

Abstract
Pancreatic cancer is characterized by a high degree of resistance to chemotherapy. Epidermal growth factor receptor (EGFR) inhibition using the small-molecule inhibitor erlotinib was shown to provide a small survival benefit in a subgroup of patients. To identify kinases whose inhibition acts synergistically with erlotinib, we employed a kinome-wide small-interfering RNA (siRNA)-based loss-of-function screen in the presence of erlotinib. Of 779 tested kinases, we identified several targets whose inhibition acted synergistically lethal with EGFR inhibition by erlotinib, among them the S6 kinase ribosomal protein S6 kinase 2 (RPS6KA2)/ribosomal S6 kinase 3. Activated RPS6KA2 was expressed in approximately 40% of 123 human pancreatic cancer tissues. RPS6KA2 was shown to act downstream of EGFR/RAS/mitogen-activated protein kinase kinase (MEK)/extracellular-signal regulated kinase (ERK) signaling and was activated by EGF independently of the presence of KRAS mutations. Knockdown of RPS6KA2 by siRNA led to increased apoptosis only in the presence of erlotinib, whereas RPS6KA2 activation or overexpression rescued from erlotinib- and gemcitabine-induced apoptosis. This effect was at least in part mediated by downstream activation of ribosomal protein S6. Genetic as well as pharmacological inhibition of RPS6KA2 by the inhibitor BI-D1870 acted synergistically with erlotinib. By applying this synergistic lethality screen using a kinome-wide RNA interference-library approach, we identified RPS6KA2 as potential drug target whose inhibition synergistically enhanced the effect of erlotinib on tumor cell survival. This kinase therefore represents a promising drug candidate suitable for the development of novel inhibitors for pancreatic cancer therapy.
Introduction
Pancreatic cancer is associated with the worst prognosis of all solid tumors [1] and is characterized by a largely drug-resistant phenotype. Histologically, the majority of pancreatic cancers are ductal adenocarcinomas that frequently express high levels of the epidermal growth factor receptor (EGFR) [2]. EGFR-dependent signaling cascades lead to enhanced cell cycle progression, cell growth, angiogenesis, and survival. To date, inhibition of the EGFR pathway by the small-molecule inhibitor erlotinib represents the only targeted therapy approved for metastatic pancreatic ductal adenocarcinoma [3]. This approval was based on a clinical trial demonstrating a significant survival benefit in patients receiving erlotinib in combination with gemcitabine compared to patients treated with gemcitabine plus placebo [4]. The benefit, however, is only marginal for the majority of patients. Interestingly, a subgroup of patients that develops a skin rash as side effect of erlotinib therapy seems to have a clinically more significant survival advantage [4]. The underlying molecular mechanism behind this observation remains to be fully elucidated [5]. Moreover, it is not entirely clear at this point whether the erlotinib-induced rash is predictive of treatment response to erlotinib or serves merely as a prognostic factor reflecting a more favorable tumor biology [5].
It is evident that the efficacy of erlotinib in combination with gemcitabine is clinically not satisfying and restricted to only a small subgroup of patients. Identifying additional targets whose inhibition might act synergistically with erlotinib thereby providing a substantial benefit for a larger group of patients is crucial to improve the devastating prognosis of pancreatic cancer.
Screening approaches to identify novel target proteins with functional impact on cardinal hallmarks of cancer such as tumor cell survival, invasiveness, and proliferation are essential to identify novel targets in an unbiased manner. Loss-of-function screens based on RNA interference (RNAi) libraries represent a powerful tool to identify new potential therapeutically relevant targets in this context [6,7]. Synthetic lethality screens aim to identify targets whose knockdown acts synergistically with another compound or a distinct genetic state [6].
Ribosomal S6 kinases (RSKs) represent a family of serine/threonine protein kinases with a molecular weight of 90 kDa. RSK was first discovered in Xenopus by Erikson and Maller who also identified the ribosomal protein S6 (rpS6) as physiological target of RSK [8]. To date, the RSK family comprises four members (RSK1 to RSK4) [9]. RSK1 and RSK2 have been described to mediate cell survival, motility, and proliferation [10,11]. RSK3, also known as ribosomal protein S6 kinase 2 (RPS6KA2), has been implicated in cell cycle progression [12]. In ovarian cancer, however, it has been reported that RSK3 may act as a tumor suppressor [13]. The RSK3 gene is located on chromosome 6q27 and highly expressed in lung, heart, muscle, and brain [9]. The RSK3 protein is composed of 733 amino acids and contains two functional domains, a carboxyl-terminal kinase domain and an amino-terminal kinase domain. RSK3 is activated by EGF-dependent signaling pathways through mitogen-activated protein kinase kinase/extracellular-signal regulated kinase (MEK)/(ERK) [12,14] and localized in its inactive form in the cytoplasm. Following activation, RSK3 is localized in the nucleus. In quiescent cells, RSK3 forms a complex with ERK1/2 [15,16]. On activation, the RSK-ERK complex shifts to the nucleus. RSK1 and RSK2 are also able to form a complex with ERK1 or ERK2, but on activation, the complex is dissociating, which could be the reason why RSK3 has a longer activation period compared to other isoforms [15,16].
The aim of this study was to identify targets acting synergistically with the EGFR inhibitor erlotinib. For this purpose, we performed a kinome-wide RNAi-based loss-of-function screen in the presence of erlotinib. Among the screen hits, we identified and characterized RSK3 as a promising druggable candidate gene whose inhibition synergistically enhanced the efficacy of erlotinib in pancreatic cancer cells.
Materials and Methods
Cell Culture
PaTu 8988t pancreatic cancer cells were obtained from the German Collection of Cell Lines [Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), Braunschweig, Germany]. BxPC-3, PANC-1, Capan-1, and HPDE cells were from the American Type Culture Collection (Rockville, MD). IMIM-PC-1 cells and Suit-2-007 cells were kind gifts from Dr F. X. Real [Centro Nacional de Investigaciones Oncologicas (CNIO), Madrid, Spain] and Takeshi Iwamura (Miyazaki, Japan), respectively. BxPC-3 cells were cultured in RPMI 1640 medium (PAA, Colbe, Germany) supplemented with 10% FBS (PAA). PaTu8988t and IMIM-PC-1 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA), also supplemented with 10% FBS. Stably transfected BxPC-3 and PaTu8988t cell clones were grown under the same conditions as the parental cell line with addition of G418 (PAA). All cells were grown at 5% CO2 and 37°C.
The Kinome RNAi Library Screen
For the screen, we used the Dharmacon (Thermo Fisher Scientific, Waltham, MA) whole-kinome RNAi library comprising 779 kinases. BxPC-3 cells were seeded into 96-well plates (7000 per well). Transfection with four pooled independent silencing sequences per gene was carried out in quadruplicate 1 day later according to the manufacturer's instructions (Dharmacon), using DharmaFECT 1 (Dharmacon) as the transfection reagent and an small interfering RNA (siRNA) concentration of 20 nM per well. Forty-eight hours after transfection, erlotinib (10 µM) or solvent was added. After incubating for another 48 hours, cell viability was measured using the CellTiter-Glo Assay from Promega (Fitchburg, WI) according to the manufacturer's instructions. As controls, we used scrambled nonsilencing siRNAs. Effects on cell viability of individual siRNAs were calculated as logarithmic z values and were normalized to the mean cell viability of all transfected wells. Three independent experiments were performed, and only targets who showed consistent results in all three runs were further validated. The complete data set of the RNAi kinome screen comprising 799 kinases is depicted in Table W1.
siRNA Transfection and Plasmids
Pooled and individual siRNA sequences for target validation as well as transfection reagents were obtained from Dharmacon. The transfection was carried out using the wet reverse transfection protocol from Dharmacon using an siRNA concentration of 25 nM. For overexpression experiments, we transfected pWZL-Neo-myr-flag/Dest and pWZL-Neo-myr-flag/RPS6KA2 plasmids from Addgene (Cambridge, MA) using the transfection reagent TransFast from Promega according to the manufacturer's instructions. For generation of stable single-cell clones, cells were seeded as single cells into 96-well plates and treated with G418 (400 µg/ml for BxPC-3 and 800 µg/ml for PaTu-8988t cells) for 2 weeks.
Reagents
The Mek1/2 inhibitor UO126 was obtained from Merck KGaA (Darmstadt, Germany), the phosphatidylinositol-4,5-bisphosphate 3 (PI3) kinase inhibitor LY294002 from Cell Signaling Technology (Danvers, MA), the EGFR inhibitor erlotinib from LC Laboratories, Inc (Woburn, MA), and the RSK inhibitor BI-D1870 from Enzo Life Sciences GmbH (Lorrach, Germany). Human recombinant EGF was purchased from R&D Systems (Minneapolis, MN). After culturing cells in serum-free medium for 24 hours, the inhibitors were added for 2 hours, and then EGF (5 ng/ml) was added for 15 minutes. Samples were lysed in buffer containing protease and phosphatase inhibitors (Roche, Basel, Switzerland).
Western Blot Analysis
Protein concentration was estimated using the Bradford method. Ten micrograms of protein sample was loaded on a 10% acrylamide gel and blotted on polyvinylidene fluoride (PVDF) membranes. The blocking reagent was 5% nonfat milk powder disolved in 0.1% trisbuffered saline supplemented with 0.1% Tween 20 (TBST), and primary antibodies were dissolved according to the manufacturer's instructions and incubated overnight at 4°C. Antibodies for pERK1/2, ERK1/2, pRSK3 (T356/S360), prpS6 (S235/236), rpS6, RSK1, RSK2, EGFR, and poly(ADP-ribose) polymerase (PARP) were obtained from Cell Signaling Technology, pRSK3 (S218) from R&D Systems, RSK3 and KRAS from Abcam (Cambridge, United Kingdom), and β-Actin from Sigma-Aldrich (St Louis, MO). The secondary anti-rabbit antibody was diluted in 5% skim milk and 1% TBST (1:10,000) and incubated for 1 hour at room temperature (RT). The ECL Substrate (PerkinElmer, Waltham, MA) and the digital imaging system from Intas (Gottingen, Germany) were used for visualization. Specificity of the RSK antibodies was confirmed by siRNA experiments (Figure W1).
Functional Assays
Cell viability was determined by using 5 mg/ml thiazolyl blue, which was incubated in serum-free medium for 1 hour at 37°C. Subsequently, cells were lysed (10% Triton X-100 with 0.1 M HCl in isopropanol) and measured in a photometer (Thermo Fisher Scientific, Waltham, MA) at 570 nm. Apoptosis was measured using a DNA fragmentation assay (Roche) according to the manufacturer's instructions or by Western blot analysis of PARP cleavage. Proliferation was determined with the bromodeoxyuridine (BrdU) cell proliferation kit (Roche) according to the manufacturer's instructions.
Immunohistochemistry
Immunohistochemistry was performed [17] using the ABC standard staining protocol. Human pancreatic cancer tissues were analyzed using a multiple tissue array (MTA), which contains 548 tissue samples from 123 patients with available clinical follow-up data. Tissues were collected according to the guidelines of the local ethics committee of the Technical University of Dresden. Murine pancreatic cancer tissues and normal pancreata were derived from KPC mice (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx1-Cre) for immunohistochemical analysis.
Statistical Analysis
Statistical analyses were performed using the Tukey-Kramer test for multiple comparisons. P values <.05 were considered significant. Data are presented as means ± SD and are representative for at least three independent experiments.
Results
Kinome-Wide Loss-of-Function Screen
To identify target genes whose knockdown would affect cell viability synergistically with erlotinib, we employed a whole-kinome siRNA library comprising 779 kinases. The pancreatic cancer cell line BxPC-3 was transfected with pooled siRNAs (four silencing sequences per gene), and the impact of the knockdown of each gene on cell viability was assessed in the presence of erlotinib. The 12 top genes whose knockdown significantly decreased cell viability are summarized in Table W1.
Subsequently, 9 of the 12 top screen hits could be validated individually by using four independent silencing sequences per gene compared to nonsilencing controls (Figure 1). Among the targets were several well-known modulators of tumor progression such as drivers of cell cycle progression [mitogen-activated protein kinase kinase kinase 3 (MAP3K3) and MAP3K2], DNA damage-associated checkpoint control [checkpoint kinase1 (CHEK1)], and cytokine signaling Janus kinase 2 ( JAK2; Figure 1]. The two proteins whose knockdown had the greatest impact on cell viability on individual validation were MAP3K3, also known as MEK kinase 3, and RPS6KA2, also known as p90 ribosomal S6 kinase RSK3. Because information on the impact of RPS6KA2/RSK3 in cancer is rudimentary, we focussed on RPS6KA2/RSK3 for further validation and characterization.
Figure 1.

Target validation of the RNAi screen results. Cell viability in BxPC-3 cells was determined in the presence of erlotinib after knockdown of the individual target genes using four independent silencing sequences or scrambled nonsilencing control siRNAs (siC).
To elucidate the role of RSK3 in pancreatic cancer, we first examined expression levels in various pancreatic cancer cell lines (Figure 2A). RSK3 protein was expressed in all examined cell lines to various extents. RSK3 is phosphorylated at several serine or threonine residues leading to its activation. Phosphorylation of both major phosphorylation sites Thr356/Ser360 and Ser218 could be detected at varying levels in all cell lines by phospho-specific antibodies (Figures 2A and W2).
Figure 2.
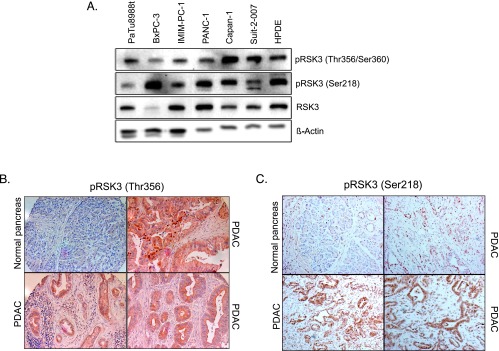
RSK3 protein expression in pancreatic cancer. (A) RSK3 protein expression in various pancreatic cancer cell lines and the pancreatic ductal epithelial cell line HPDE, as assessed by Western blot analysis. (B) Representative immunohistochemical analysis of phospho-RSK3 (Thr356/Ser360) expression indicative of active RSK3 in human normal pancreatic tissue and pancreatic cancers. (C) RSK3 expression in murine pancreatic cancer tissue derived from the KPC mouse model, as assessed by immunohistochemistry using the phospho-RSK3 (Ser2218) antibody.
Interestingly, BxPC-3 cells that had been used in the RNAi screen showed relatively low expression levels of RSK3 protein, although phosphorylation at both Thr356/Ser360 and Ser218 sites could be readily detected. We therefore decided to use two pancreatic cell lines, BxPC-3 and additionally PaTu-8988t, which exhibits high basal RSK3 expression levels, for further validation experiments. Moreover, PaTu-8988t cells harbor an activating KRAS mutation, whereas wild-type KRAS is found in BxPC-3 cells. This is particularly important because RSK3 is activated by signaling pathways downstream EGF that might involve KRAS. However, similar to the effects seen in BxPC-3 cells, we could confirm that knockdown of RSK3 significantly decreased cell viability in PaTu-8988t cells (Figure W3).
RSK3 Is Expressed in Human and Murine Pancreatic Cancer
To assess RSK3 activation levels in pancreatic cancer tissues, we performed immunohistochemical analysis of RSK3 on human and murine paraffin-embedded pancreatic sections. Human tissue was analyzed in anMTA comprising 548 tissue samples from 123 patients. In 220 of 548 tissue samples (49 of 123 patients, 40%), we found high signals after staining for pRSK3 (Thr353/Thr356), indicative for active RSK3 (Figure 2B). Murine pancreatic tissues were obtained from the KPC mouse model (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx1-Cre), which reliably recapitulates human pancreatic carcinogenesis on both molecular and histologic level [18]. In pancreatic cancers derived from this mouse model, strong signals of active RSK could be confirmed (Figure 2C).
RSK3 Protects Cells from Apoptosis
To functionally dissect the impact of RSK3 knockdown on cell viability detected in the screen, we assessed its effect on cell proliferation and apoptosis.
Interestingly, knockdown of RSK3 had only a minor effect on cell proliferation in the presence of erlotinib in both PaTu-8988t and BxPC-3 cells, as assessed by BrdU assays (Figure W4). On the basis of these assays, it appears that the effect on cell viability, as detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays, is not mediated through modulation of cell proliferation.
Therefore, we examined the impact of RSK3 modulation on both basal and drug-induced apoptosis. Knockdown of RSK3 in PaTu-8988t cells significantly increased apoptosis in the presence of erlotinib. In the absence of erlotinib, however, apoptosis was unaffected (Figure 3A), indicating a synergistic action between erlotinib and RSK3 inhibition. Similar results were obtained in BxPC-3 cells (Figure W5). Furthermore, we generated PaTu-8988t cells stably overexpressing RSK3 to examine whether RSK3 expression is able to rescue from drug-induced apoptosis (Figure 3B). RSK3 overexpression resulted in a slight but nonsignificant reduction in apoptosis induced by erlotinib alone. However, when apoptosis was induced by erlotinib in combination with the nucleoside analog gemcitabine, which is commonly used in pancreatic cancer therapy, or by gemcitabine alone, RSK3 completely abolished apoptosis to baseline levels (Figure 3B). These data suggest that RSK3 overexpression is able to rescue from drug-induced apoptosis and inhibition of RSK3 acts synergistically with EGFR inhibition to induce apoptosis.
Figure 3.

RSK3 inhibition induces apoptosis synergistically with erlotinib, and RSK3 mediates resistance to apoptosis. (A) PaTu-8988t cells were transiently transfected with siRNA against RSK3 (siRSK3) or siC and treated with erlotinib or solvent DMSO. siRSK3 sensitizes PaTu-8988t cells to erlotinib treatment but is ineffective in the absence of erlotinib. (B) Clones stably overexpressing RSK3 or empty vector were incubated with gemcitabine and/or erlotinib. Apoptosis was determined by a DNA fragmentation assay. *P < .05 compared to siC (A) or Mock-transfected clones (B). Results are representative for three independent experiments.
RSK3 Activity Is Regulated by EGF/MEK/ERK Signaling
Several reports indicate that EGF-dependent signaling pathways function as major activators of RSK proteins in various cell types [19]. To examine the upstream activation pathways of RSK3 in pancreatic cancer cells, we incubated serum-starved PaTu-8988t and BxPC-3 cells with recombinant EGF and examined phosphorylation of RSK3 at residues Thr356/Ser360 and Ser218 with phospho-specific antibodies. Phosphorylation of both residues was detected 5 to 10 minutes after addition of EGF, peaking at 15 minutes (Figure W6).
EGF-dependent signaling pathways are mediated through two main downstream cascades involving the kinases MEK-ERK or phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-AKT. Phosphorylation at residue Thr356/Ser360 has been attributed to MEK/ERK signaling, whereas Ser218 has been associated with phosphorylation through 3′-phosphoinositide-dependent protein kinase-1 (PDK1), which, in addition, acts upstream of PI3K-AKT [11]. To further elucidate the upstream signaling events involved in RSK3 activation, we coincubated EGF-activated BxPC-3 and PaTu-8988t cells with erlotinib as well as the MEK inhibitor UO126 and the PI3K inhibitor LY294002.
We could demonstrate that EGF induces phosphorylation of RSK3 at residues Thr356/Ser360 and Ser218, both of which could be reverted by erlotinib (Figure 4A). MEK inhibition by UO126 abolished basal and EGF-induced phosphorylation at residues Thr356/Ser360 but only EGF-induced phosphorylation at Ser218, indicating that EGF-induced phosphorylation at Ser218 involves MEK-dependent mechanisms but basal phosphorylation occurs independently of MEK. PI3K inhibition by LY294002 had no effect on RSK phosphorylation at either residue, suggesting that PI3K/AKT signaling is not able to modulate RSK activation in these cell lines. Similar results were obtained in KRAS mutated PaTu-8988t (Figure 4A) and KRAS wild-type BxPC-3 cells (Figure 4C). To rule out cell line-dependent differences and to further investigate the impact of constitutively active KRAS in RSK phosphorylation, we generated BxPC-3 cells harboring a constitutively active KRAS. In these cells, we detected elevated basal RSK3 phosphorylation levels at both Thr356/Ser360 and Ser218, but EGF stimulation as well as inhibition by erlotinib and UO126 was still effective (Figure 4C). Both data in PaTu-8988t and mutant KRAS-expressing BxPC-3 cells indicate that EGF stimulation and downstream signaling to RSK3 is still active despite the presence of a constitutively active KRAS protein.
Figure 4.

RSK3 is activated by EGF/EGFR through the MEK/ERK signaling cascade and modulates rpS6 phosphorylation. Serum-starved cells were incubated with the EGFR inhibitor erlotinib, the MEK inhibitor UO126, or the PI3K inhibitor LY294002 for 2 hours before stimulation with EGF for 15 minutes. Phosphorylation status of ERK and RSK3 (A) as well as rpS6 (B) was determined using specific antibodies by Western blot analysis in PaTu-8988t cells harboring mutant KRAS. (C and D) BxPC-3 cells with wild-type KRAS (Mock) and BxPC-3 cells stably transfected with constitutively active KRAS (KRAS) were incubated with the indicated compounds as above. Phosphorylation status of ERK and RSK3 (C) as well as rpS6 (D) was determined using specific antibodies by Western blot analysis in BxPC-3 cells stably expressing wild-type (Mock) or mutant KRAS (KRAS). Note that the PI3K inhibitor LY294002 has a profound impact on rpS6 phosphorylation but not on RSK3 phosphorylation, suggesting that PI3K/mTOR signaling affects rpS6 phosphorylation independently of RSK3.
rpS6 Is a Downstream Target for RSK3
On the basis of our functional data demonstrating a strong antiapoptotic phenotype induced by RSK3 after activation by EGF/MEK/ERK signaling, we were interested in which downstream effectors are employed by RSK3 to mediate its prosurvival effects. Reports in various cell systems suggest several downstream effectors such as 5′ AMP-activated protein kinase β (AMPKβ), a key regulator of energy homeostasis, the tumor suppressor LKB1, and the rpS6 as putative phosphorylation targets of RSK3. In pancreatic cancer cells, we could not detect a consistent modulation of AMPKβ and/or LKB1 phosphorylation by RSK3 (data not shown). The rpS6, however, which has been linked to tumor cell growth and survival [20], could be verified as target of RSK3 in pancreatic cancer cells: EGF-induced RSK3 phosphorylation and its inhibition by erlotinib or UO126 were accompanied by concordant phosphorylation patterns of rpS6 in both PaTu-8988t (Figure 4B) and BxPC-3 cells with or without KRAS mutation (Figure 4D). Furthermore, knockdown of RSK3 by siRNA markedly decreased phosphorylation of rpS6 (Figures 5A and W7). In addition, we used the specific RSK inhibitor BI-D1870 that potently diminishes RSK3 activity but also affects other RSK isoforms [21]. Incubation with BI-D1870 decreased basal phosphorylation of rpS6, as did the addition of erlotinib (Figures 5B and W8). Simultaneous incubation with both BI-D1870 and erlotinib decreased rpS6 phosphorylation even further, indicating a synergistic action of the RSK inhibitor BI-D1870 and the EGFR inhibitor erlotinib in decreasing rpS6 activity (Figures 5B and W8). These data indicate that rpS6 is phosphorylated by RSK3 in pancreatic cancer cells and RSK inhibition and EGFR inhibition synergistically affect rpS6 phosphorylation.
Figure 5.

RSK3 phosphorylates rpS6. (A) PaTu-8988t cells were transiently transfected with siRSK3 or siC at two different siRNA concentrations (25 and 50 nM). Phosphorylated rpS6 and total rpS6 were detected with specific antibodies. (B) PaTu-8988t cells were treated with the RSK inhibitor BI-D1870 (5 µM) and/or erlotinib (10 µM) for the indicated time points. Phospho-rpS6 and total rpS6 were detected by specific antibodies.
RSK Inhibition by BI-D1870 Acts Synergistically with Erlotinib
In addition to our genetic screening approach, we were interested in if pharmacological RSK inhibition using BI-D1870 is also acting synergistically with erlotinib. A dose-response experiment revealed that both BxPC-3 and PaTu-8988t cells were sensitive to BI-D1870. PaTu-8988t cells, however, required an approximately 5x higher concentration to achieve a 50% reduction in cell viability (Figure W9).
Treatment of both PaTu-8988t and BxPC-3 cells with BI-D1870 in the presence or absence of erlotinib resulted in a synergistic decrease of cell viability after combined treatment (Figure 6).
Figure 6.

Pharmacological inhibition of RSK3 acts synergistically with erlotinib. BxPC-3 (A) and PaTu-8988t (B) cells were treated with the RSK inhibitor BI-D1870 in the presence or absence of erlotinib for 48 hours. Cell viability of cells was assessed using MTT assays. Results are representative for three independent experiments. *P < .05 compared to erlotinib only and to erlotinib + BI-D (1 µM, A or 5 µM, B).
As seen after RNAi-mediated knockdown of RSK3 in the presence of erlotinib, pharmacological inhibition of RSK in the presence of erlotinib resulted in a marked increase in apoptosis, determined by PARP cleavage (Figures 7A and W10). Conversely, apoptosis induced by BI-D1870 or by combined treatment with BI-D1870 and erlotinib was abolished by overexpression of RSK3 in stable clones of PaTu-8988t cells (Figure 7B). This indicates that pharmacological RSK inhibition and EGFR inhibition act together by inducing apoptosis and RSK3 overexpression might confer resistance to drug-induced apoptosis.
Figure 7.

Synergistic effect of RSK inhibition by BI-D1870 and EGFR inhibition by erlotinib. (A) PaTu-8988t cells were treated with BI-D1870 (5 µM) and/or erlotinib (10 µM), and PARP cleavage was determined by Western blot analysis after 24 hours. (B) PaTu-8988t cells stably overexpressing RSK3 or empty vector (Mock) were treated with BI-D1870 and/or erlotinib, and apoptosis was determined by DNA fragmentation assays. *P < .05 compared to either Mock clone. Data are representative for three independent experiments.
Discussion
In this study, we employed an RNAi-based loss-of-function screen to identify mediators of tumor progression whose therapeutic inhibition would act synergistically with the EGFR inhibitor erlotinib. We identified the p90 S6 kinase RSK3 as potent mediator of resistance to apoptosis whose genetic silencing or pharmacological inhibition synergistically enhanced the effect of erlotinib. RSK3 is predominantly activated by EGF signaling through MEK/ERK-dependent pathways, both in KRAS wild-type and KRAS mutated pancreatic cancer cells. The rpS6 was identified as downstream target of RSK3 in pancreatic cancer cells, mediating its effects on tumor cell survival.
Increased expression of RSKs has been reported in several tumors such as breast [22] and prostate cancers [23]. RSKs are also expressed in pancreatic cancer cells, and their activation following EGF stimulation has been shown to be dependent on MEK/ERK signaling [24]. So far, data on RSK expression levels in pancreatic cancer tissues are missing. Our data in a large MTA indicate that RSK3 is expressed in approximately 40% of pancreatic cancer tissues. Interestingly, our expression data with pancreatic cancer cell lines harboring mutant or wild-type KRAS indicate that the protein expression is regulated by signaling pathways independent of KRAS.
Most functional data on RSK family members have been obtained with RSK1 and RSK2 [11]. Reports on the functional effects of RSK3 are limited. Generally, RSKs are known to promote cell cycle progression through the regulation of mediators of the cell cycle, e.g., by controlling the activity of p27 [11,25]. In addition, RSK1 and RSK2 were found to promote survival in various cell types, e.g., by phosphorylation of the proapoptotic protein Bad, which enhances its binding and inactivation by 14-3-3 proteins [26]. Furthermore, RSK1 and RSK2 have been shown to phosphorylate and thereby inactivate proapoptotic death-associated protein kinase (DAPK), resulting in increased cell survival in response to mitogenic stimulation [11,27].
The few reports on the putative role of RSK3 in cancer show the following conflicting results: In malignant mesotheliomas, RSK3 activity has been shown to decrease on ERK2 inhibition [28]. In breast cancer, v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 (ERBB2) has been reported to activate RSK3 in a membrane-associated complex together with the estrogen receptor [12]. Both reports suggest a tumor-promoting role of RSK3. In contrast, Bignone et al. describe RSK3 as putative tumor suppressor with reduced expression in ovarian cancer and show that RSK3 overexpression is associated with reduced proliferation and increased apoptosis in ovarian cancer cell lines [13]. Our data based on both genetic and pharmacological inhibition as well as overexpression experiments clearly indicate that RSK3 plays a tumor-promoting role and acts as strong mediator of survival and resistance to drug-induced apoptosis in pancreatic cancer. The reasons for these discrepant findings remain to be elucidated. Different epigenetic methylation patterns might be responsible for the varying expression patterns. Furthermore, the different RSK3 isoforms detected in the ovarian cancer cell lines [13] may explain some of the functional differences. It appears evident that the role of RSK3 is cell type and context dependent.
RSKs are known to be activated by EGF-dependent pathways through MEK/ERK [11,29]. Therefore, at first glance, it appears surprising that RSK inhibition acts synergistically with the EGFR inhibitor erlotinib, because both EGFR and MEK/ERK signal within one major pathway. Moreover, this synergism appears to be functional in both KRAS wild-type and KRAS mutant cells. On the basis of our data, it is likely that RSKs are activated by several EGFR-dependent mechanisms, among them KRAS-dependent, but also KRAS-independent complementary oncogenic signaling pathways. Recent reports in pancreatic cancer cell lines and genetic mouse models corroborate this hypothesis: In vitro, the unique activities of EGFR have been shown to promote cell proliferation and invasion even when KRAS is mutated [30,31]. In vivo, genetic ablation or pharmacological inhibition of EGFR by erlotinib effectively eliminated KRAS-driven tumorigenesis in a mouse model of pancreatic cancer. Without EGFR activity, KRAS was not sufficient to induce robust MEK/ERK activity. In addition, EGFR activity was required for acinar-ductal metaplasia [32]. Moreover, active KRAS has been shown to induce EGFR expression, which underlines the complex interplay between KRAS and EGFR signaling and provides an additional rational for EGFR inhibition even in the presence of mutated KRAS. Our data indicate that RSK3 activity is induced by EGFR signaling through substantial amplification of MEK-ERK activation through KRAS-dependent and KRAS-independent pathways. The exact mechanisms of KRAS-independent MEK-ERK activation leading to subsequent RSK induction remain to be elucidated. Phosphorylation of RSK3 at Thr356/Ser360 has been associated with ERK activity. In contrast, Ser218 is phosphorylated by PDK1 [11]. We could show that MEK inhibition by UO126 abolished basal and EGF-induced phosphorylation at residues Thr356/Ser360 and EGF-induced phosphorylation at Ser218. This is consistent with the notion that phosphorylation by ERK creates a docking site for PDK1 that, in turn, phosphorylates Ser218 in the activation loop [11]. The PI3K inhibitor LY294002 had no effect on either phosphorylation site, indicating that PI3K/Akt signaling, which represents an additional downstream target of PDK1, is not involved in activation of RSK3.
Numerous downstream targets of RSKs have been described, most of them for RSK1 and RSK2. The rpS6 represents one of these targets and plays a crucial role within the translation machinery as component of the 40S ribosomal subunit [33]. rpS6 is phosphorylated by RSKs at Ser235/236 in vitro and in vivo through mechanistic target of rapamycin (mTOR)-independent mechanisms. Our data provide evidence that rpS6 represents a crucial downstream target of RSK3 mediating at least partly its effects on tumor cell survival. In addition to its role in enhancing protein translation, several reports confirm that rpS6 is important for cell survival [20]. Interestingly, rpS6 has been identified as target in an siRNA-based synthetic lethal screen for modifiers of insulin-like growth factor 1 receptor (IGF1R) kinase inhibitor activity in sarcomas [20]. In this report, an IGF1R kinase inhibitor failed to block rpS6 activation in resistant sarcoma cell lines, indicating that rpS6 represents an important mediator of resistance to IGF1R inhibition and therefore a potential target for the implementation of synergistically acting inhibitor strategies [20].
Our data clearly indicate that RSK inhibition and EGFR inhibition is synthetically lethal in pancreatic cancer cells. In contrast to erlotinib that is used routinely in patients, available RSK inhibitors are limited to in vitro use so far, mainly due to solubility restrictions. On the basis of our data, the development of novel RSK inhibitors or the chemical modification of existing RSK inhibitors are warranted and currently ongoing in our laboratory. This will facilitate the in vivo evaluation of simultaneous RSK- and EGFR-targeting strategies in preclinical trials using genetic mouse models of pancreatic cancer as well as clinical trials that hopefully will be able to improve the devastating prognosis and therapy resistance of pancreatic cancer.
Supplementary Material
Acknowledgments
We thank Michael Krause, Genomics Unit of the University of Marburg, for his help with the screen setup and Martin Eilers, University of Wurzburg, for helpful discussions.
Abbreviations
- EGFR
epidermal growth factor receptor
- RNAi
RNA interference
- RSK
ribosomal S6 kinase
- PARP
poly(ADP-ribose) polymerase
Footnotes
This work was supported by grants of the Deutsche Krebshilfe/Mildred Scheel Stiftung (to P.M.), Deutsche Forschungsgemeinschaft (DFG; to P.M.), the von Behring-Rüntgen Foundation (to P.M.), and the European Commisson FP7 grant (Collaborative Project “EPC-TM-Net”, to P.M. and T.G.). This publication reflects only the authors' views. The European Community is not liable for any use that may be made of the information herein.
This article refers to supplementary materials, which are designated by Table W1 and Figures W1 to W10 and are available online at www.neoplasia.com.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F, Ciardiello F. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets. 2012;13(6):802–810. doi: 10.2174/138945012800564158. [DOI] [PubMed] [Google Scholar]
- 3.Michl P, Gress TM. Current concepts and novel targets in advanced pancreatic cancer. Gut. 2013;62:317–326. doi: 10.1136/gutjnl-2012-303588. [DOI] [PubMed] [Google Scholar]
- 4.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 5.Heinemann V, Haas M, Boeck S. Systemic treatment of advanced pancreatic cancer. Cancer Treat Rev. 2012;38(7):843–853. doi: 10.1016/j.ctrv.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Michl P, Ripka S, Gress T, Buchholz M. Screening technologies for target identification in pancreatic cancer. Cancers. 2011;3:79–90. doi: 10.3390/cancers3010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Downward J. RNA interference-based functional genomics in cancer research—an introduction. Oncogene. 2004;23:8334–8335. doi: 10.1038/sj.onc.1208121. [DOI] [PubMed] [Google Scholar]
- 8.Maller JL, Foulkes JG, Erikson E, Baltimore D. Phosphorylation of ribosomal protein S6 on serine after microinjection of the Abelson murine leukemia virus tyrosine-specific protein kinase into Xenopus oocytes. Proc Natl Acad Sci USA. 1985;82:272–276. doi: 10.1073/pnas.82.2.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moller DE, Xia CH, Tang W, Zhu AX, Jakubowski M. Human rsk isoforms: cloning and characterization of tissue-specific expression. Am J Physiol. 1994;266:C351–C359. doi: 10.1152/ajpcell.1994.266.2.C351. [DOI] [PubMed] [Google Scholar]
- 10.De Cesare D, Jacquot S, Hanauer A, Sassone-Corsi P. Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc Natl Acad Sci USA. 1998;95:12202–12207. doi: 10.1073/pnas.95.21.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romeo Y, Zhang X, Roux PP. Regulation and function of the RSK family of protein kinases. Biochem J. 2012;441:553–569. doi: 10.1042/BJ20110289. [DOI] [PubMed] [Google Scholar]
- 12.Pancholi S, Lykkesfeldt AE, Hilmi C, Banerjee S, Leary A, Drury S, Johnston S, Dowsett M, Martin LA. ERBB2 influences the subcellular localization of the estrogen receptor in tamoxifen-resistant MCF-7 cells leading to the activation of AKT and RPS6KA2. Endocr Relat Cancer. 2008;15:985–1002. doi: 10.1677/ERC-07-0240. [DOI] [PubMed] [Google Scholar]
- 13.Bignone PA, Lee KY, Liu Y, Emilion G, Finch J, Soosay AE, Charnock FM, Beck S, Dunham I, Mungall AJ, et al. RPS6KA2, a putative tumour suppressor gene at 6q27 in sporadic epithelial ovarian cancer. Oncogene. 2007;26:683–700. doi: 10.1038/sj.onc.1209827. [DOI] [PubMed] [Google Scholar]
- 14.Ikuta M, Kornienko M, Byrne N, Reid JC, Mizuarai S, Kotani H, Munshi SK. Crystal structures of the N-terminal kinase domain of human RSK1 bound to three different ligands: implications for the design of RSK1 specific inhibitors. Protein Sci. 2007;16:2626–2635. doi: 10.1110/ps.073123707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roux PP, Richards SA, Blenis J. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol Cell Biol. 2003;23:4796–4804. doi: 10.1128/MCB.23.14.4796-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith JA, Poteet-Smith CE, Malarkey K, Sturgill TW. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J Biol Chem. 1999;274:2893–2898. doi: 10.1074/jbc.274.5.2893. [DOI] [PubMed] [Google Scholar]
- 17.Griesmann H, Ripka S, Pralle M, Ellenrieder V, Baumgart S, Buchholz M, Pilarsky C, Aust D, Gress TM, Michl P. WNT5A-NFAT signaling mediates resistance to apoptosis in pancreatic cancer. Neoplasia. 2013;15:11–22. doi: 10.1593/neo.121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 19.Pasic L, Eisinger-Mathason TS, Velayudhan BT, Moskaluk CA, Brenin DR, Macara IG, Lannigan DA. Sustained activation of the HER1-ERK1/2-RSK signaling pathway controls myoepithelial cell fate in human mammary tissue. Genes Dev. 2011;25:1641–1653. doi: 10.1101/gad.2025611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potratz JC, Saunders DN, Wai DH, Ng TL, McKinney SE, Carboni JM, Gottardis MM, Triche TJ, Jurgens H, Pollak MN, et al. Synthetic lethality screens reveal RPS6 and MST1R as modifiers of insulin-like growth factor-1 receptor inhibitor activity in childhood sarcomas. Cancer Res. 2010;70:8770–8781. doi: 10.1158/0008-5472.CAN-10-1093. [DOI] [PubMed] [Google Scholar]
- 21.Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005;65:1027–1034. [PubMed] [Google Scholar]
- 23.Clark DE, Errington TM, Smith JA, Frierson HF Jr, Weber MJ, Lannigan DA. The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res. 2005;65:3108–3116. doi: 10.1158/0008-5472.CAN-04-3151. [DOI] [PubMed] [Google Scholar]
- 24.Seufferlein T, Van Lint J, Liptay S, Adler G, Schmid RM. Transforming growth factor α activates Ha-Ras in human pancreatic cancer cells with Ki-ras mutations. Gastroenterology. 1999;116:1441–1452. doi: 10.1016/s0016-5085(99)70509-3. [DOI] [PubMed] [Google Scholar]
- 25.Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003;278:49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- 26.Shimamura A, Ballif BA, Richards SA, Blenis J. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr Biol. 2000;10:127–135. doi: 10.1016/s0960-9822(00)00310-9. [DOI] [PubMed] [Google Scholar]
- 27.Anjum R, Roux PP, Ballif BA, Gygi SP, Blenis J. The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr Biol. 2005;15:1762–1767. doi: 10.1016/j.cub.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 28.Shukla A, Hillegass JM, MacPherson MB, Beuschel SL, Vacek PM, Butnor KJ, Pass HI, Carbone M, Testa JR, Heintz NH, et al. ERK2 is essential for the growth of human epithelioid malignant mesotheliomas. Int J Cancer. 2011;129:1075–1086. doi: 10.1002/ijc.25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douville E, Downward J. EGF induced SOS phosphorylation in PC12 cells involves P90 RSK-2. Oncogene. 1997;15:373–383. doi: 10.1038/sj.onc.1201214. [DOI] [PubMed] [Google Scholar]
- 30.Pino MS, Balsamo M, Di Modugno F, Mottolese M, Alessio M, Melucci E, Milella M, McConkey DJ, Philippar U, Gertler FB, et al. Human Mena+11a isoform serves as a marker of epithelial phenotype and sensitivity to epidermal growth factor receptor inhibition in human pancreatic cancer cell lines. Clin Cancer Res. 2008;14:4943–4950. doi: 10.1158/1078-0432.CCR-08-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS One. 2011;6:e19605. doi: 10.1371/journal.pone.0019605. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Ardito CM, Grüner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.