazine 1 with O-nucleophiles have been studied to a lesser extent (Yeh et al., 1994; Ishmetova et al., 2009). Here, we wish to report the unexpected discovery of an unusual dimethylpyrazolyl moiety displacement of compound 1 when it is treated with N-nucleophile, such as 3-amino-1-tret-butyl-1,2,4-triazole 2.
Upon a refluxing of compound 1 with amine 2 and K2CO3 in acetonitrile and following treatment with water, an step required to workup of the reaction mixture, it was found that instead of introduction of N-nucleophile (with a formation of compound 3) the reaction gave the hydroxy derivative 4' (Figure 1).
According to X-ray data, the title compound adopts nearly planar structure (r.m.s. deviation of the non-hydrogen atoms is 0.072 Å) (Figure 2). In general, it can exist in two tautomeric forms (Stanovnik et al., 2006) as shown in Figure 1. The hydrogen atom H4 was localized and refined at the N4 nitrogen atom that means that the title compound exists in keto-form, and the equilibrium favors the tautomer 4'. This is also supproted by the bond length distribution in the tetrazin-3-one: the C7—O1 (1.215 (3) Å) bond corresponds to normal carbonyl bond (standard X-ray C═O value is in the range of 1.192–1.235 Å (Allen et al. 1987), while the N3—C6 (1.300 (2) Å) and N5—N6 (1.286 (2) Å) bonds are significantly shorter than the C7—N4 (1.368 (3) Å), N3—N4 (1.384 (2) Å), C6—N5 (1.377 (2) Å) and N6—C7 (1.427 (2) Å) bonds.
The crystal structure of 4' is stabilized by intermolecular N—H···N and C—H···O hydrogen bonds (Table 1). By means of the N4—H4···N1i hydrogen bond, molecules are connected into spiral chains along the c axis (Figure 3). Those chains are linked into 3D-framework (Figure 3) by the C2—H2A···O1ii contacts. Symmetry codes: (i) –x+2, –y+1, z+1/2; (ii) –x+3/2, y–1/2, z–3/2.
2. Experimental
The X-ray quality crystals of the title compound were grown by slow evaporation of acetonitrile solution.
All the reagents were of analytical grade, purchased from commercial sources, and used as received. Infrared spectra were determined in KBr pellets on a Perkin-Elmer Model 577 spectrometer. Mass-spectra were recorded on a Varian MAT-311 A instrument. The 1H, 13C and 15N NMR spectra (external standard: CH3NO2) were recorded at 300.13, 75.47 and 50.7 MHz, respectively. The chemical shift values (δ, p.p.m.) are expressed relative to the chemical shift of the solvent-d or to external standard without correction nitromethane (14N and 15N). Melting points were determined on Gallenkamp melting point apparatus and are uncorrected.
6-(3,5-Dimethyl-1H-pyrazol-1-yl)-1,2,4,5-tetrazin-3(2H)-one (4'). A mixture of 3,6-bis(3,5-dimethylpyrazol-1-yl)-1,2,4,5-tetrazine 1 (0.27 g, 1 mmol), 3-amino-1-tretbutyl-1,2,4-triazole 2 (0.14 g, 1 mmol), and K2CO3 (0.14 g, 1 mmol) was refluxed in dry acetonitrile (10 mL) for 3 d. The reaction mixture was then cooled and a violet solid formed was collected by filtration. The solid was dissolved in water (10 mL), acidifed with 3% HCl to pH 1 and cooled to 278 K. A solid was collected by filtration, washed with 1% HCl and hexane and crystallized from MeCN/H2O, yield 0.15 g (82%), mp 483–484 K (dec.); IR (KBr, cm-1): ν = 3135–2823, 1744, 1706, 1693, 1553, 1506, 1435, 1405, 1373, 1263, 1161, 1138, 1105, 1063, 1025, 993, 975, 836, 808. 1H MNR (DMSO-d3, 293 K): δ = 2.20 and 2.38 (3H+3H, C1—CH3 and C3—CH3), 6.17 (c, 1H, CH), 8.51 (br, 1H, OH); 13C MNR (DMSO-d3, 293 K): δ = 13.3, 13.2, 108.9, 141.9, 147.7, 150.7, 152.0. 15N MNR (DMSO-d3, 293 K): δ = –1.87, –35.0, –77.6, –173.3. Anal. Calcd. for C7H8N6O (192.17): C, 43.75; H, 4.20; N, 43.73. Found: C, 43.81; H, 4.24; N, 43.61.
3. Refinement
The hydrogen atom of the NH group was found in difference Fourier synthesis and refined in isotropic approximation. The H(C) atomic positions were calculated and refined in isotropic approximation in riding model with the Uiso(H) parameters equal to 1.5 Ueq(Ci), 1.2 Ueq(Cj), where Ueq(Ci) and Ueq(Cj) are the equivalent thermal parameters of the methyl carbon atoms and all the other carbon atoms, respectively, to which corresponding H atoms are bonded.
Figures
Fig. 1.

Synthesis of the title compound.
Fig. 2.

General view of the structure of the title compound with the atomic numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. H atoms are presented as small spheres of arbitrary radius.
Fig. 3.

Crystal packing fragment of the title compound. Hydrogen atoms of the methyl groups are omitted for clarity. Dashed lines indicate the intermolecular N—H···N and C—H···O hydrogen bonds.
Crystal data
| C7H8N6O | Dx = 1.541 Mg m−3 |
| Mr = 192.19 | Melting point = 474–473 K |
| Orthorhombic, Pna21 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2c -2n | Cell parameters from 4489 reflections |
| a = 12.1431 (11) Å | θ = 2.3–30.2° |
| b = 12.6551 (12) Å | µ = 0.11 mm−1 |
| c = 5.3907 (5) Å | T = 120 K |
| V = 828.40 (13) Å3 | Prizm, red |
| Z = 4 | 0.28 × 0.22 × 0.20 mm |
| F(000) = 400 |
Data collection
| Bruker APEXII CCD diffractometer | 1228 reflections with I > 2σ(I) |
| Radiation source: sealed tube | Rint = 0.050 |
| Graphite monochromator | θmax = 30.2°, θmin = 2.3° |
| φ and ω scans | h = −17→17 |
| 14157 measured reflections | k = −17→17 |
| 1353 independent reflections | l = −7→7 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.037 | Hydrogen site location: mixed |
| wR(F2) = 0.094 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0529P)2 + 0.2463P] where P = (Fo2 + 2Fc2)/3 |
| 1353 reflections | (Δ/σ)max < 0.001 |
| 133 parameters | Δρmax = 0.40 e Å−3 |
| 1 restraint | Δρmin = −0.28 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.86033 (12) | 0.65658 (12) | 1.3631 (3) | 0.0238 (3) | |
| N1 | 0.85424 (13) | 0.38253 (13) | 0.4472 (3) | 0.0155 (3) | |
| N2 | 0.76721 (12) | 0.41809 (12) | 0.5895 (3) | 0.0144 (3) | |
| N3 | 0.89653 (13) | 0.50888 (12) | 0.8195 (3) | 0.0158 (3) | |
| N4 | 0.91664 (13) | 0.57202 (12) | 1.0123 (3) | 0.0155 (3) | |
| H4 | 0.989 (2) | 0.583 (2) | 1.029 (7) | 0.034 (7)* | |
| N5 | 0.70626 (13) | 0.52560 (13) | 0.9220 (3) | 0.0188 (4) | |
| N6 | 0.72745 (13) | 0.58417 (14) | 1.1110 (4) | 0.0202 (4) | |
| C1 | 0.80934 (15) | 0.31818 (14) | 0.2799 (4) | 0.0148 (3) | |
| C2 | 0.69351 (15) | 0.31263 (15) | 0.3124 (4) | 0.0173 (4) | |
| H2A | 0.6432 | 0.2729 | 0.2149 | 0.021* | |
| C3 | 0.66818 (15) | 0.37567 (14) | 0.5118 (4) | 0.0156 (4) | |
| C4 | 0.87981 (15) | 0.26232 (15) | 0.0946 (4) | 0.0186 (4) | |
| H4A | 0.9493 | 0.3008 | 0.0741 | 0.028* | |
| H4B | 0.8413 | 0.2591 | −0.0650 | 0.028* | |
| H4C | 0.8951 | 0.1905 | 0.1529 | 0.028* | |
| C5 | 0.55864 (15) | 0.39560 (17) | 0.6288 (4) | 0.0227 (4) | |
| H5A | 0.5624 | 0.3785 | 0.8059 | 0.034* | |
| H5B | 0.5028 | 0.3512 | 0.5489 | 0.034* | |
| H5C | 0.5389 | 0.4702 | 0.6085 | 0.034* | |
| C6 | 0.79310 (14) | 0.48639 (14) | 0.7861 (4) | 0.0139 (3) | |
| C7 | 0.83879 (16) | 0.60685 (15) | 1.1765 (4) | 0.0168 (4) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0219 (7) | 0.0287 (7) | 0.0207 (7) | 0.0014 (6) | −0.0019 (6) | −0.0093 (6) |
| N1 | 0.0105 (6) | 0.0199 (7) | 0.0162 (7) | −0.0001 (6) | 0.0027 (6) | −0.0025 (6) |
| N2 | 0.0093 (6) | 0.0197 (7) | 0.0144 (7) | −0.0004 (5) | 0.0012 (6) | −0.0029 (6) |
| N3 | 0.0123 (7) | 0.0206 (7) | 0.0145 (8) | 0.0001 (6) | −0.0012 (6) | −0.0022 (6) |
| N4 | 0.0100 (6) | 0.0205 (7) | 0.0161 (7) | 0.0001 (6) | −0.0020 (6) | −0.0016 (6) |
| N5 | 0.0125 (7) | 0.0223 (8) | 0.0216 (8) | 0.0002 (6) | 0.0015 (7) | −0.0056 (7) |
| N6 | 0.0145 (7) | 0.0240 (8) | 0.0221 (9) | −0.0001 (6) | 0.0015 (7) | −0.0072 (7) |
| C1 | 0.0141 (8) | 0.0154 (7) | 0.0147 (8) | −0.0008 (6) | 0.0000 (7) | 0.0013 (7) |
| C2 | 0.0133 (8) | 0.0194 (8) | 0.0192 (9) | −0.0028 (6) | −0.0020 (7) | −0.0022 (8) |
| C3 | 0.0118 (8) | 0.0187 (8) | 0.0164 (8) | −0.0010 (6) | −0.0013 (7) | −0.0003 (7) |
| C4 | 0.0160 (8) | 0.0227 (9) | 0.0171 (8) | 0.0004 (7) | 0.0022 (8) | −0.0029 (7) |
| C5 | 0.0092 (8) | 0.0326 (10) | 0.0263 (11) | −0.0013 (7) | −0.0008 (8) | −0.0071 (9) |
| C6 | 0.0131 (8) | 0.0162 (8) | 0.0125 (8) | 0.0001 (6) | 0.0008 (7) | −0.0003 (7) |
| C7 | 0.0149 (8) | 0.0180 (8) | 0.0176 (9) | 0.0011 (6) | −0.0013 (7) | −0.0004 (7) |
Geometric parameters (Å, º)
| O1—C7 | 1.215 (3) | C1—C2 | 1.419 (2) |
| N1—C1 | 1.332 (3) | C1—C4 | 1.494 (3) |
| N1—N2 | 1.381 (2) | C2—C3 | 1.373 (3) |
| N2—C3 | 1.382 (2) | C2—H2A | 0.9500 |
| N2—C6 | 1.403 (2) | C3—C5 | 1.494 (3) |
| N3—C6 | 1.300 (2) | C4—H4A | 0.9800 |
| N3—N4 | 1.334 (2) | C4—H4B | 0.9800 |
| N4—C7 | 1.368 (3) | C4—H4C | 0.9800 |
| N4—H4 | 0.89 (3) | C5—H5A | 0.9800 |
| N5—N6 | 1.286 (2) | C5—H5B | 0.9800 |
| N5—C6 | 1.377 (2) | C5—H5C | 0.9800 |
| N6—C7 | 1.427 (2) | ||
| C1—N1—N2 | 105.18 (15) | C1—C4—H4A | 109.5 |
| N1—N2—C3 | 111.78 (16) | C1—C4—H4B | 109.5 |
| N1—N2—C6 | 116.66 (14) | H4A—C4—H4B | 109.5 |
| C3—N2—C6 | 131.54 (16) | C1—C4—H4C | 109.5 |
| C6—N3—N4 | 114.58 (15) | H4A—C4—H4C | 109.5 |
| N3—N4—C7 | 124.80 (16) | H4B—C4—H4C | 109.5 |
| N3—N4—H4 | 111 (2) | C3—C5—H5A | 109.5 |
| C7—N4—H4 | 124 (2) | C3—C5—H5B | 109.5 |
| N6—N5—C6 | 118.45 (15) | H5A—C5—H5B | 109.5 |
| N5—N6—C7 | 120.11 (17) | C3—C5—H5C | 109.5 |
| N1—C1—C2 | 110.64 (17) | H5A—C5—H5C | 109.5 |
| N1—C1—C4 | 120.53 (17) | H5B—C5—H5C | 109.5 |
| C2—C1—C4 | 128.82 (17) | N3—C6—N5 | 125.96 (16) |
| C3—C2—C1 | 106.84 (17) | N3—C6—N2 | 117.11 (15) |
| C3—C2—H2A | 126.6 | N5—C6—N2 | 116.91 (15) |
| C1—C2—H2A | 126.6 | O1—C7—N4 | 123.63 (18) |
| C2—C3—N2 | 105.54 (16) | O1—C7—N6 | 120.87 (18) |
| C2—C3—C5 | 128.92 (17) | N4—C7—N6 | 115.47 (18) |
| N2—C3—C5 | 125.54 (18) | ||
| C1—N1—N2—C3 | 0.3 (2) | C6—N2—C3—C5 | 0.5 (3) |
| C1—N1—N2—C6 | 178.57 (15) | N4—N3—C6—N5 | 3.3 (3) |
| C6—N3—N4—C7 | 4.1 (3) | N4—N3—C6—N2 | −178.47 (16) |
| C6—N5—N6—C7 | −0.3 (3) | N6—N5—C6—N3 | −5.2 (3) |
| N2—N1—C1—C2 | 0.4 (2) | N6—N5—C6—N2 | 176.59 (18) |
| N2—N1—C1—C4 | −178.72 (16) | N1—N2—C6—N3 | −1.0 (2) |
| N1—C1—C2—C3 | −1.0 (2) | C3—N2—C6—N3 | 176.88 (19) |
| C4—C1—C2—C3 | 178.11 (19) | N1—N2—C6—N5 | 177.36 (16) |
| C1—C2—C3—N2 | 1.0 (2) | C3—N2—C6—N5 | −4.7 (3) |
| C1—C2—C3—C5 | −178.20 (19) | N3—N4—C7—O1 | 173.22 (18) |
| N1—N2—C3—C2 | −0.8 (2) | N3—N4—C7—N6 | −8.9 (3) |
| C6—N2—C3—C2 | −178.82 (18) | N5—N6—C7—O1 | −175.41 (19) |
| N1—N2—C3—C5 | 178.44 (18) | N5—N6—C7—N4 | 6.6 (3) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N4—H4···N1i | 0.89 (3) | 2.00 (3) | 2.863 (2) | 161 (3) |
| C2—H2A···O1ii | 0.95 | 2.40 | 3.193 (3) | 141 |
Symmetry codes: (i) −x+2, −y+1, z+1/2; (ii) −x+3/2, y−1/2, z−3/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: KQ2010).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bruker (2009). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Clavier, G. & Audebert, P. (2010). Chem. Rev. 110, 3299–3314. [DOI] [PubMed]
- Ishmetova, R. I., Latosh, N. I., Ganebnykh, I. N., Ignatenko, N. K., Tolshchina, S. G. & Rusinov, G. L. (2009). Russ. J. Org. Chem. 45, 1102–1107 [translated from (2009). Zh. Org. Khim. 45, 1113–1118].
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheremetev, A. B., Palysaeva, N. V., Struchkova, M. I. & Suponitskii, K. Yu. (2012a). Russ. Chem. Bull. 61, 121–130 [translated from (2012). Izv. Akad. Nauk, Ser. Khim. 119–128].
- Sheremetev, A. B., Palysaeva, N. V., Struchkova, M. I. & Suponitsky, K. Yu. (2012b). Mendeleev Commun. 22, 302–304.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Stanovnik, B., Tisler, M., Katritzky, A. R. & Denisko, O. V. (2006). Adv. Heterocycl. Chem. 91, 1–134.
- Tolshchina, S. G., Rusinov, G. L. & Charushin, V. N. (2013). Chem. Heterocycl. Compd, 49, 66–91 [translated from (2013). Khim. Geterotsikl. Soedin. pp. 75–101].
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Yeh, M.-Y., Huang, C.-Y., Ueng, C.-H. & Wang, Y. (1994). Acta Cryst. C50, 1781–1784.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S1600536813027360/kq2010sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536813027360/kq2010Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536813027360/kq2010Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report