

Abstract
Nontransformed breast epithelial cells that are adjacent to tumor cells are constantly exposed to tumor necrosis factor-α (TNFα) and interleukin-1β (IL-1β), two inflammatory cytokines identified as having pro-tumoral causative roles. We show that continuous stimulation of nontransformed breast epithelial cells by TNFα + IL-1β for 2 to 3 weeks induced their spreading and epithelial-to-mesenchymal transition (EMT). The mechanistic bases for this slow induction of EMT by TNFα + IL-1β are: 1) it took 2 to 3 weeks for the cytokines to induce the expression of the EMT activators Zeb1 and Snail; 2) although Twist has amplified the EMT-inducing activities of Zeb1 + Snail, its expression was reduced by TNFα + IL-1β; however, the lack of Twist was compensated by prolonged stimulation with TNFα + IL-1β that has potentiated the EMT-inducing activities of Zeb1 + Snail. Stimulation by TNFα + IL-1β has induced the following dissemination-related properties in the nontransformed cells: 1) up-regulation of functional matrix metalloproteinases; 2) induction of migratory and invasive capabilities; 3) disruption of the normal phenotype of organized three-dimensional acini structures typically formed only by nontransformed breast cells and spreading of nontransformed cells out of such acini. Our findings suggest that TNFα + IL-1β induce dissemination of nontransformed breast epithelial cells and their reseeding at the primary tumor site; if, then, such detached cells are exposed to transforming events, they may form secondary malignant focus and lead to disease recurrence. Thus, our study reveals novel pathways through which the inflammatory microenvironment may contribute to relapsed disease in breast cancer.
Introduction
Breast cancer evolves out of multifactorial and dynamic processes, which are greatly influenced by the tumor microenvironment. Dominated by inflammatory traits, the tumor milieu in breast cancer is enriched with inflammatory cytokines that very often fail to induce immune protective mechanisms but rather skew the balance toward tumor-promoting events. Thus, multifaceted activities exerted by inflammatory cytokines on stroma cells, leukocytes, and the tumor cells themselves lead to increased angiogenesis, tumor growth and progression, and eventually aggravate disease course [1–4].
In this context, major tumor-promoting roles were recently attributed to the cytokines tumor necrosis factor-α (TNFα) and interleukin-1β (IL-1β). These two cytokines are minimally expressed by normal breast epithelial cells; however, both TNFα and IL-1β are expressed by breast tumor cells in the majority of breast cancer patients, with pronounced expression of both cytokines in >80% of patients who have experienced relapse [5–16]. In murine breast model systems, TNFα induces many cancer-promoting functions, and inhibiting TNFα expression leads to reduced breast malignancy [17–26]. Thus, in contrast to previous studies suggesting that local administration of TNFα directly into tumors may kill cancer cells, many researchers now consider TNFα a factor whose chronic expression at the tumor microenvironment leads to a more aggressive tumor phenotype, including in breast cancer [26–29]. Similarly, IL-1β, shown to upregulate a variety of processes that contribute to higher angiogenesis, tumor growth, and progression in breast cancer, is considered a strong and causative pro-malignancy factor whose expression is associated with advanced disease [5–9,16,30–37].
Within the large diversity of the tumor-promoting functions exerted by TNFα and IL-1β, their ability to induce cell remodeling in breast cancer cells was recently discovered. Several publications, including from our group, indicate that TNFα and IL-1β increase tumor cell spreading, epithelial-to-mesenchymal transition (EMT), and invasiveness of breast tumor cells [16,38–43]. EMT is a process in which the tumor cells lose epithelial markers that are required for cell-to-cell adhesion, such as E-cadherin, and acquire mesenchymal properties that promote tumor cell motility and invasiveness, like vimentin [44–48]. In breast cancer, EMT was strongly linked to tumor aggressiveness and metastasis [49–52]; thus by inducing EMT, TNFα and IL-1β manifest yet another level by which the inflammatory microenvironment can promote disease course.
TNFα and IL-1β, both expressed simultaneously in the majority of breast cancer patients with relapsed disease [16], may affect not only cancer cells and cells of the tumor microenvironment but also breast epithelial cells that are present in proximity to the tumor cells and are yet nontransformed. Recent findings have provided sporadic evidence to the ability of TNFα to affect cell morphology and possibly EMT in nontransformed breast epithelial cells [53–55]; however, these studies were performed mainly when TNFα was combined with the strong EMT inducer transforming growth factor-β, they did not address the important and most clinically relevant aspect of combined TNFα + IL-1β activities, and they did not provide profound systematic analysis of the mechanisms involved in cell remodeling induced by the cytokines in breast epithelial nontransformed cells.
In the present study, we have addressed these issues and provided in-depth understanding of the effects of TNFα, IL-1β, and both cytokines together on cell plasticity, EMT, and dissemination of nontransformed breast epithelial cells. Briefly, in response to TNFα + IL-1β stimulation, nontransformed breast epithelial cells acquired high spreading capabilities and underwent EMT. Mechanistic analysis indicated that induction of EMT required prolonged stimulation of the cells by TNFα + IL-1β and this was because of complex regulatory processes of the EMT activators Zeb1, Snail, and Twist. When analyzing the functional implications of cell remodeling and EMT induced by TNFα + IL-1β stimulation, we found that the two cytokines together have induced high release of matrix metalloproteinases (MMPs) by the cells, migration, invasion, distortion of three-dimensional (3D) acini structures that are typically formed only by nontransformed breast epithelial cells [56,57], and spreading of the nontransformed cells out of such ordered acini structures.
Overall, the findings of this study suggest that nontransformed breast epithelial cells located in TNFα + IL-1β-enriched tumors respond to the two cytokines by increased EMT, migration, and invasion. As a result of these processes, nontransformed breast epithelial cells may detach and migrate out of normally organized breast structures that may have still remained in proximity to the tumor, disseminate, and reseed at the primary tumor site. Such cells may be then exposed to transforming events prevalent at the inflammatory microenvironment of breast tumors, such as nitric oxide (NO) that induces mutagenesis [58–60]. Such a process may consecutively lead to formation of a new tumor focus adjacent to the primary focus, where the tumor was initiated. These events may stand on the basis of cases of breast cancer recurrence, which have been highly correlated with elevated expression of TNFα + IL-1β [16] and thus have major clinical implications.
Materials and Methods
Cells
The nontransformed human breast epithelial MCF-10A cells [61,62] (kindly provided by Prof. Berger, Chaim Sheba Medical Center, Tel-Hashomer, Israel) were maintained in Dulbecco's modified Eagle's medium-F12 medium, supplemented with 5% horse serum, 10 µg/ml insulin, 2 ng/ml epidermal growth factor, 100 U/ml penicillin, 100 µg/ml streptomycin (all purchased from Biological Industries, Beit Haemek, Israel), 100 ng/ml cholera toxin (Sigma-Aldrich, St Louis, MO), and 0.5 µg/ml hydrocortisone (Sigma). The nontransformed human mammary epithelial HB2 cells (a clonal derivative of the nontumorigenic mammary epithelial cell line, MTSV1-7 [63,64]; kindly provided by Prof. Tsarfaty, Tel Aviv University, Tel Aviv, Israel) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 10 µg/ml insulin, and 0.5 µg/ml hydrocortisone (Biological Industries). In some of the experiments (see details below), the cells were transferred to serum-free medium before the actual tests were performed: MCF-10A cells were transferred to LPM medium (Biological Industries), and HB2 cells were transferred to their corresponding serum-free medium.
Stimulation by Cytokines
In all parts of this study, the cells were stimulated by recombinant human (rh) TNFα at 50 ng/ml (Cat. No. 300-01A; PeproTech, Rocky Hill, NJ), rhIL-1β at 500 pg/ml (Cat. No. 200-01B; PeproTech), or both cytokines together (termed TNFα + IL-1β; concentrations as above). These cytokine concentrations were selected on the basis of dose-response analyses that were performed in our laboratory, using induction of inflammatory chemokines by breast tumor cells as readouts of TNFα- and IL-1β-induced effects (data not shown). On the basis of these analyses, we chose TNFα and IL-1β doses that gave prominent impact on chemokine release but were not at the highest end of the concentration range. So far, published reports on TNFα and IL-1β levels in breast tumors in patients presented the data as “cytokine concentration per tissue weight” [5,6] (at the low range of dose spectrum), thus we cannot conclude whether the concentrations used in our study are within the amounts existing in tumors (as we used them in “cytokine concentration per volume” values). Nevertheless, it is important to mention that the concentrations of TNFα and IL-1β that we used are relatively low, are not inducing death or necrosis of the cells (high TNFα doses of ∼1–4 mg, which are used for loco-regional treatments, cause necrosis [65]), and are within the conventional scale used by other investigators. Control nonstimulated cells were exposed to the solubilizer of the cytokines (0.1% BSA).
In experiments analyzing chemokine induction by ELISA, the cells were stimulated by the cytokines for 24 hours only, in serum-free medium, allowing processes of chemokine production and accumulation in conditioned media (CM) to take place. In cell remodeling and EMT-related studies, the cells were stimulated with the cytokines for 3 to 4 weeks in growth medium containing serum. Fresh cytokines were replenished every 3 to 4 days (after removal of previous cytokine-containing medium). Twenty-four to 48 hours before the cell remodeling and EMT-related assays (e.g., flow cytometry, MMP production, invasion, and migration), the growth medium was replaced by serum-free medium containing the cytokines (unless otherwise indicated). Control cells were grown under similar conditions, albeit in the absence of cytokines (they were replaced by their solubilizer). In specific cases that are mentioned in the text and figure legends, a short stimulation of 3 days was also included in the cell remodeling analyses.
Enzyme-Linked Immunosorbent Assays
To determine the release of CXCL8 and CCL2 by TNFα- and IL-1β-stimulated cells, the nontransformed breast epithelial cells were stimulated by the cytokines for 24 hours, as described above. Then, ELISA analyses were performed, and CXCL8 and CCL2 levels in CM were determined using standard curves with rhCXCL8 or rhCCL2 (Cat. No. 200-08 and No. 300-04, respectively; PeproTech), at the linear range of absorbance. The following antibodies were used (all from PeproTech): for CXCL8—coating polyclonal antibodies (Cat. No. 500-P28), detecting biotinylated rabbit polyclonal antibodies (Cat. No. 500-P28Bt); for CCL2—coating monoclonal antibodies (Cat. No. 500-M71), detecting biotinylated rabbit polyclonal antibodies (Cat. No. 500-P34Bt). After the addition of streptavidin-HRP (Jackson ImmunoResearch Laboratories, West Grove, PA), the substrate TMB/E solution (Chemicon, Temecula, CA) was added. The reaction was stopped by the addition of 0.18 M H2SO4 and was measured at 450 nm.
Determination of Cell Morphology
Cells were stimulated by the cytokines for up to 3 weeks, as described above (except that the growth medium was not exchanged to serum-free medium). During this time course, cell morphology was determined by microscopy at 1-week intervals (see figures; Eclipse Ti; Nikon, Melville, NY), and the cells were photographed (NIS-Elements version 4.0). Magnification of x10 to x20 was used, as indicated in the figure legends.
Formation of 3D Acini Structures
MCF-10A and HB2 cells were infected to express mCherry (with pQC-mCherry retroviral vector), and formation of 3D acini structures was performed as previously described [56,57]. Briefly, eight-well chamber slides (Nunc, Rochester, NY) were coated with 40 to 50 µl of ice-cold liquid Matrigel (Cat. No. 356234; BD Biosciences, Bedford, MA) that has solidified for 15 minutes at 37°C. Later, 5 x 103 MCF-10A cells or 2.4 x 103 HB2 cells were seeded in each well of the chamber, in 400 µl of 2% Matrigel-containing medium. Because acini have not been previously described in HB2 cells, we have verified that these cells indeed formed the hollow structures that are typical of acini [56,57]. To this end, fluorescence z-stack images were captured with a confocal laser scanning microscope, using two-photon laser for optical imaging (LSM 510; Carl Zeiss, Jena, Germany). Three-dimensional surface renderings were generated from the z-stack confocal images, using ImageJ software.
In this study, 3D acini structures were formed under two cytokine-stimulatory conditions in which the cells were exposed to the cytokines for 3 weeks (without exchange to serum-free medium) under the following stimulatory setups: 1) Formation of acini by cells that have been already stimulated by TNFα + IL-1β: Cells were stimulated by the cytokines (concentrations as above) for 3 weeks, in 2D standard cultures. Control nonstimulated cells (exposed to the solubilizer of the cytokines) and the cytokine-stimulated cells were seeded on top of Matrigel-coated wells to allow for acini structures to be formed. Pictures of the cells were taken 7 days after seeding, by laser scanning confocal microscope (CLSM510, Zeiss). 2) Stimulation of pre-formed acini by TNFα + IL-1β: Nonstimulated cells were seeded on top of Matrigel-coated wells and were allowed to grow for 6 days, until 3D structures were starting to form. Then, the cells were either stimulated by TNFα + IL-1β (concentrations as above) or not (control cells, stimulated by the solubilizer of the cytokines) for additional 2 weeks. At that point, pictures of the structures were taken by laser scanning confocal microscope (CLSM510, Zeiss).
Flow Cytometry
Cells were stimulated by the cytokines for up to 3 to 4 weeks in serum-containing growth medium. At specific time points (see figures), the cells were analyzed by flow cytometry. Before this analysis, the cells were incubated overnight in serum-free medium in the presence of the cytokines (or their solubilizer), and then the expression of E-cadherin and vimentin was determined as previously described [16]. Briefly, E-cadherin surface expression was determined by mouse IgG1 antibodies against human E-cadherin (Cat. No. SC-21791; Santa Cruz Biotechnology, Santa Cruz, CA). Determination of vimentin expression was performed in methanol-permeabilized cells using mouse IgG1 antibodies against human vimentin (Cat. No. SC-6260; Santa Cruz Biotechnology). Baseline staining was obtained by nonrelevant isotype control IgG1 antibodies (Cat. No. 400101; BioLegend, San Diego, CA). Then, the cells were stained by fluorescein isothiocyanate-conjugated goat anti-mouse IgG1 (Cat. No. 115-095-003; Jackson ImmunoResearch Laboratories). Staining was determined by flow cytometry with a Becton Dickinson FACSort (Mountain View, CA) and the win MDI software.
Vimentin expression score was calculated as (mean fluorescence) x (percentage of positive cells). The score obtained for control non-stimulated cells was given the value of 1, and the value for TNFα + IL-1β-stimulated cells was calculated relative to the control cells.
shRNAs for Zeb1 and Snail and Overexpression of Zeb1, Snail, and Twist
Down-regulation of Zeb1 and Snail expression was performed by infection with Zeb1 short hairpin RNA (shRNA; Cat. No. RHS4529-NM_030751; Open Biosystems, Huntsville, AL; kindly provided by Prof. Rotter, Weizmann Institute of Science, Rehovot, Israel) or with Snail shRNA (Cat. No. SHCLNG-NM_005985; Sigma-Aldrich; kindly provided by Prof. Reich, The Hebrew University of Jerusalem, Jerusalem, Israel). For both targets, several different shRNAs were assayed and the most effective ones were chosen for use throughout the study. Following infection, selection was performed with 6 µg/ml puromycin (A.G. Scientific, San Diego, CA). Then, the cell population was used as a whole to prevent bias toward specific cell clones. Control cells were infected with control shRNA vectors carrying similar antibiotic resistance. Down-regulation of Zeb1 and Snail was verified by quantitative real-time polymerase chain reaction (qPCR) analyses, using the primers and conditions indicated below in the qPCR Analyses section.
To induce overexpression of Zeb1, Zeb1 cDNA (Cat. No. MHS4426-98361372; Open Biosystems) was obtained and was then amplified by using the following sequences of primer pairs: HA tag and the Age1 restriction site were added to the forward primer 5′-CACACAACCGGTATGTACCCTTACGACGTTCCTGATTACGCTAGCCTCATGGCGGATGGCCCCAGGTGTAA-3′, and the BamH1 restriction site was added to the reverse primer 5′-GTGTGTGGATCCCGATTAGGCTTCATTTGTCTTTTCTTCA-3′. The reaction conditions were given as follows: 94°C for 3 minutes, and then two cycles of 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 4 minutes. Annealing was performed at 68°C for 30 seconds, followed by 30 amplification cycles of 72°C for 10 minutes. The amplified Zeb1 fragment was inserted to the pQCXIN vector (neomycin resistant).
To induce overexpression of Snail and of Twist1 (to be hereby termed Twist), total RNA was isolated from human HB2 cell line. Full-length cDNA was amplified by reverse transcriptase-PCR. The sequences of the primer pairs used for Snail fragment amplification were given as follows:Myc tag and the Age1 restriction site were added to the forward primer 5′-TCTCTCACCGGTATGGAACAAAAACTCATCTCAGAAGAGGATCTGATGCCGCGCTCTTTCCTCGTCAGGAA-3′, and the BamH1 restriction site was added to the reverse primer 5′-AAATCTGGATCCTCAGCGGGGACATCCTGAGCAG-3′. The sequences of the primers used for Twist fragment amplification were given as follows: HA tag and the Age1 restriction site were added to the forward primer 5′-AGAGAGACCGGTATGGGATCCTACCCTTACGACGTTCCTGATTAC-3′, and the BamH1 restriction site was added to the reverse primer 5′-AGGAGAGGATCCCTAGTGGGACGCGGACATGGA-3′. The reaction conditions were given as follows: 94°C for 3 minutes, and then two cycles of 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 4 minutes. Annealing was performed at 68°C for 30 seconds, followed by 40 amplification cycles of 72°C for 10 minutes. The amplified Snail fragment was inserted to the pQCXIH vector (hygromycin resistant), and the Twist fragment was inserted into the pQCXIP vector (puromycin resistant).
Following the above, the cloned sequences of Zeb1, Snail, and Twist were verified by GenBank sequences. Then, the cells were infected with the relevant vectors or with control vectors, and following selection (500 µg/ml neomycin, 200 µg/ml hygromycin, or 6 µg/ml puromycin, as appropriate), in each infection type, the cell population was used as a whole to prevent bias toward specific cell clones. The overexpression of Zeb1, Snail, and Twist was verified by qPCR, using the primers and conditions described below in the qPCR Analyses section.
qPCR Analyses
qPCR analyses of Zeb1, Snail, and Twist were performed in two conditions: 1) following stimulation by the cytokines; in this case, the cells were stimulated for 3 days or 3 weeks in serum-containing growth medium, which was replaced by serum-free medium at the last 48 hours of stimulation; 2) following down-regulation or overexpression of Zeb1, Snail, or Twist (in different combinations, as appropriate; see Results section).
Total RNA was isolated from the cells using the EZ-RNA kit (Biological Industries), and first-strand cDNA was produced using the M-MLV reverse transcriptase (Ambion, Austin, TX). Quantification of cDNA targets by qPCR was performed using Rotor Gene 6000 (Corbett Life Science, Sydney, Australia) and the Rotor Gene 6000 series software. Transcripts were detected using SYBR Green I (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. The primers were given as follows: Zeb1: forward 5′-TGCAGCTGACTGTGAAGGTGT-3′, reverse 5′-CTTGCCCTTCCTTTCTGTCATC-3′; Snail: forward 5′-CTAATCCAGAGTTTACCTTCCAGCA-3′, reverse 5′-AGTCCCAGATGAGCATTGGC-3′; Twist: forward 5′-GGCCGGAGACCTAGATGTCA-3′, reverse 5′-CCACGCCCTGTTTCTTTGAATT-3′; rS9 (normalizing gene): forward 5′-TTACATCCTGGGCCTGAAGAT-3′, reverse 5′-GGGATGTTCACCACCTGCTT-3′. PCR amplification was performed over 40 cycles (95°C for 15 seconds, 59°C for 20 seconds, 72°C for 15 seconds). Dissociation curves for each primer set indicated a single product, and “no template” controls were negative after 40 cycles. Quantification was performed by standard curves on the linear range of quantification.
Gelatin Substrate Zymography
The cells were stimulated for 3 weeks by the cytokines and then were plated onto 24-well plates in serum-containing growth medium. After an overnight incubation in serum-free medium in the presence of the cytokines, CM were collected for determination of MMP activities. In parallel, cells in each treatment were counted for further per-cell-basis quantitation of MMP secretion (see below). CM were centrifuged and then were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis containing 0.1% gelatin substrate. After electrophoresis, gels were washed in 50 mM Tris HCl, pH 7.5, containing 2.5% Triton X-100. The gels were then washed three times in 50 mM Tris HCl buffer, pH 7.4, followed by incubation in buffer consisting of 50 mM Tris HCl, pH 7.4, 0.02% NaN3, and 10 mM CaCl2 for 48 hours at 37°C. After three washes in double-distilled H2O, the gels were stained with 0.1% Coomassie blue and destained in 20% methanol and 10% acetic acid. Then, clear bands of protein degradation, indicating the presence of enzymatically active MMPs, were visualized.
Invasion Assays
The invasion potential of the cells was determined in transwell migration chambers with 8-µm pore size, polycarbonate membrane plates (Cat. No. 3422; Costar, Cambridge, MA), as previously described [16]. Briefly, the membranes located in the upper wells were pre-coated with Matrigel (as above) for 1 hour at 37°C, then blocking was performed for 1 hour with 0.1% heat-inactivated BSA. Cells that have been either stimulated by the cytokines or nonstimulated (exposed to the solubilizer of the cytokines) for 3 weeks in serum-containing medium were suspended in serum-free medium and were added to the upper wells of the chamber on top of the membrane, in the presence or absence of cytokine stimulation, as appropriate. Following 2 hours of incubation in 37°C, the upper wells were put on the lower wells of the chamber, containing medium supplemented with 10% serum. Both the upper and lower wells of the chamber included cytokines, thus preventing formation of a cytokine gradient in the course of the assay. Following incubation for 8 hours at 37°C, the nonmigrated cells on the upper surface of the membrane were completely removed by wiping with a cotton swab. Next, the filters were fixed and were stained with Hemacolor for microscopy (Cat. No. 1.11661; Merck, Darmstadt, Germany). The invaded cells were photographed (Olympus DP70 Digital Camera; Olympus America Inc, Center Valley, PA) and then counted in high power fields by light microscopy (Eclipse TE2000-S; Nikon).
Wound Healing Assays
The nontransformed cells, stimulated for 3 weeks by the cytokines or nonstimulated (exposed to the solubilizer of the cytokines) in serum-containing growth medium, were seeded in six-well tissue culture plates and have reached confluence in the presence of the cytokines. A wound was incised in the central area of the confluent culture using a plastic tip, followed by careful two washing steps of the detached cells and addition of fresh serum-free medium. Closure of the denuded area was monitored using an inverted microscope (Eclipse TS-100; Nikon) fitted with a digital camera (DX-FI1; Nikon). The wounded area was followed and documented at the time of the scratching and following 20 hours. Of note, in separate analyses, cell counts indicated that TNFα and IL-1β did not induce growth of the nontransformed breast epithelial cells.
Statistical Analyses
Statistical analyses were done using Student's t tests. Values of P < .05 were considered statistically significant. In each type of experimental analysis, the results are of a representative experiment of at least three independent repeats, showing similar results (in rare cases, when more appropriate, the results were presented as average of several experiments). In each representative experiment, data are presented as means ± SD of technical repeats within the experiment (when appropriate). When required, adjustment for multiplicity of comparisons was done using the Benjamini-Hochberg procedure. Using this procedure, all the significant results that are presented in the manuscript remained statistically significant after correcting for their multiplicity.
Results
Continuous Stimulation by TNFα + IL-1β Leads to Cell Remodeling and EMT in Nontransformed Breast Epithelial Cells
Primary tumors in breast cancer are enriched with both TNFα and IL-1β [5–16], which may affect adjacent nontransformed cells in a cooperative manner. Before determining the impact of the two cytokines on cell remodeling and EMT properties in the nontransformed cells, we asked to what extent these cells respond to TNFα and IL-1β, and if the two cytokines can act in concert, as may be expected by their joint expression in the majority of breast cancer patients [16].
As proxies for responsiveness to TNFα and IL-1β, we used induction of the inflammatory chemokines CXCL8 and CCL2. In the immune context, TNFα and IL-1β are powerful inducers of inflammatory chemokines, as is the case in tumor cells, including breast cancer cells [26,66–72]. In many malignancies, including breast cancer, CXCL8 and CCL2 are well identified as potent tumor-promoting factors [73–78], and thus their induction provides a relevant readout for responsiveness of the nontransformed breast epithelial cells to TNFα and IL-1β. The results presented in Figure 1 indicate that nontransformed MCF-10A and HB2 breast epithelial cells indeed responded to TNFα and IL-1β stimuli by significant elevations in CXCL8 and CCL2. Moreover, cooperativity was observed between TNFα and IL-1β in inducing CXCL8 in both cell types and also in CCL2 induction in HB2 cells (Figure 1). Of note, MCF-10A cells responded to TNFα and IL-1β stimulation more potently than HB2, and this may be due to intrinsic differences in the expression levels of receptors for the cytokines or of the downstream signaling pathways that evolved.
Figure 1.

Nontransformed breast epithelial cells respond to TNFα and IL-1β by increased release of the pro-malignancy chemokines CXCL8 and CCL2. MCF-10A cells (A, B) and HB2 cells (C, D) were stimulated by TNFα (50 ng/ml), IL-1β (500 pg/ml), or both cytokines together (TNFα + IL-1β; concentrations as above) for 24 hours, in serum-free medium. Cytokine concentrations for stimulation were selected on the basis of previous titration analyses (see Materials and Methods section for details; data not shown). Control, cells not stimulated by the cytokines (exposed to the solubilizer of the cytokines). The expression levels of CXCL8 (A, C) and CCL2 (B, D) were determined in cell supernatants by ELISA, in the linear range of absorbance. ***P < .001 for cytokine-stimulated cells compared to nonstimulated cells. In all panels, a representative experiment of n = 3 is presented.
To follow on the above findings, we determined the joint impact of TNFα and IL-1β together (namely, TNFα + IL-1β) and of each cytokine alone on induction of spreading and invasion-related properties in the nontransformed cells. First, we performed kinetics analyses in which we followed the morphology of stimulated and nonstimulated cells for 3 weeks. We found that IL-1β, but much more strongly TNFα and TNFα + IL-1β, induced cell remodeling and morphology changes in both MCF-10A cells (Figure 2A) and HB2 cells (Figure 2B). However, in contrast to tumor cells in which it took 1 to 3 days for such a process to develop (e.g., in our studies of MCF-7 cells [16] and in other investigations [38,42,43]), in the nontransformed cells 2 to 3 weeks of stimulation were necessary for induction of cell remodeling, visualized by cell spreading and formation of cellular protrusions (Figure 2).
Figure 2.

Prolonged stimulation by TNFα and IL-1β induces morphologic changes, spreading, and formation of cellular protrusions in nontransformed breast epithelial cells. MCF-10A cells (A) and HB2 cells (B) were stimulated by TNFα, IL-1β, or TNFα + IL-1β (concentrations as in Figure 1). Control, cells not stimulated by the cytokines (exposed to the solubilizer of the cytokines). Cell images were taken immediately after cell culturing (time 0) and then after 1, 2, and 3 weeks of stimulation, at x10 to x20 magnification (x10 magnification was used only for HB2 cells, in which case photographs were adjusted to x20 magnification digitally). Arrows point to some of the cells in which cellular protrusions and spreading have been observed. In all panels, the images are representatives of many pictures taken in n = 3 experiments.
In view of the above results and of the ability of TNFα and IL-1β to induce EMT in breast tumor cells, we performed a detailed analysis of the ability of the two cytokines to induce EMT in the two nontransformed breast epithelial cells, MCF-10A and HB2, analyzing each cytokine alone and both of them together. To determine the impact of the cytokines on EMT, we began by using the following two most typical EMT markers: 1) reduction in cell surface expression of E-cadherin, indicative of loss of cell-to-cell contacts and 2) up-regulation of vimentin expression, manifesting acquisition of a mesenchymal phenotype by the cells [44–48]. After 3 weeks of stimulation, both MCF-10A cells and HB2 cells manifested reduced expression levels of E-cadherin, particularly following stimulation by TNFα and TNFα + IL-1β (Figure 3, A and B). In parallel, vimentin expression was elevated in the two cell lines following TNFα and TNFα + IL-1β stimulations, whereas IL-1β alone was a weak/noninducer of vimentin expression (Figure 3, C and D). Of interest, following TNFα + IL-1β exposure a certain part of the cell population of MCF-10A cells has gained increased expression of vimentin per cell (indicated by increased mean fluorescence values), whereas in HB2 cells a new subpopulation has emerged, which was characterized with particularly high expression levels of vimentin (indicated in Figure 3D by an arrow). In view of its separable and very definitive phenotype, this high vimentin-expressing sub-population of HB2 cells had a clear advantage in analyzing EMT processes in a quantitative manner and thus was used along the study as a proxy for measuring EMT events. Using this high vimentin-expressing subpopulation of HB2 cells, we found that stimulation of the nontransformed cells by TNFα + IL-1β for 3 days indeed was not sufficient for induction of EMT in these cells, but rather 2 to 3 weeks were required (data not shown), as was also indicated by the morphology observations presented in Figure 2B.
Figure 3.

Continuous stimulation by TNFα and IL-1β leads to reduced surface expression of E-cadherin and increased expression of vimentin in nontransformed breast epithelial cells. MCF-10A cells (A, C) and HB2 cells (B, D) were stimulated by TNFα, IL-1β, or TNFα + IL-1β (concentrations as in Figure 1) for 3 weeks. Control, cells not stimulated by the cytokines (exposed to the solubilizer of the cytokines). Surface expression of E-cadherin was determined in intact cells (A, B), while intracellular expression of vimentin was determined in permeabilized cells (C, D), using flow cytometry. Isotype, nonrelevant isotype control. The arrow points out the new subpopulation of cells with high expression of vimentin that has appeared following TNFα + IL-1β stimulation in HB2 cells. In all panels, a representative experiment of at least n = 3 is presented.
Thus, the above results indicate that TNFα was a more prominent inducer of cell remodeling and EMT-related characteristics in non-transformed cells than IL-1β. Furthermore, highly potent activities were also induced by the combination of TNFα + IL-1β, which recapitulates the high co-expression of the two cytokines together in the majority of breast cancer patients, particularly in >80% patients with recurrent disease [16]. In view of the high clinical relevance of the combined TNFα + IL-1β stimulation, and due to the advantage expressed by the joint stimulation in induction of CXCL8 and CCL2 compared to stimulation by each cytokine alone (Figure 1), further analyses focused on the combined stimulation of the cells by TNFα + IL-1β together.
Specifically, we asked whether nontransformed epithelial breast cells that were stimulated by the cytokines will be able to form multicellular organized structures that are typical of normal breast cells. To investigate this issue, we employed the commonly used model of 3D acini structures, formed in nonadherent conditions on Matrigel by nontransformed breast epithelial cells but not by malignant cells [56,57]. Formation of 3D acini structures has been well described for the MCF-10A cells [56,57], and we have followed published protocols to establish such acini of MCF-10A cells in our current study. Using acini structures as platform to decipher perturbations of normal textures, we investigated the ability of nontransformed cells that have been stimulated for 3 weeks by TNFα + IL-1β and have undergone EMT to form acini. To this end, MCF-10A cells were stimulated for 3 weeks by the cytokines and then were allowed to form acini on Matrigel, in comparison to nonstimulated cells. We found that the control nonstimulated cells have established multicellular organized structures, whereas the cytokine-stimulated cells did not form such structures in the majority of cases (Figure 4A). Moreover, the cytokine-stimulated cells—which have been already exposed to TNFα + IL-1β for 3 weeks—have gained very potent spreading abilities and degraded the matrix when they were plated on Matrigel to form acini. Therefore, the acini formation process in this type of experiment could not be followed for more than 6 days because the process of acini establishment has began (instead of 2 to 3 weeks that are usually required), and this is why the control nonstimulated cells did not form yet the hollow structures typical of acini [56,57], although as indicated, they already made organized multicellular structures (Figure 4A).
Figure 4.

Nontransformed breast epithelial cells stimulated by TNFα + IL-1β are incapable of forming organized acini structures. MCF-10A cells (A) and HB2 cells (B) were stimulated by TNFα + IL-1β (concentrations as in Figure 1) for 3 weeks. Control, cells not stimulated by the cytokines (exposed to the solubilizer of the cytokines). Then, the cells were plated onto nonadherent conditions on Matrigel to allow for acini formation. After 7 days, pictures of the structures were taken by confocal microscopy. In all panels, the images are representatives of many pictures taken in n = 3 experiments. See text (Results section) for explanation on the acini structures formed by the control nonstimulated cells.
In parallel to MCF-10A cells, we have established acini with HB2 cells. Because this is the first demonstration of acini formation by HB2 cells, we show in Figure W1 that these cells indeed formed hollow structures as they should (Figure W1). After the method was set, we have tested the ability of cytokine-stimulated HB2 cells to form acini. The findings with HB2 cells (Figure 4B) were similar to those obtained with MCF-10A cells (Figure 4A): Instead of adhering to each other and forming cell-to-cell contacts required for acini formation, the cells have detached from each other and have formed cellular protrusions, characteristic of cells undergoing EMT (as was shown in Figure 2B). Note that as with MCF-10A cells, the control nonstimulated cells did not have enough time (only 6 days) to form hollow acini structures, but they already established multicellular textures, as expected (Figure 4B).
Taken together, the above results indicate that TNFα + IL-1β stimulation has induced cell plasticity, spreading and EMT in the non-transformed breast epithelial cells. This has been visualized by extensive formation of cell protrusions, down-regulation of the epithelial marker E-cadherin, elevation in expression of the mesenchymal marker vimentin, and inability to form acini.
The Need for Continuous and Prolonged Stimulation by TNF α + IL-1β for Induction of EMT in Breast Nontransformed Cells Is due to Imbalance in the Activation Profiles of EMT Inducers
As indicated above, the fact that full-blown EMT was induced in nontransformed breast epithelial cells only after 2 to 3 weeks of stimulation by TNFα + IL-1β contrasted the rapid kinetics of the same process induced by the cytokines (each alone) in malignant breast cells that took 1 to 3 days to develop [16,38,42,43]. Furthermore, using the subpopulation of high vimentin-expressing HB2 cells as proxy for EMT, we found that EMT induction by the cytokines required persistent presence of TNFα + IL-1β (Figure W2). This was indicated by the fact that when the stimulation by TNFα + IL-1β was removed, the EMT phenotype has been gradually extinguished (Figure W2).
To provide mechanistic insights into the regulatory events dictating the slow EMT process in the nontransformed cells, we focused on Zeb1, Snail, and Twist, known as key inducers of EMTin breast tumor cells [46–52,79–89]. Time-dependent analyses of HB2 cells, using the high vimentin-expressing subpopulation as indicator of EMT, have shown that following 3 days of stimulation by TNFα + IL-1β—which is a time point that is insufficient for induction of EMT in these cells (Figure 2B)—none of these three EMT activators was induced (Figure 5A). Here, it is important to indicate that in breast tumor cells, in which EMT was induced already after 1 to 3 days [16,38,42,43], we found that Zeb1 and Snail were elevated in much more rapid kinetics and were increased within 3 days of stimuli (data not shown). In contrast to the 3-day stimulation process that did not promote any of the EMT inducers, a totally different pattern was revealed after 2 to 3 weeks of stimulation by TNFα + IL-1β, which are the conditions leading to EMT in these cells: Zeb1 and Snail were eventually upregulated, and in parallel, down-regulation in the expression of Twist was detected (Figure 5B).
Figure 5.

Continuous stimulation by TNFα + IL-1β induces upregulation in Zeb1 and Snail expression and downregulates Twist expression in nontransformed breast epithelial cells. HB2 cells were stimulated by TNFα, IL-1β, or TNFα + IL-1β (concentrations as in Figure 1) for 3 days (A) or for 2 to 3 weeks (B). Control, cells not stimulated by the cytokines (exposed to the solubilizer of the cytokines). At each time point, the expression of Zeb1, Snail, and Twist was determined by qPCR. **P < .01, ***P < .001; NS, not significant for cytokine-stimulated cells compared to nonstimulated cells. Nonstimulated cells were given the value of 1. In all panels, a representative experiment of n = 3 is presented.
These results suggested that the delayed activation of Zeb1 and Snail and down-regulation of Twist stand on the basis of the slow activation process of EMT that was induced by TNFα + IL-1β in the nontransformed cells. To see if this was the case, first we analyzed the regulation of the EMT process in these cells by Zeb1 and Snail. As a result of Zeb1 or Snail down-regulation by shRNAs (Figure W3), the TNFα + IL-1β-induced EMT process was blocked even when 4 weeks of cytokine stimulation were introduced (Figure 6, A–D for Zeb1 and Figure 6, E–H for Snail). This was indicated by the inability of TNFα + IL-1β to induce up-regulation of the high vimentin-expressing subpopulation in cells experiencing Zeb1 or Snail down-regulation (due to expression of shRNAs). Furthermore, we found that overexpression of Zeb1 + Snail in the cells (Figure W4A) has potently induced the EMT process, as indicated by elevation in vimentin expression (Figure 7A). These findings indicated that Zeb1 and Snail were EMT inducers in the nontransformed breast epithelial cells, they were essential for TNFα + IL-1β-induced EMT in these cells, and thus their delayed induction following cytokine stimulation prevented earlier activation of the EMT process (Figure 5B).
Figure 6.

Zeb1 and Snail are essential for EMT induction in nontransformed breast epithelial cells stimulated by TNFα + IL-1β. HB2 cells were infected to express shRNA to Zeb1, shRNA to Snail, or control shRNA vector (down-regulation of Zeb1 and Snail expression was verified by qPCR, as shown in Figure W3). The cells were either stimulated by TNFα + IL-1β (concentrations as in Figure 1) for 1 or 4 weeks or nonstimulated by the cytokines (exposed to the solubilizer of the cytokines) for the same time course. Following downregulation of these factors and cytokine stimulation, the expression of vimentin was determined by flow cytometry in permeabilized cells, as proxy for cells undergoing EMT. Isotype, nonrelevant isotype control. (A–D) The impact of Zeb1 down-regulation on EMT. (A, C) Cells grown with or without cytokine stimulation for 1 week. (B, D) Cells grown with or without cytokine stimulation for 4 weeks. (A, B) Cells infected with control vector. (C, D) Cells infected with shRNA for Zeb1. (E–H) The impact of Snail down-regulation on EMT. (E, G) Cells grown with or without cytokine stimulation for 1 week. (F, H) Cells grown with or without cytokine stimulation for 4 weeks. (E, F) Cells infected with control vector. (G, H) Cells infected with shRNA for Snail. The arrows point out the new subpopulation of cells with high expression of vimentin that has appeared following cytokine stimulation in control HB2 cells and has disappeared upon down-regulation of Zeb1 and Snail. In all panels, a representative experiment of at least n = 3 is presented.
Figure 7.
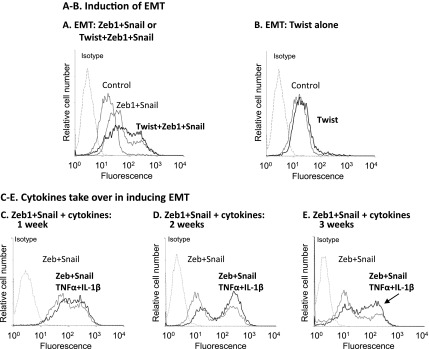
The EMT-inducing activities of Zeb1 and Snail on nontransformed breast epithelial cells are potentiated by Twist or alternatively by prolonged stimulation with TNFα + IL-1β. HB2 cells were infected to overexpress Zeb1+ Snail or Twist + Zeb1+ Snail (A) or Twist alone (B) (the increased expression of these EMT inducers following infection was verified by qPCR, as shown in FigureW4). Control, cells infected with control vector. Following expression of these factors, the expression of vimentin was determined by flow cytometry in permeabilized cells, as proxy for cells undergoing EMT. Isotype, nonrelevant isotype control. In all panels, a representative experiment of n = 3 is presented. (C–E) HB2 cells were infected to overexpress Zeb1 + Snail (termed “Zeb + Snail”; the expression of these EMT inducers following infection was verified by qPCR as shown in Figure W4). The cells were either not stimulated by the cytokines (exposed to the solubilizer of the cytokines) or stimulated for 1 week (C), 2 weeks (D), or 3 weeks (E) by TNFα + IL-1β (concentrations as in Figure 1; termed “Zeb + Snail, TNFα + IL-1β”). At each time point, the expression of vimentin was determined by flow cytometry in permeabilized cells, as proxy for cells undergoing EMT. Isotype, nonrelevant isotype control. In all panels, a representative experiment of n = 3 is presented.
In parallel to Zeb1 and Snail, we analyzed the roles played by Twist in EMT induction in the nontransformed cells in response to TNFα + IL-1β. Because prolonged stimulation of the cells by TNFα + IL-1β has led to down-regulation of Twist expression (Figure 5B), we decided to learn more on the roles of Twist in EMT induction in the nontransformed cells by inducing its overexpression in these cells and determining its impact on EMT, using vimentin expression as readout. The results of Figure 7B indicate that the overexpression of Twist in the cells (Figure W4B) has not induced EMT. However, the roles of Twist in EMT induction were revealed when it was combined with Zeb1 and Snail. The addition of Twist to Zeb1 + Snail overexpression has amplified the EMT process that now exceeded the levels obtained by Zeb1 + Snail by themselves (Figure 7A).
The reduction in Twist expression by the prolonged stimulation with TNFα + IL-1β has led us to ask if the two cytokines may compensate for the lack of Twist expression. Indeed, we found that TNFα + IL-1β stimulation has amplified the ability of Zeb1 + Snail to induce EMT (Figure 7, C–E), as was previously noticed for Twist when it was combined with Zeb1 + Snail (Figure 7A). To potentiate the activities of Zeb1 + Snail, 2 to 3 weeks of stimulation by TNFα + IL-1β were required, indicating that while the cytokines have downregulated Twist expression, they have recapitulated its missing activities in a process requiring prolonged stimulation.
Taken together, the findings obtained in this part of the study indicate that to induce EMT, TNFα + IL-1β had to stimulate the cells for prolonged durations of 2 to 3 weeks and that the delayed EMT process was due to the following two reasons: 1) It took 2 to 3 weeks for the cytokines to induce the expression of Zeb1 and Snail, which were required for TNFα + IL-1β-induced EMT processes in these cells (Figures 5, 6, and 7A); 2) 2 to 3 weeks of stimulation were required for the cytokines to take over the missing activities of Twist and to further amplify the EMT-inducing activities of Zeb1 + Snail (Figure 7, A and C–E).
Stimulation of Nontransformed Breast Epithelial Cells by TNFα + IL-1β Leads to Dissemination-Related Functions and to Cell Spreading Out of Organized Acini Structures
To follow on the cell remodeling and EMT processes induced by TNFα + IL-1β in the nontransformed cells (Figures 2–4), we have determined the functional implications of cytokine stimulation. Here, we focused on activities that may lead to dissemination of the nontransformed cells at the primary tumor site, turning these cells later to potential targets for transformation and formation of a new tumor focus.
The results of Figure 8A indicate that TNFα + IL-1β stimulation of the nontransformed cells has given rise to pronounced induction of active MMP2 and MMP9, in levels similar to those induced by TNFα alone, and much more pronounced than the levels induced by IL-1β alone. Furthermore, nontransformed cells stimulated by the cytokines have gained potent invasive properties, as indicated by invasion assays performed through Matrigel-coated membranes in response to serum-containing medium in transwells. The invasion assays emphasized once again the functional benefit of the combined stimulation by the two cytokines together compared to each alone (Figure 8, B and C; note that none of the stimulations affected significantly the basal migration phenotype of the cells, determined in response to serum-free medium; Figure W5). IL-1β was a weak inducer of invasion (Figure 8, B and C), agreeing well with its lower ability to induce spreading and EMT in these cells (Figures 2–4). Although TNFα has induced invasion more efficiently than IL-1β, the greatest impact on invasion was revealed upon combined stimulation by TNFα + IL-1β, being in line with the most effective ability of these two cytokines to act together and lead to preferential induction of CXCL8 and CCL2 (Figure 1). This combined stimulation has also promoted considerably the migratory properties of the nontransformed cells, as indicated by wound closure assays of motility (Figure 8D; note that cytokine stimulation did not induce the growth of the cells; data not shown).
Figure 8.

TNFα + IL-1β induce high MMP levels and increased invasive and migratory phenotype in nontransformed breast epithelial cells. HB2 cells were stimulated by TNFα, IL-1β, or TNFα + IL-1β (concentrations as in Figure 1) for 3 weeks. Control, cells not stimulated by the cytokines (exposed to the solubilizer of the cytokines). (A) Release of functional MMPs. CM were collected, and the presence of active MMPs was determined by zymography. A representative experiment of n = 3 is presented. (B, C) Invasion through Matrigel, determined after 8 hours in response to serum-containing medium, in transwells. Of note, the constitutive level of basal migration (no serum included) was not affected by cytokine stimulation (Figure W5). In cytokine-stimulated groups, TNFα and/or IL-1β were present throughout the time of assay in the upper wells to enable constant stimulation and also in the bottom wells of the chamber to prevent cytokine gradients. (B) Representative images of cells that have invaded, in control and cytokine-stimulated groups. In all panels, the images are representatives of many pictures taken in n = 3 experiments. (C) Quantitative analysis of the number of cells that have invaded in each of the groups. **P < .01, ***P < .001 for cytokine-stimulated cells compared to nonstimulated cells. In all panels, a representative experiment of n = 3 is presented. (D) Migration in wound closure assays. Following plating, the cells reached full confluence and a scratch was performed. Images of the cells were taken at the time of scratching (wound: 0 hour) and after 20 hours (wound: 20 hours). TNFα + IL-1β were present throughout the time of assay, and in parallel analyses were not found to induce cell growth (data not shown). In all panels, the images are representatives of many pictures taken in n = 3 experiments.
The above findings provide evidence to the eminent power of the combined stimulation by TNFα + IL-1β in inducing dissemination-related properties in the nontransformed breast epithelial cells. The question that followed was whether cytokine stimulation would disrupt those structures that are normally established in the breast by nontransformed cells. To answer this question, we used again the acini structures, as they represent the organized cellular texture established by normal breast epithelial cells, and their perturbation is often used to investigate mechanisms involved in tumor initiation and progression [56,57]. Specifically, we asked how the cytokines will affect the organization of acini that have been already formed. Thus, acini were allowed to start forming for 1 week without cytokine stimulation, and then the structures were grown for additional 2 weeks with or without TNFα + IL-1β stimulation. The findings of Figure 9 indicate that the control nonstimulated cells have established organized multicellular hollow acini structures as expected, while the stimulation by the cytokines has led to total disruption of the well-ordered acini structures: In the majority of the acini that were stimulated by TNFα + IL-1β, the nontransformed cells have spread out of the acini, demonstrating an unorganized distribution of cells and collapse of the previously organized acini structure (Figure 9).
Figure 9.

TNFα + IL-1β stimulation of nontransformed breast epithelial cells ruins pre-formed acini structures and is accompanied by cell spreading out of acini. HB2 cells were plated onto nonadherent conditions on Matrigel to allow for acini formation. After 6 days, when acini have formed, the cells were stimulated for 2 weeks by TNFα + IL-1β (concentrations as in Figure 1). Control, cells not stimulated by the cytokines (exposed to the solubilizer of the cytokines). Pictures of structures were taken by confocal microscopy. The arrows point to cells that have spread out of pre-formed acini, as a result of cytokine stimulation. In all panels, the images are representatives of many pictures taken in n = 3 experiments. See text (Results section) for explanation on the acini structures formed by the control nonstimulated cells.
Overall, the results presented in this part of the study indicate that TNFα + IL-1β, which have been shown to be highly expressed in the breast tumor microenvironment [5–16], induced in nontransformed breast epithelial cell processes of motility and invasion, leading to cell migration out of normally organized breast textures of acini. The outcome of such events may be dissemination of the yet nontransformed epithelial cells at the tumor site, and if these cells are then exposed to transforming events, they may be the “cornerstone” of new tumor focus that will be formed in close proximity to the primary tumor site.
Discussion
The tumor microenvironment of breast tumors is enriched with inflammatory mediators, including the cytokines TNFα and IL-1β that are highly released by the cancer cells [5–16]. The two cytokines are expressed together in tumors of most patients with recurrent disease [16] and have been shown to have causative pro-tumoral activities. TNFα and IL-1β promote breast malignancy by many different functions that are exerted on leukocytes and stroma cells and on the tumor cells themselves [16–26,30–37]. Between others, the two cytokines were found to induce EMT in breast tumor cells and causatively contributed to increased disease course [16,38–43].
The novel findings obtained in this study demonstrate the strong impact of joint TNFα + IL-1β stimulation on cell remodeling and plasticity of nontransformed breast epithelial cells; it provides mechanistic understanding of the events mediating these activities of the two cytokines and illustrates the functional implications and the clinical relevance of these findings. Our results show that in response to the two cytokines together, these nontransformed cells have spread considerably, underwent EMT, and exhibited the important functional readouts of MMP production, increased motility, and invasiveness. Of major importance are the findings showing that the stimulation of the nontransformed cells by TNFα + IL-1β has led to spreading of cells out of the organized structures that are typical of normal breast cells (acini). Thus, it is possible that nontransformed cells that have remained in the breast tissue in proximity to tumor cells would disseminate at the primary tumor site and may be the target of transformation thereafter.
Here, it is interesting to indicate that in contrast to breast tumor cells in which the cytokines induced EMT rapidly following 1 to 3 stimulation days [16,38,42,43], in the nontransformed cells the process was characterized by much slower kinetics, and only after 2 to 3 weeks of stimulation by TNFα + IL-1β cell remodeling and EMT were induced (Figures 2 and 5). In our investigation, we provided mechanistic explanation to the delayed EMT kinetics and to nonconventional regulatory events that took place in the nontransformed breast epithelial cells following stimulation by TNFα + IL-1β. We demonstrated that the delayed induction of EMT was the direct result of imbalance in the regulation of Zeb1, Snail, and Twist, indicated by 1) delayed activation of Zeb1 and Snail induced by TNFα + IL-1β stimulation and 2) induction of cooperativity between TNFα + IL-1β and Zeb1 + Snail that took time to develop. These results add another layer of complexity to the regulation of EMT processes that have been already revealed in breast tumor cells. Indeed, in breast cancer cells, these three regulators were found to have cardinal involvement in inducing EMT, invasiveness, and metastasis, and the majority of studies of breast cancer patients provide evidence to correlations between high expression levels of Zeb1, Snail, and/or Twist with EMT and with aggressive tumor phenotype [46–52,79–89]. However, in parallel, complex interactions between these three regulators were revealed in several studies, suggesting that the equilibrium between them may dictate the extent of EMT, invasiveness, and disease course [46–48,90,91].
The TNFα + IL-1β-induced processes of EMT and plasticity in nontransformed breast epithelial cells may have high relevance to what may be happening in breast tumors. In patients' tumors, nontransformed epithelial cells that are still present in the tissue are constantly exposed to TNFα + IL-1β [5–16], expressed together at high levels in >80% of the patients with relapsed disease [16]. One of the characteristics of such patients is local recurrence of tumor growth in the breast. With the improvement of diagnostic measures, nowadays it is known that recurrent ipsilateral breast tumors may be due to one of two scenarios: 1) true local recurrence—regrowth of malignant cells that were not killed by radiotherapy or not removed by surgery; 2) new primary tumors—cases that have developed de novo at the primary tumor site. In new primary tumors, the cancer cells may exhibit different markers and genetic setup compared to the original primary tumor [92–95]. Such a phenomenon can take place if nontransformed cells that were present at the initial tumor focus have migrated away from the primary site and have been exposed later to transforming events, for example, the mutagenizing factor nitric oxide that is prevalent at the inflammatory microenvironment of breast tumors [58–60].
On the basis of our results, we suggest that such a possibility may evolve when TNFα and IL-1β are expressed at high levels at the tumor site and induce EMT, MMP production, and invasion out of ordered breast structures in nontransformed breast epithelial cells. As a result, the nontransformed cells may acquire high migratory and invasive properties, may disseminate in the tissue, and seed themselves in proximal sites. If then, following the TNFα + IL-1β-induced dissemination of the nontransformed cells and their reseeding in the vicinity to the primary tumor site, they would be exposed to mutagenizing agents, they may undergo malignant transformation. Under such circumstances, a new tumor focus will develop in proximity to the primary focus, carrying genetic properties and markers that are different from the first tumor (as has been described in breast cancer; [92–95]). The continuous need for TNFα + IL-1β stimulation to induce invasive properties and consequently dissemination of the nontransformed cells may be one of the reasons of long duration until recurrence occurs in some of the patients.
In this way, a devastating interaction would exist between high TNFα and IL-1β levels, plasticity and EMT-related processes taking place in breast epithelial cells that have not been transformed as yet, disease recurrence and relapse. Such mechanisms may act in parallel to processes taking place in breast tumor cells, in which EMT events were suggested to promote tumor recurrence [96,97]. Most importantly, our study suggests that it is the inflammatory microenvironment that can promote plasticity, EMT, spreading, and invasion of nontransformed cells. Eventually, by acting in such a manner, the inflammatory microenvironment would dictate not only the elevated growth of primary tumors and their metastatic dissemination to remote organs but also whether the patients will develop secondary tumors and will succumb to disease recurrence.
Supplementary Material
Abbreviations
- 3D
three-dimensional
- CM
conditioned medium
- EMT
epithelial-to-mesenchymal transition
- IL-1β
interleukin-1β
- MMPs
matrix metalloproteinases
- qPCR
quantitative real-time polymerase chain reaction
- rh
recombinant human
- TNFα
tumor necrosis factor-α
Footnotes
The authors acknowledge the financial support provided to this study by Federico Foundation. The authors disclose no potential conflicts of interest.
This article refers to supplementary materials, which are designated by Figures W1 to W5 and are available online at www.neoplasia.com.
References
- 1.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 2.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2007;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Jiang X, Shapiro DJ. The immune system and inflammation in breast cancer. Mol Cell Endocrinol. 2013;382:673–682. doi: 10.1016/j.mce.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldberg JE, Schwertfeger KL. Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Curr Drug Targets. 2013;11:1133–1146. doi: 10.2174/138945010792006799. [DOI] [PubMed] [Google Scholar]
- 5.Jin L, Yuan RQ, Fuchs A, Yao Y, Joseph A, Schwall R, Schnitt SJ, Guida A, Hastings HM, Andres J, et al. Expression of interleukin-1beta in human breast carcinoma. Cancer. 1997;80:421–434. doi: 10.1002/(sici)1097-0142(19970801)80:3<421::aid-cncr10>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 6.Chavey C, Bibeau F, Gourgou-Bourgade S, Burlinchon S, Boissiere F, Laune D, Roques S, Lazennec G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007;9:R15. doi: 10.1186/bcr1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pantschenko AG, Pushkar I, Anderson KH, Wang Y, Miller LJ, Kurtzman SH, Barrows G, Kreutzer DL. The interleukin-1 family of cytokines and receptors in human breast cancer: implications for tumor progression. Int J Oncol. 2003;23:269–284. [PubMed] [Google Scholar]
- 8.Abrahamsson A, Morad V, Saarinen NM, Dabrosin C. Estradiol, tamoxifen, and flaxseed alter IL-1β and IL-1Ra levels in normal human breast tissue in vivo. J Clin Endocrinol Metab. 2012;97:E2044–E2054. doi: 10.1210/jc.2012-2288. [DOI] [PubMed] [Google Scholar]
- 9.Kurtzman SH, Anderson KH, Wang Y, Miller LJ, Renna M, Stankus M, Lindquist RR, Barrows G, Kreutzer DL. Cytokines in human breast cancer: IL-1alpha and IL-1beta expression. Oncol Rep. 1999;6:65–70. doi: 10.3892/or.6.1.65. [DOI] [PubMed] [Google Scholar]
- 10.Leek RD, Landers R, Fox SB, Ng F, Harris AL, Lewis CE. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br J Cancer. 1998;77:2246–2251. doi: 10.1038/bjc.1998.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miles DW, Happerfield LC, Naylor MS, Bobrow LG, Rubens RD, Balkwill FR. Expression of tumour necrosis factor (TNF alpha) and its receptors in benign and malignant breast tissue. Int J Cancer. 1994;56:777–782. doi: 10.1002/ijc.2910560603. [DOI] [PubMed] [Google Scholar]
- 12.García-Tuñón I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. Role of tumor necrosis factor-α and its receptors in human benign breast lesions and tumors (in situ and infiltrative) Cancer Sci. 2006;97:1044–1049. doi: 10.1111/j.1349-7006.2006.00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El Agouza IM, Eissa SS, El Houseini MM, El-Nashar DE, Abd El Hameed OM. Taurine: a novel tumor marker for enhanced detection of breast cancer among female patients. Angiogenesis. 2011;14:321–330. doi: 10.1007/s10456-011-9215-3. [DOI] [PubMed] [Google Scholar]
- 14.Cui LF, Guo XJ, Wei J, Liu FF, Fan Y, Lang RG, Gu F, Zhang XM, Fu L. Overexpression of TNF-α and TNFRII in invasive micropapillary carcinoma of the breast: clinicopathological correlations. Histopathology. 2008;53:381–388. doi: 10.1111/j.1365-2559.2008.03128.x. [DOI] [PubMed] [Google Scholar]
- 15.Sheen-Chen SM, Chen WJ, Eng HL, Chou FF. Serum concentration of tumor necrosis factor in patients with breast cancer. Breast Cancer Res Treat. 1997;43:211–215. doi: 10.1023/a:1005736712307. [DOI] [PubMed] [Google Scholar]
- 16.Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, Weitzenfeld P, Meshel T, Shabtai E, Gutman M, et al. Inflammatory mediators in breast cancer: coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer. 2011;11:130–149. doi: 10.1186/1471-2407-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagemann T, Robinson SC, Schulz M, Trümper L, Balkwill FR, Binder C. Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-α dependent up-regulation of matrix metalloproteases. Carcinogenesis. 2004;25:1543–1549. doi: 10.1093/carcin/bgh146. [DOI] [PubMed] [Google Scholar]
- 18.Warren MA, Shoemaker SF, Shealy DJ, Bshar W, Ip MM. Tumor necrosis factor deficiency inhibits mammary tumorigenesis and a tumor necrosis factor neutralizing antibody decreases mammary tumor growth in neu/erbB2 transgenic mice. Mol Cancer Ther. 2009;8:2655–2663. doi: 10.1158/1535-7163.MCT-09-0358. [DOI] [PubMed] [Google Scholar]
- 19.Sangaletti S, Tripodo C, Ratti C, Piconese S, Porcasi R, Salcedo R, Trinchieri G, Colombo MP, Chiodoni C. Oncogene-driven intrinsic inflammation induces leukocyte production of tumor necrosis factor that critically contributes to mammary carcinogenesis. Cancer Res. 2010;70:7764–7775. doi: 10.1158/0008-5472.CAN-10-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romieu-Mourez R, Francois M, Abate A, Boivin MN, Birman E, Bailey D, Bramson JL, Forner K, Young YK, Medin JA, et al. Mesenchymal stromal cells expressing ErbB-2/neu elicit protective antibreast tumor immunity in vivo, which is paradoxically suppressed by IFN-γ and tumor necrosis factor-α priming. Cancer Res. 2010;70:7742–7747. doi: 10.1158/0008-5472.CAN-10-0296. [DOI] [PubMed] [Google Scholar]
- 21.Hamaguchi T, Wakabayashi H, Matsumine A, Sudo A, Uchida A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem Biophys Res Commun. 2011;407:525–530. doi: 10.1016/j.bbrc.2011.03.051. [DOI] [PubMed] [Google Scholar]
- 22.Rubio MF, Werbajh S, Cafferata EG, Quaglino A, Colo GP, Nojek IM, Kordon EC, Nahmod VE, Costas MA. TNF-α enhances estrogen-induced cell proliferation of estrogen-dependent breast tumor cells through a complex containing nuclear factor-kappa B. Oncogene. 2006;25:1367–1377. doi: 10.1038/sj.onc.1209176. [DOI] [PubMed] [Google Scholar]
- 23.Rivas MA, Tkach M, Beguelin W, Proietti CJ, Rosemblit C, Charreau EH, Elizalde PV, Schillaci R. Transactivation of ErbB-2 induced by tumor necrosis factor α promotes NF-κB activation and breast cancer cell proliferation. Breast Cancer Res Treat. 2009;122:111–124. doi: 10.1007/s10549-009-0546-3. [DOI] [PubMed] [Google Scholar]
- 24.Rivas MA, Carnevale RP, Proietti CJ, Rosemblit C, Beguelin W, Salatino M, Charreau EH, Frahm I, Sapia S, Brouckaert P, et al. TNFα acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-κB-dependent pathways. Exp Cell Res. 2008;314:509–529. doi: 10.1016/j.yexcr.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Purohit A, Newman SP, Reed MJ. The role of cytokines in regulating estrogen synthesis: implications for the etiology of breast cancer. Breast Cancer Res. 2002;4:65–69. doi: 10.1186/bcr425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben-Baruch A. The tumor-promoting flow of cells into, within and out of the tumor site: regulation by the inflammatory axis of TNFα and chemokines. Cancer Microenviron. 2012;5:151–164. doi: 10.1007/s12307-011-0094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balkwill F, Mantovani A. Cancer and inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Ther. 2010;87:401–406. doi: 10.1038/clpt.2009.312. [DOI] [PubMed] [Google Scholar]
- 28.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Mocellin S, Nitti D. TNF and cancer: the two sides of the coin. Front Biosci. 2008;13:2774–2783. doi: 10.2741/2884. [DOI] [PubMed] [Google Scholar]
- 30.Reed JR, Leon RP, Hall MK, Schwertfeger KL. Interleukin-1beta and fibroblast growth factor receptor 1 cooperate to induce cyclooxygenase-2 during early mammary tumourigenesis. Breast Cancer Res. 2009;11:R21. doi: 10.1186/bcr2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmid MC, Avraamides CJ, Foubert P, Shaked Y, Kang SW, Kerbel RS, Varner JA. Combined blockade of integrin-α4β1 plus cytokines SDF-1α or IL-1β potently inhibits tumor inflammation and growth. Cancer Res. 2011;71:6965–6975. doi: 10.1158/0008-5472.CAN-11-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou W, Guo S, Gonzalez-Perez RR. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br J Cancer. 2011;104:128–137. doi: 10.1038/sj.bjc.6606013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naldini A, Filippi I, Miglietta D, Moschetta M, Giavazzi R, Carraro F. Interleukin-1β regulates the migratory potential of MDAMB231 breast cancer cells through the hypoxia-inducible factor-1α. Eur J Cancer. 2010;46:3400–3408. doi: 10.1016/j.ejca.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 34.Palmieri C, Roberts-Clark D, Assadi-Sabet A, Coope RC, O'Hare M, Sunters A, Hanby A, Slade MJ, Gomm JJ, Lam EW, et al. Fibroblast growth factor 7, secreted by breast fibroblasts, is an interleukin-1β-induced paracrine growth factor for human breast cells. J Endocrinol. 2003;177:65–81. doi: 10.1677/joe.0.1770065. [DOI] [PubMed] [Google Scholar]
- 35.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25:387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 36.Voronov E, Carmi Y, Apte RN. Role of IL-1-mediated inflammation in tumor angiogenesis. Adv Exp Med Biol. 2007;601:265–270. doi: 10.1007/978-0-387-72005-0_28. [DOI] [PubMed] [Google Scholar]
- 37.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–329. doi: 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong R, Wang Q, He XL, Chu YK, Lu JG, Ma QJ. Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-alpha-induced epithelial-mesenchymal transition of MCF-7 cells. Braz J Med Biol Res. 2007;40:1071–1078. doi: 10.1590/s0100-879x2007000800007. [DOI] [PubMed] [Google Scholar]
- 39.Zhou C, Nitschke AM, Xiong W, Zhang Q, Tang Y, Bloch M, Elliott S, Zhu Y, Bazzone L, Yu D, et al. Proteomic analysis of tumor necrosis factor-α resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008;10:R105. doi: 10.1186/bcr2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72:1290–1300. doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang FM, Liu HQ, Liu SR, Tang SP, Yang L, Feng GS. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1beta in vivo and in vitro. Breast Cancer Res Treat. 2005;89:5–14. doi: 10.1007/s10549-004-1002-z. [DOI] [PubMed] [Google Scholar]
- 42.Pérez-Yépez EA, Ayala-Sumuano JT, Reveles-Espinoza AM, Meza I. Selection of a MCF-7 breast cancer cell subpopulation with high sensitivity to IL-1β: characterization of and correlation between morphological and molecular changes leading to increased invasiveness. Int J Breast Cancer. 2012;2012:609148. doi: 10.1155/2012/609148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franco-Barraza J, Valdivia-Silva JE, Zamudio-Meza H, Castillo A, Garcia-Zepeda EA, Benitez-Bribiesca L, Meza I. Actin cytoskeleton participation in the onset of IL-1beta induction of an invasive mesenchymal-like phenotype in epithelial MCF-7 cells. Arch Med Res. 2010;41:170–181. doi: 10.1016/j.arcmed.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 44.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 45.Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–3456. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomaskovic-Crook E, Thompson EW, Thiery JP. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 2009;11:213. doi: 10.1186/bcr2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vincent-Salomon A, Thiery JP. Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res. 2003;5:101–106. doi: 10.1186/bcr578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kokkinos MI, Wafai R, Wong MK, Newgreen DF, Thompson EW, Waltham M. Vimentin and epithelial-mesenchymal transition in human breast cancer—observations in vitro and in vivo. Cells Tissues Organs. 2007;185:191–203. doi: 10.1159/000101320. [DOI] [PubMed] [Google Scholar]
- 49.Jeong H, Ryu YJ, An J, Lee Y, Kim A. Epithelial-mesenchymal transition in breast cancer correlates with high histological grade and triple-negative phenotype. Histopathology. 2012;60:E87–E95. doi: 10.1111/j.1365-2559.2012.04195.x. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 51.Karihtala P, Auvinen P, Kauppila S, Haapasaari KM, Jukkola-Vuorinen A, Soini Y. Vimentin, zeb1 and Sip1 are up-regulated in triple-negative and basal-like breast cancers: association with an aggressive tumour phenotype. Breast Cancer Res Treat. 2013;138:81–90. doi: 10.1007/s10549-013-2442-0. [DOI] [PubMed] [Google Scholar]
- 52.Liu T, Zhang X, Shang M, Zhang Y, Xia B, Niu M, Liu Y, Pang D. Dysregulated expression of Slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J Surg Oncol. 2013;107:188–194. doi: 10.1002/jso.23240. [DOI] [PubMed] [Google Scholar]
- 53.Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFβ/TNFα-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011;71:4707–4719. doi: 10.1158/0008-5472.CAN-10-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto H, Mukaisho K, Sugihara H, Hattori T, Asano S. Down-regulation of FXYD3 is induced by transforming growth factor-β signaling via ZEB1/δEF1 in human mammary epithelial cells. Biol Pharm Bull. 2011;34:324–329. doi: 10.1248/bpb.34.324. [DOI] [PubMed] [Google Scholar]
- 55.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–724. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- 56.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 57.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korde Choudhari S, Chaudhary M, Bagde S, Gadbail AR, Joshi V. Nitric oxide and cancer: a review. World J Surg Oncol. 2011;11:118. doi: 10.1186/1477-7819-11-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grimm EA, Sikora AG, Ekmekcioglu S. Molecular pathways: inflammation-associated nitric-oxide production as a cancer-supporting redox mechanism and a potential therapeutic target. Clin Cancer Res. 2013;19:5557–5563. doi: 10.1158/1078-0432.CCR-12-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yakovlev VA. Nitric oxide-dependent downregulation of BRCA1 expression promotes genetic instability. Cancer Res. 2013;73:706–715. doi: 10.1158/0008-5472.CAN-12-3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 62.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 63.Medrek C, Landberg G, Andersson T, Leandersson K. Wnt-5a-CKIα signaling promotes β-catenin/E-cadherin complex formation and intercellular adhesion in human breast epithelial cells. J Biol Chem. 2009;284:10968–10979. doi: 10.1074/jbc.M804923200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berdichevsky F, Alford D, D'Souza B, Taylor-Papadimitriou J. Branching morphogenesis of human mammary epithelial cells in collagen gels. J Cell Sci. 1994;107(pt 12):3557–3568. doi: 10.1242/jcs.107.12.3557. [DOI] [PubMed] [Google Scholar]
- 65.Bertazza L, Mocellin S, DeWitte M, Nitti D. TNF and cancer: friends and/or foes? In: Ben-Baruch A, editor. The Inflammatory Milieu of Tumors: Cytokines and Chemokines that Affect Tumor Growth and Metastasis. United Arab Emirates: Bentham Science Publishers; 2012. pp. 29–56. [Google Scholar]
- 66.Neumark E, Cohn MA, Lukanidin E, Witz IP, Ben-Baruch A. Possible co-regulation of genes associated with enhanced progression of mammary adenocarcinomas. Immunol Lett. 2002;82:111–121. doi: 10.1016/s0165-2478(02)00026-3. [DOI] [PubMed] [Google Scholar]
- 67.Neumark E, Sagi-Assif O, Shalmon B, Ben-Baruch A, Witz IP. Progression of mouse mammary tumors: MCP-1-TNFα cross-regulatory pathway and clonal expression of promalignancy and antimalignancy factors. Int J Cancer. 2003;106:879–886. doi: 10.1002/ijc.11337. [DOI] [PubMed] [Google Scholar]
- 68.Seeger H, Wallwiener D, Mueck AO. Different effects of estradiol and various antiestrogens on TNF-α-induced changes of biochemical markers for growth and invasion of human breast cancer cells. Life Sci. 2006;78:1464–1468. doi: 10.1016/j.lfs.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 69.Seeger H, Wallwiener D, Mueck AO. Effects of estradiol and progestogens on tumor-necrosis factor-alpha-induced changes of biochemical markers for breast cancer growth and metastasis. Gynecol Endocrinol. 2008;24:576–579. doi: 10.1080/09513590802288267. [DOI] [PubMed] [Google Scholar]
- 70.Soria G, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, Weitzenfeld P, Meshel T, Shabtai E, Gutman M, Ben-Baruch A. An inflammatory network in breast cancer: coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer. 2011;11:130–149. doi: 10.1186/1471-2407-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeLarco JE, Wuertz BR, Rosner KA, Erickson SA, Gamache DE, Manivel JC, Furcht LT. A potential role for interleukin-8 in the metastatic phenotype of breast carcinoma cells. Am J Pathol. 2001;158:639–646. doi: 10.1016/S0002-9440(10)64005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pantschenko AG, Pushkar I, Miller LJ, Wang YP, Anderson K, Peled Z, Kurtzman SH, Kreutzer DL. In vitro demonstration of breast cancer tumor cell sub-populations based on interleukin-1/tumor necrosis factor induction of interleukin-8 expression. Oncol Rep. 2003;10:1011–1017. [PubMed] [Google Scholar]
- 73.Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008;267:271–285. doi: 10.1016/j.canlet.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 74.Yadav A, Saini V, Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin Chim Acta. 2010;411:1570–1579. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 75.Conti I, Rollins BJ. CCL2 (monocyte chemoattractant protein-1) and cancer. Semin Cancer Biol. 2004;14:149–154. doi: 10.1016/j.semcancer.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 76.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 77.Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008;267:226–244. doi: 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 78.Ali S, Lazennec G. Chemokines: novel targets for breast cancer metastasis. Cancer Metastasis Rev. 2007;26:401–420. doi: 10.1007/s10555-007-9073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teng Y, Mei Y, Hawthorn L, Cowell JK. ASF3 regulates miR-200 inactivation by ZEB1 through suppression of KISS1 leading to increased invasiveness in breast cancer cells. Oncogene. 2013 doi: 10.1038/onc.2012.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, Shang Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012;31:110–123. doi: 10.1038/emboj.2011.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875–883. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun M, Guo X, Qian X, Wang H, Yang C, Brinkman KL, Serrano-Gonzalez M, Jope RS, Zhou B, Engler DA, et al. Activation of the ATM-Snail pathway promotes breast cancer metastasis. J Mol Cell Biol. 2012;4:304–315. doi: 10.1093/jmcb/mjs048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yuen HF, Chan YK, Grills C, McCrudden CM, Gunasekharan V, Shi Z, Wong AS, Lappin TR, Chan KW, Fennell DA, et al. Polyomavirus enhancer activator 3 protein promotes breast cancer metastatic progression through Snail-induced epithelial-mesenchymal transition. J Pathol. 2011;224:78–89. doi: 10.1002/path.2859. [DOI] [PubMed] [Google Scholar]
- 85.Riaz M, Sieuwerts AM, Look MP, Timmermans MA, Smid M, Foekens JA, Martens JW. High TWIST1 mRNA expression is associated with poor prognosis in lymph node-negative and estrogen receptor-positive human breast cancer and is co-expressed with stromal as well as ECM related genes. Breast Cancer Res. 2013;14:R123. doi: 10.1186/bcr3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372–386. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Banerjee A, Wu ZS, Qian P, Kang J, Pandey V, Liu DX, Zhu T, Lobie PE. ARTEMIN synergizes with TWIST1 to promote metastasis and poor survival outcome in patients with ER negative mammary carcinoma. Breast Cancer Res. 2011;13:R112. doi: 10.1186/bcr3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Nes JG, de Kruijf EM, Putter H, Faratian D, Munro A, Campbell F, Smit VT, Liefers GJ, Kuppen PJ, van de Velde CJ, et al. Co-expression of SNAIL and TWIST determines prognosis in estrogen receptor-positive early breast cancer patients. Breast Cancer Res Treat. 2012;133:49–59. doi: 10.1007/s10549-011-1684-y. [DOI] [PubMed] [Google Scholar]
- 89.Yuen HF, Zhang SD, Wong AS, McCrudden CM, Huang YH, Chan KY, El-Tanani M, Khoo US. Regarding “Co-expression of SNAIL and TWIST determines prognosis in estrogen receptor-positive early breast cancer patients”. Breast Cancer Res Treat. 2012;131:351–352. doi: 10.1007/s10549-011-1831-5. [DOI] [PubMed] [Google Scholar]
- 90.Roxanis I. Occurrence and significance of epithelial-mesenchymal transition in breast cancer. J Clin Pathol. 2013;66:517–521. doi: 10.1136/jclinpath-2012-201348. [DOI] [PubMed] [Google Scholar]
- 91.Montserrat N, Gallardo A, Escuin D, Catasus L, Prat J, Gutierrez-Avigno FJ, Peiro G, Barnadas A, Lerma E. Repression of E-cadherin by SNAIL, ZEB1, and TWIST in invasive ductal carcinomas of the breast: a cooperative effort? Hum Pathol. 2011;42:103–110. doi: 10.1016/j.humpath.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 92.Sirohi B, Leary A, Johnston SR. Ipsilateral breast tumor recurrence: is there any evidence for benefit of further systemic therapy? Breast J. 2009;15:268–278. doi: 10.1111/j.1524-4741.2009.00716.x. [DOI] [PubMed] [Google Scholar]
- 93.Abd-Alla HM, Lotayef MM, Abou Bakr A, Moneer MM. Ipsilateral in-breast tumor relapse after breast conservation therapy: true recurrence versus new primary tumor. J Egypt Natl Canc Inst. 2006;18:183–190. [PubMed] [Google Scholar]
- 94.Nishimura R, Osako T, Okumura Y, Tashima R, Toyozumi Y, Arima N. Changes in the ER, PgR, HER2, p53 and Ki-67 biological markers between primary and recurrent breast cancer: discordance rates and prognosis. World J Surg Oncol. 2011;9:131. doi: 10.1186/1477-7819-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.West NR, Panet-Raymond V, Truong PT, Alexander C, Babinszky S, Milne K, Ross LA, Loken S, Watson PH. Intratumoral immune responses can distinguish new primary and true recurrence types of ipsilateral breast tumor recurrences, (IBTR) Breast Cancer (Auckl) 2011;5:105–115. doi: 10.4137/BCBCR.S7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8:197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 97.Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009;14:29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.