

Abstract
Activating mutations of the epidermal growth factor receptor (EGFR) occur in multiple tumor types, including non-small cell lung cancer (NSCLC) and malignant glioma, and have become targets for therapeutic intervention. The determination of EGFR mutation status using a noninvasive, molecular imaging approach has the potential for clinical utility. In this study, we investigated [11C]-erlotinib positron emission tomography (PET) imaging as a tool to identify activating mutations of EGFR in both glioma and NSCLC xenografts. Radiotracer specific binding was determined for high and low specific activity (SA) [11C]-erlotinib PET scans in mice bearing synchronous human cancer xenografts with different EGFR expression profiles (PC9, HCC827, U87, U87 ΔEGFR, and SW620). Although xenograft immunohistochemistry demonstrated constitutive EGFR phosphorylation, PET scan analysis using the Simplified Reference Tissue Model showed that only kinase domain mutant NSCLC (HCC827 and PC9) had significantly greater binding potentials in high versus low SA scans. Xenografts with undetectable EGFR expression (SW620), possessing wild-type EGFR (U87), and expressing an activating extracellular domain mutation (U87 ΔEGFR) were indistinguishable under both high and low SA scan conditions. The results suggest that [11C]-erlotinib is a promising radiotracer that could provide a novel clinical methodology for assessing EGFR and erlotinib interactions in patients with tumors that harbor EGFR-activating kinase domain mutations.
Introduction
The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein commonly overexpressed by malignant tumors. Ligand stimulation of the EGFR extracellular domain causes receptor dimerization, activates the intracellular kinase domain, and drives downstream signal transduction cascades that promote proliferation, migration, angiogenesis, and survival. In addition to ligand-induced activation, genetic mutation or deletion also produces constitutively active forms of the receptor that drive tumor signaling [1,2]. In malignant glioma, in-frame deletion of EGFR exons 2 to 7 removes extracellular domain constraints for receptor activation and is found in 40% of patients [3]. In non-small cell lung cancer (NSCLC), tyrosine kinase domain deletions (or point mutations) increase kinase activity and are found in 10% to 15% and up to 50% of Western and Asian populations, respectively [4,5]. The relative frequency of activating EGFR mutations found in tumors along with the central role of EGFR in stimulating cell signaling have therefore identified this receptor as a therapeutic target in oncology.
Successful EGFR targeting has been achieved clinically with either receptor-specific antibodies such as cetuximab [6], which depletes EGFR from the cell surface, or small molecule tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib, which are ATP-competitive inhibitors of the kinase domain active site [7]. Both antibody and TKI-based EGFR-specific therapies have been exploited and adapted as potential imaging agents in the diagnosis and management of patients with malignant tumors [8–10]. Recently, EGFR TKIs with either reversible or irreversible binding characteristics have been radio-labeled and investigated as potential radiotracers in animal models [11–13]. Of these tracers, the most promising appears to be [11C]-erlotinib, which has a structure identical to the clinically used drug. This tracer has been advanced to clinical investigations in patients with NSCLC and has been proposed to detect tumors bearing EGFR mutations [14,15].
Although preliminary work on this positron emission tomography (PET) imaging strategy is encouraging, the extent to which [11C]-erlotinib can be used to image tumors with activated EGFR remains undefined. In addition, the optimal method for kinetic modeling and image analysis for [11C]-erlotinib PET also requires further investigation. In the present report, we tested the ability of [11C]-erlotinib to selectively detect EGFR expression in mouse tumor xenograft models. Using NSCLC and glioma xenografts expressing different EGFR-activating gene mutations, we performed kinetic modeling analysis using the Simplified Reference Tissue Model (SRTM) under conditions of both high and low specific activities of the radiotracer. Our analysis and quantitative comparisons of PET scans between NSCLC and glioma xenografts suggest that [11C]-erlotinib imaging provides a noninvasive method for determining the density of kinase domain mutant EGFR expression in tumors.
Materials and Methods
Western Blot Analysis
Western blots were performed by standard methods. The C-terminal EGFR antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA; SC-03). EGF was purchased from Biomedical Technologies (Stoughton, MA) and used to stimulate cells for 10 minutes at a final concentration of 10 ng/ml.
Xenografts and Immunohistochemistry
Xenografts were initiated in athymic, nude-Foxn1nu mice (Harlan, South Easton, MA) by subcutaneous injection of 1 x 107 cells in the flank or shoulder. The SW620 cell line was purchased from American Tissue Culture Collection (ATCC, Manassas, VA). The PC9 and HCC827 cell lines were gifts from Katie Politi (Yale University, New Haven, CT) and Jeff Engelman (Harvard Medical School, Boston, MA), respectively. The U87 control and U87 ΔEGFR (EGFR VIII expression) cell lines were a gift from Webster Cavanee (University of California, San Diego, CA). Tumors were grown over 10 to 14 days until a maximal dimension of ∼1.0 cm for PET imaging is reached. For immunohistochemistry (IHC), xenografts were dissected and fixed in formalin-buffered saline for 24 hours followed by 70% ethanol before paraffin embedding. Tumor sections were deparaffinized and rehydrated in distilled water with 3% hydrogen peroxide to block endogenous peroxidase. Antigenic sites were exposed using Proteinase K. Primary antibody incubation was then performed for 30 minutes followed by incubation with secondary antibody conjugates. The primary EGFR antibody (clone 111.5) recognizing the extracellular domain was from Thermo Fisher Scientific (Lafayette, CO), and the EGFR Y1173-specific antibody was from Cell Signaling Technology (Danvers, MA). Immunoreactivity was then detected with HRP-conjugated secondary antibodies using Envision+ and DAB (Dako, Carpenteria, CA). Slides were subsequently counterstained with hematoxylin and permanently cover-slipped in resin mounting media.
[11C]-Erlotinib Synthesis
Radiosynthesis of [11C]-erlotinib was carried out with a labeling method similar to that reported previously [9]. In brief, 1 to 5 mg of 6-O-desmethyl-erlotinib was dissolved in N,N-dimethylformamide (0.3 ml) and mixed with sodium hydride. The solution was degassed with argon for 5 minutes before bubbling with [11C]-methyl iodide generated from [11C]-CO2 in a General Electric FX MeI module. The resulting solution was heated at 120°C for 5 minutes, diluted with the semi-preparative HPLC mobile phase (1.5 ml), and loaded onto the HPLC column (Phenomenex Luna C18, 10 µm, 10 x 250 mm). The column was eluted with a mixture of 45% acetonitrile and 55% 0.1 M ammonium acetate with 0.1% acetic acid (pH = 5.5), at a flow rate of 5 ml/min. The desired product fraction (eluting at 15-16 minutes) was collected, diluted with water (50 ml), and passed through a C18 SepPak cartridge (Waters, Milford, MA). The cartridge was rinsed with 0.001 N HCl (10 ml). The radioactive product, trapped on the SepPak, was recovered by elution with 1 ml of United States Pharmacopeial (USP) Convention absolute ethanol, followed by 3 ml of USP saline, into a product vial containing 7 ml of USP saline and 40 µl of USP 4.2% NaH2CO3 solution. This mixture was then passed through a sterile membrane filter (0.22 µm) for terminal sterilization and collected in an empty sterile vial to afford a formulated solution ready for injection. Radiochemical purity of the final product was >99%, and the specific activity (SA) was 4.3 ± 1.3 mCi/nmol (n = 9) at the end of synthesis.
Identity of the product was confirmed by co-elution of the radioactive product with an authentic reference standard, as determined by HPLC analysis (Column: Luna, C18, 5 µm, 4.6 x 250 mm; mobile phase: mixture of 42% acetonitrile and 58% 0.1 M ammonium acetate with 0.1% acetic acid, pH = 5.5; flow rate = 2 ml/min). The retention time for [11C] erlotinib was ∼7.5 minutes.
[11C]-Erlotinib PET Scans
Mice bearing xenografts were anesthetized with inhaled isoflurane (2%) throughout the course of image acquisition. The mice were imaged in pairs with torsos positioned in the scanner's field of view to optimize resolution of the flanks and shoulders. A 100-W heating lamp was positioned above the scan bed to maintain body temperature. Mice were imaged with a Siemens Focus 220 PET scanner, which has a spatial resolution of 1.4 mm at the field of view center. Before imaging, a computed tomography (CT) transmission scan using a rotating Cobalt-57 point source was performed for attenuation correction and to establish mouse orientation. Image acquisition was initiated immediately after delivery of a retro-orbital sinus injection of the tracer. High SA scans delivered [11C]-erlotinib at an activity of 0.16 ± 0.12 mCi and mass of 9.31 ± 5.16 nmol. Low SA scans, where unlabeled erlotinib (10 mg/kg; 465 µmol) was combined with the [11C]-erlotinib tracer, had an injected activity of 0.06 ± 0.03 mCi. Low SA scans were performed on the same day 4 hours (>10 half-lives) after the high SA scans. During emission scans, three-dimensional (3D) data were acquired in list mode for up to 120 minutes and Fourier rebinned as two-dimensional (2D) sinograms with time frames of 7 x 30 seconds, 5 x 90 seconds, and 21 x 300 seconds. Dynamic images were reconstructed with ordered subset expectation maximization at a voxel size of 0.949 x 0.949 x 0.796 mm. Corrections for attenuation, scatter, dead time, detector sensitivity, and randoms were applied to emission data. Images were reconstructed in volumes of 256 x 256 x 95 voxels.
Kinetic Modeling
Regions of interest (ROIs) for xenografts were manually drawn using anatomic location and summed images for late (95–120 minutes) or total (0–120 minutes) [11C]-erlotinib PET scans. In larger tumors, round central areas devoid of tracer uptake were thought to be consistent with tumor necrosis and were excluded from analysis. [11C]-erlotinib time-activity curves (TACs) were generated for each ROI by averaging voxel intensity at each time point. TACs are given in units of radioactivity concentration. For the purpose of display, we used standard uptake value (SUV) units. That is, we normalized the TACs by the injected activity per body weight so that curves from different animals could be properly displayed on one scale. The SRTM [16] was used to fit the TACs of each region, and the shoulder muscle without a xenograft was used as the reference region. SRTM fitting of TACs yielded an estimate of non-displaceable binding potential (BPND, abbreviated here as BP), perfusion rate (R1), and efflux rate constant (k2a) in the ROI. BP is the primary parameter of interest as it approximates the steady-state ratio of free to bound tracer in the tissue. BP also depicts the ratio of available receptors (Bavail) to the tracer's affinity for the receptors (Kd). R1 describes the regional tracer delivery to the ROI relative to the reference region. The [11C]-erlotinib BP was averaged, and comparisons were made across xenograft types (e.g., PC9 vs HCC827) or SA conditions (e.g., high SA vs low SA). As an alternate end point, SUV was calculated for each ROI and condition by averaging the TAC values in SUV units from the first 90 minutes. All statistical comparisons were made using the Student's t test (one-tailed). Comparisons with P < .05 were considered to be statistically significant.
Results
EGFR Phosphorylation in Tumor Xenograft Models
We used both lung and glioma xenograft tumor models to investigate [11C]-erlotinib imaging of EGFR-activating mutations in vivo. Xenografts were first evaluated for EGFR activation with IHC using an EGFR-specific antibody that recognizes phosphorylation of tyrosine 1173 (Y1173), a primary EGFR auto-phosphorylation site. HCC827 and PC9 NSCLC xenografts, both containing in-frame EGFR kinase domain deletions (E746-A750), displayed high levels of phospho-Y1173 immunoreactivity consistent with receptor activation (Figure 1A). In contrast, the SW620 colon cancer cell line, which has virtually undetectable levels of EGFR, had no phospho-Y1173 immunoreactivity. The control U87 glioma and ΔEGFR-expressing U87 glioma xenografts were also analyzed by IHC (Figure 1B). The ΔEGFR-expressing U87 xenografts showed a high level of EGFR Y1173 phosphorylation compared to the low level seen in control U87 tumors. These results are consistent with in vitro experiments using these cell lines, which demonstrate high levels of constitutive ΔEGFR Y1173 phosphorylation and low but EGF-inducible Y1173 phosphorylation of the endogenous, wild-type receptor (Figure 1C). These IHC experiments demonstrate EGFR activation in HCC827 and PC9 NSCLC as well as ΔEGFR-expressing U87 xenograft model systems.
Figure 1.

Xenograft tumor models for [11C]-erlotinib PET imaging. (A) Xenograft IHC using EGFR and tyrosine (Y) 1173 phosphorylation-specific antibodies. Representative images from colon cancer-derived (SW620) and NSCLC-derived (HCC827 and PC9) xenografts are shown. (B) IHC of EGFR-phosphorylated Y1173 in U87 glioma xenografts with and without expression of the extracellular domain-truncated ΔEGFR. (C) Western blot analysis demonstrating constitutive or EGF-induced EGFR Y1173 phosphorylation in U87 cell lines.
[11C]-Erlotinib PET Imaging
[11C]-erlotinib PET imaging was performed as described in Materials and Methods section. Examples of a transmission scan (Figure 2A) and summed PET images (Figure 2, B and C) used to define the anatomic locations of xenografts for ROI definitions (Figure 2D) are presented. ROI parameters for data acquisition are presented in Table 1 for scans with both high and low SA images in planes containing the xenografts, and the reference ROIs demonstrate considerable [11C]-erlotinib accumulation at the site of flank xenografts as well as a central accumulation of the tracer within the mouse. The latter finding corresponds to hepatic metabolism and gastrointestinal tract excretion of erlotinib [17,18].
Figure 2.

[11C]-erlotinib imaging of a mouse bearing bilateral HCC827 and PC9 bilateral flank xenografts. (A) Transmission scan of the mouse used to gain anatomic information for ROI placement. (B) [11C]-erlotinib PET early time image summed from 0 to 10 minutes. PET images are smoothed with a 1.0-mm3 kernel for illustrative purposes. (C) [11C]-erlotinib PET late time image summed from 95 to 120 minutes. (D) ROIs (blue) corresponding to flank xenograft location used for data acquisition and TAC analysis.
Table 1.
[11C]-Erlotinib Tumor Imaging and Data Acquisition Parameters for Both High and Low SA Conditions.
| Xenograft | EGFR Properties | SA | n | Voxels | R1 | BP | SUV | ||||
| SW620 | WT* | High | 1 | 100 | n/a | 1 | n/a | -1 | n/a | 0.28 | n/a |
| HCC827 | Exon 19 del (E746-A750) | 3 | 168 | (13) | 1.12 | (0.22) | 6.38 | (1.90) | 0.91 | (0.60) | |
| PC9 | Exon 19 del (E746-A750) | 3 | 82 | (50) | 0.787 | (0.27) | 2.45 | (1.72) | 0.45 | (0.31) | |
| U87 | WT | 3 | 102 | (16) | 1.3 | (0.07) | -0.69 | (0.53) | 0.51 | (0.56) | |
| U87Δ | ΔEGFR | 3 | 124 | (23) | 0.23 | (0.08) | -0.13 | (0.20) | 0.46 | (0.29) | |
| SW620 | WT* | Low | 1 | 42 | n/a | 1.24 | n/a | -1 | n/a | 0.08 | n/a |
| HCC827 | Exon 19 del (E746-A750) | 3 | 153 | (57) | 0.49 | (0.35) | 0.38 | (0.27) | 0.93 | (1.33) | |
| PC9 | Exon 19 del (E746-A750) | 3 | 72 | (49) | 0.53 | (0.53) | 0.12 | (0.40) | 0.48 | (0.67) | |
| U87 | WT | 2 | 100 | (25) | 1.75 | (0.58) | -0.43 | (0.81) | 0.33 | (0.30) | |
| U87Δ | ΔEGFR | 2 | 106 | (62) | 0.57 | (0.27) | 0.16 | (0.79) | 0.24 | (0.16) | |
The mean number of voxels per ROI is reported along with SRTM calculated parameters and SUVs calculated at 90 minutes. Average R1 and BP values are used to quantify delivery of [11C]-erlotinib and receptor density, respectively. All values are shown as an average across sample size n with the SD in parentheses.
The WT EGFR of SW620 is not expressed at detectable levels.
[11C]-Erlotinib Selectivity
To evaluate the selectivity of [11C]-erlotinib for imaging EGFR-activating mutations, TACs were generated for each xenograft type using both high and low SA tracer preparations. Sample TACs for a mouse bearing synchronous HCC827 and PC9 NSCLC xenografts show sustained accumulation of tracer over time in the high SA condition (Figure 3A). HCC827 xenografts show quantitatively higher tracer concentrations than PC9 xenografts for comparable doses. In contrast, the SW620 xenograft shows only an initial uptake of tracer followed by a washout phase that begins approximately 20 minutes into the scan. This pattern of rapid uptake and washout was identical to the muscle TAC (the curves are superimposable on Figure 3A). In contrast to the high SA scans, scans with concurrent administration of excess unlabeled erlotinib (“low SA scans”) did not display prolonged tissue uptake over time or appreciable differences between any of the xenografts and the reference ROI (Figure 3C).
Figure 3.
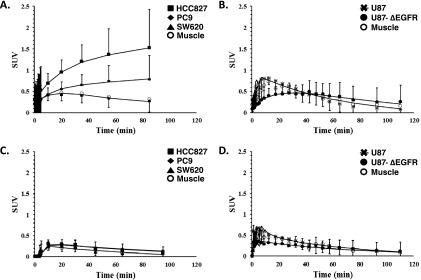
[11C]-erlotinib TACs in mice bearing multiple tumor xenografts for mice imaged under high (A and B) or low (C and D) SA conditions. TACs for HCC827 (squares), PC9 (diamonds), and SW620 (triangles) or U87 (asterisks) and U87Δ (closed circles) xenografts with muscle (open circles) used as reference. Data points are SUVs with SD, and overlaid lines depict SRTM curve fits.
In PET imaging experiments performed with U87 glioma xenografts, specific [11C]-erlotinib tracer uptake was not observed irrespective of ΔEGFR expression (Figure 3B). The early increase in tracer uptake (0-20 minutes) in tumor was not different from the reference ROI, and the same pattern for all curves was seen in low SA imaging experiments (Figure 3D) as well. Together, these data suggested that [11C]-erlotinib accumulates in NSCLC tumors with activating kinase domain mutations but does not accumulate in gliomas with an extracellular domain-activating mutation. Further kinetic modeling was therefore performed.
[11C]-Erlotinib Kinetic Modeling
TACs for each xenograft type were generated and fit according to the SRTM to estimate [11C]-erlotinib BPs for each tumor type. The calculated BP values are presented graphically in Figure 4 for both high and low SA scan conditions. For high SA conditions, the HCC827 and PC9 NSCLC xenografts had the highest BP values: 6.38 (±1.9) and 2.45 (±1.72), respectively. HCC827 BP values were significantly greater in the high SA scans than in low SA scans (P < .02). Differences between high SA and low SA BPs for PC9 trended toward significance (P = .075). BP values of HCC827 (high SA) were also significantly greater than those for PC9 (P < .03). There was no significant difference in BP estimates for U87, U87 ΔEGFR, or SW620 xenografts demonstrating the absence of selective [11C]-erlotinib accumulation in these tumor types.
Figure 4.

Average BP of [11C]-erlotinib determined by SRTM in both high SA (black) and low SA (white) conditions. (A) Comparison of average BPs for SW620, HCC827, and PC9 xenografts. (B) Average BPs for U87 and U87 ΔEGFR tumors. Error bars represent the SD. Significant differences are reported in Results section.
Discussion
Small molecule inhibitors of protein kinases, enzymes that drive survival signaling in tumors, are a relatively new class of therapeutics that have been developed in oncology over the past two decades. The sheer number and availability of these targeted inhibitors have provided considerable opportunity for exploring their utility as molecular imaging agents. In the present work, we investigated the potential for imaging the biology of EGFR using 11C-labeled erlotinib, an EGFR-specific TKI. [11C]-erlotinib PET imaging was tested in xenograft tumor models expressing common EGFR mutations that cause receptor activation and signaling. Our results demonstrate that [11C]-erlotinib binds to a target in xenografts expressing EGFR with kinase domain mutations but does not bind in those bearing an activating mutation in the extra-cellular domain (ΔEGFR). Further modeling of dynamic PET data using SRTM, a standard kinetic model of tracer uptake, demonstrated significant amounts of specific tracer binding to xenografts containing kinase domain mutant EGFR. This finding is important as it suggests that [11C]-erlotinib PET images show the density of one type of mutant receptors expressed in a tumor. Together, these data support the validity of this new molecular imaging tool and provide mechanistic insights for the clinical implementation of [11C]-erlotinib for tumor imaging.
[11C]-erlotinib PET scans of NSCLC and glioma xenograft tumors only demonstrated specific radiotracer binding in NSCLC xenografts. Although EGFR is active and expressed at high levels in the U87 ΔEGFR cell line, we were not able to detect specific binding of the tracer. Instead, [11C]-erlotinib PET results for U87 ΔEGFR were similar to those of control U87 xenografts that express only the endogenous, wild-type receptor. The discrepant results between EGFR mutants expressed in NSCLC tumors versus malignant gliomas are best explained by the nature of NSCLC EGFR kinase domain mutations, which have increased affinities for ATP-competitive small molecule inhibitors [19,20]. Our data strongly suggest that the selectivity of [11C]-erlotinib imaging for NSCLC xenografts is independent of wild-type EGFR expression or activation and is instead dependent on the molecular and biophysical alteration caused by EGFR kinase domain mutations.
EGFR is a prototypical receptor tyrosine kinase implicated in tumorigenesis, proliferation, and survival signaling and has served as a model for kinase targeting and the development of novel therapeutics. However, because the utility of [11C]-erlotinib PET appears to be limited to detection of mutant EGFR with increased inhibitor affinity, kinase inhibitor radiolabeling may not be a generally applicable approach for imaging other clinically relevant kinases. We predict, for example, that radiolabeled kinase inhibitors would not be suitable for imaging other types of kinase activating mutations that do not change receptor affinity, such as those seen with Kit in gastrointestinal stromal tumors [21]. Furthermore, reversible kinase inhibitors are likely to be poor imaging agents for kinase domain-intact receptors such as the fibroblast growth factor receptor and ErbB2 that are amplified and overexpressed in 15% of squamous NSCLC and 30% of breast cancers, respectively. New PET imaging strategies to image the amplification of EGFR or other protein kinases will thus require an approach that exploits the differential expression levels between tumors and normal tissues, such as the investigation of irreversible kinase inhibitors [22,23] or through the design of high affinity and mutation-specific kinase inhibitors [24] as PET radiotracers.
A major advantage of our experimental design is the use of synchronous flank xenografts. This approach reduced animal-specific variability in tracer bioavailability and metabolism, which often confound interanimal comparisons. Furthermore, our experiments were performed in both high and low SA conditions. In the low SA condition, excess unlabeled erlotinib leads to a measure of nonspecific (“non-displaceable”) tracer uptake only. Recently, Bahce et al. emphasized the need for low SA studies to confirm their clinical observations of [11C]-erlotinib uptake by NSCLC tumors [15], because performing both high and low SA injections in patients is impractical. Thus, our animal experiments take on even higher significance by demonstrating both [11C]-erlotinib selectivity for EGFR-mutant NSCLC and receptor-mediated binding through the comparison between high SA (tracer) and low SA (blocking) studies. Our study design also facilitates comparisons between HCC827 and PC9 xenografts, where a significant difference in BP between the two tumor types was observed. Because the two NSCLC xenografts express identical EGFR kinase domain deletions (E746-A750), we assume that the affinity of the tracer for the receptor is the same in each tumor. Thus, differences in BP between the NSCLC tumor types can be interpreted to reflect unequal levels of expression of mutant EGFR. The results from IHC presented herein as well as DNA copy number analysis demonstrating greater gene amplification and protein expression in HCC827 versus PC9 provide additional support for this interpretation [2,25].
The use of the SRTM leads to estimation of BP without the need for arterial blood sampling. However, SRTM must be applied with care as it assumes that the reference region differs only in its lack of binding sites for the tracer and is otherwise identical to the target region. Said another way, the nonspecific volume of distribution is assumed to be the same in the target and reference. In our experiments, we initially used a synchronous SW620 xenograft, a tumor with no detectable EGFR expression, as the preferred reference tissue. However, once subsequent analysis using the shoulder muscle as a reference was found to be comparable to that using SW620, the easier approach using healthy muscle as a reference was adopted. SRTM also neglects vascular-borne activity in the target and reference tissues [16]. As we anticipate human imaging, we must keep in mind that differential vascular growth between tumors and healthy normal tissue may lead to biases in SRTM-based estimates of BP. It will be necessary to validate the use of SRTM against models using arterial- or image-derived input functions [15]. Provided that assumptions of the model are met and biases are minimal, it is always preferable to estimate BP rather than simply SUV. Whereas SUV makes no distinction between nonspecific versus specific (receptor-mediated) uptake, BP is a measure of the latter only. Thus, BP will be more sensitive than SUV to differences in expression level of the target (i.e., differences in receptor density). This advantage is reflected in the statistical significance of the difference in BP values across tumor types but lack of significance when looking at SUV.
Further clinical experiments to validate [11C]-erlotinib as a sensitive radiotracer to detect kinase domain mutations of the EGFR are required, but our results strongly suggest that this tracer may present applications for the management of patients with NSCLC. Notwithstanding the greater convenience of a tracer labeled with fluorine-18 for use in the clinic, [11C]-erlotinib PET could be a useful diagnostic tool for select patients undergoing surveillance of enlarging lung nodules that are negative on fluorodeoxyglucose (FDG)-PET or those patients unable to undergo standard biopsy because of risks associated with the procedure. In addition, [11C]-erlotinib PET may provide clinically relevant information that complements molecular pathology analyses by simultaneous quantification of kinase domain mutation density at multiple involved or metastatic sites. The predicted and reported absence [10] of selective tracer accumulation for labeled erlotinib analogs with T790M mutations (secondary EGFR kinase domain mutations that mediate therapeutic resistance by decreasing affinity for erlotinib) suggests that this EGFR imaging strategy could detect either primary or acquired resistance to EGFR inhibitors. In the clinic, [11C]-erlotinib PET has the potential to define anatomic sites of resistance and even alter treatment recommendations. However, we recognize that [11C]-erlotinib does have anatomic limitations, as demonstrated in our study by hepatic metabolism and gastrointestinal excretion of the tracer, which would limit its use in imaging tumors in these sites.
In summary, we have performed [11C]-erlotinib PET imaging in NSCLC and glioma xenografts and found specific binding of the tracer for activating mutations of the kinase domain but no specific binding for activating mutations of the extracellular domain of the EGFR. Together, our work further characterizes the mechanistic basis for translation of this PET imaging modality for clinical use.
Footnotes
This work was supported by a grant from the Yale Clinical Center for Investigation Scholar Award (UL1RR024139/KL2RR024138) to J.N.C., the Kalimeris fund, and support from the Yale PET Center. J.R.P. is supported by the National Science Foundation Graduate Research Fellowship under grant DGE-1122492. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation.
References
- 1.Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart Salmon J, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–235. [PubMed] [Google Scholar]
- 2.Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, Coldren C, Barón A, Zeng C, Franklin WA, et al. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res. 2006;12:7117–7125. doi: 10.1158/1078-0432.CCR-06-0760. [DOI] [PubMed] [Google Scholar]
- 3.Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, Bhat K, McDonald JM, Yung WK, Colman H, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25:2288–2294. doi: 10.1200/JCO.2006.08.0705. [DOI] [PubMed] [Google Scholar]
- 4.Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Jänne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 5.Yang CH, Yu CJ, Shih JY, Chang YC, Hu FC, Tsai MC, Chen KY, Lin ZZ, Huang CJ, Shun CT, et al. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J Clin Oncol. 2008;26:2745–2753. doi: 10.1200/JCO.2007.15.6695. [DOI] [PubMed] [Google Scholar]
- 6.Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11:21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 7.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 8.Nayak TK, Garmestani K, Milenic DE, Baidoo KE, Brechbiel MW. HER1-targeted 86Y-panitumumab possesses superior targeting characteristics than 86Y-cetuximab for PET imaging of human malignant mesothelioma tumors xenografts. PLoS One. 2011;6:e18198. doi: 10.1371/journal.pone.0018198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E. Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009;69:873–878. doi: 10.1158/0008-5472.CAN-08-3118. [DOI] [PubMed] [Google Scholar]
- 10.Yeh HH, Ogawa K, Balatoni J, Mukhapadhyay U, Pal A, Gonzalez-Lepera C, Shavrin A, Soghomonyan S, Flores L, II, Young D, et al. Molecular imaging of active mutant L858R EGF receptor (EGFR) kinase-expressing nonsmall cell lung carcinomas using PET/CT. Proc Natl Acad Sci USA. 2011;108:1603–1608. doi: 10.1073/pnas.1010744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vasdev N, Dorff PN, O'Neil JP, Chin FT, Hanrahan S, VanBrocklin HF. Metabolic stability of 6, 7-dialkoxy-4-(2-, 3- and 4-[18 F]fluoroanilino) quinazolines, potential EGFR imaging probes. Bioorg Med Chem. 2011;19:2959–2965. doi: 10.1016/j.bmc.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pisaneschi F, Nguyen QD, Shamsaei E, Glaser M, Robins E, Kaliszczak M, Smith G, Spivey AC, Aboagye EO. Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: synthesis and biological evaluation. Bioorg Med Chem. 2010;18:6634–6645. doi: 10.1016/j.bmc.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Samen E, Thorell JO, Fredriksson A, Stone-Elander S. The tyrosine kinase inhibitor PD153035: implication of labeling position on radiometabolites formed in vitro. Nucl Med Biol. 2006;33:1005–1011. doi: 10.1016/j.nucmedbio.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Memon AA, Weber B, Winterdahl M, Jakobsen S, Meldgaard P, Madsen HH, Keiding S, Nexo E, Sorensen BS. PET imaging of patients with non-small cell lung cancer employing an EGF receptor targeting drug as tracer. Br J Cancer. 2011;105:1850–1855. doi: 10.1038/bjc.2011.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahce I, Smit EF, Lubberink M. Development of [11C] erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status. Clin Cancer Res. 2013;19:183–193. doi: 10.1158/1078-0432.CCR-12-0289. [DOI] [PubMed] [Google Scholar]
- 16.Lammertsma AA, Bench CJ, Hume SP, Osman S, Gunn K, Brooks DJ, Frackowiak RS. Comparison of methods for analysis of clinical [11C]raclopride studies. J Cereb Blood Flow Metab. 1996;16:42–52. doi: 10.1097/00004647-199601000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13:3731–3737. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 18.Ling J, Johnson KA, Miao Z, Rakhit A, Pantze MP, Hamilton M, Lum BL, Prakash C. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab Dispos. 2006;34:420–426. doi: 10.1124/dmd.105.007765. [DOI] [PubMed] [Google Scholar]
- 19.Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N, Sliwkowski MX. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–8171. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]
- 20.Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, Anderson KS, Settleman J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67:2325–2330. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 21.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- 22.Mishani E, Abourbeh G, Jacobson O, Dissoki S, Ben Daniel R, Rozen Y, Shaul M, Levitzki A. High-affinity epidermal growth factor receptor (EGFR) irreversible inhibitors with diminished chemical reactivities as positron emission tomography (PET)-imaging agent candidates of EGFR overexpressing tumors. J Med Chem. 2005;48:5337–5348. doi: 10.1021/jm0580196. [DOI] [PubMed] [Google Scholar]
- 23.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 25.Okabe T, Okamoto I, Tamura K, Terashima M, Yoshida T, Satoh T, Takada M, Fukuoka M, Nakagawa K. Differential constitutive activation of the epidermal growth factor receptor in non-small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res. 2007;67:2046–2053. doi: 10.1158/0008-5472.CAN-06-3339. [DOI] [PubMed] [Google Scholar]