

Abstract
Adenosine monophosphate-activated protein kinase (AMPK), a master regulator of cellular energy homeostasis, has emerged as a promising molecular target in the prevention of breast cancer. Clinical trials using the United States Food and Drug Administration (FDA)-approved, AMPK-activating, antidiabetic drug metformin are promising in this regard, but the question of why metformin is protective for some women but not others still remains. Breast cancer associated gene 2 (BCA2/Rabring7/RNF115), a novel Really Interesting New Gene (RING) finger ubiquitin E3 ligase, is overexpressed in >50% of breast tumors. Herein, we report that BCA2 is an endogenous inhibitor of AMPK activation in breast cancer cells and that BCA2 inhibition increases the efficacy of metformin. BCA2 overexpression inhibited both basal and inducible Thr172 phosphorylation/activation of AMPKα1, while BCA2-specific small interfering RNA (siRNA) enhanced phosphorylated AMPKα1 (pAMPKα1). The AMPK-suppressive function of BCA2 requires its E3 ligase-specific RING domain, suggesting that BCA2 targets some protein controlling (de)phosphorylation of AMPKα1 for degradation. Activation of AMPK by metformin triggered a growth inhibitory signal but also increased BCA2 protein levels, which correlated with AKT activation and could be curbed by an AMPK inhibitor, suggesting a potential feedback mechanism from pAMPKα1 to pAkt to BCA2. Finally, BCA2 siRNA, or inhibition of its upstream stabilizing kinase AKT, increased the growth inhibitory effect of metformin in multiple breast cancer cell lines, supporting the conclusion that BCA2 weakens metformin's efficacy. Our data suggest that metformin in combination with a BCA2 inhibitor may be a more effective breast cancer treatment strategy than metformin alone.
Introduction
The ubiquitin-proteasome system (UPS) is the cell's primary means by which protein homeostasis is maintained; specific protein substrates such as those that are misfolded, damaged, or mutated and may otherwise be harmful to the cell are ubiquitinated and degraded continuously [1]. Cancer can develop as a result of disruption of the UPS, including dysregulated degradation of specific protein targets. Characteristic of many cancer types is the stabilization of oncoproteins and destabilization of tumor suppressors; in this way, the balance is tipped to allow for maximal cell survival. For example, in some cancers, such tumor suppressor proteins like p53 and p27 are overdegraded by the UPS, whereas growth-promoting receptors like epidermal growth factor receptor and transforming growth factor-β receptor are underdegraded [1]. While the proteasome has been extensively studied as an anticancer therapeutic target, from which bortezomib and several other effective proteasome inhibitors have emerged [2], the roles of the individual components in the ubiquitin-mediated steps preceding degradation, which are still not well characterized, may provide us with not only more insight into how this equilibrium is disturbed but also with novel and specific molecular drug targets.
Ubiquitin E3 ligases provide substrate specificity to the UPS and their imbalance has been implicated in the pathogenesis of breast cancer [3]. Therefore, this group of enzymes has much potential as effective and specific drug targets for the treatment of breast cancer. An example of one such approach is the well-studied E3 ligase, Mdm2, and its interaction with p53 for which antitumor agents have been developed [4]. Breast cancer associated gene 2 (BCA2), like Mdm2, belongs to the Really Interesting New Gene (RING) finger containing subset of ubiquitin E3 ligases. The RING finger is a specialized zinc finger critical for its intrinsic autoubiquitination activity, a trademark of all E3 ligases. Upon its discovery, BCA2 was found to be overexpressed in breast cancer [5], linked to breast cancer cell proliferation in vitro, and correlative to clinical disease outcome, suggesting its importance in cancer progression [6]. BCA2 is expressed in both the nucleus and cytoplasm of breast cancer cells, implying multiple functions [6]. In hormone-responsive breast tumors, nuclear BCA2 expression appears to be under the control of estrogen, although its tumor-promoting function may not be completely dependent on estrogen signaling [7]. In the cytoplasm, several binding partners of BCA2 have been reported, including ubiquitin, UBC9, 14-3-3σ, tethrin, and hHR23a [8,9]. While these studies have great implications in the understanding of how BCA2 is regulated, its downstream targets, which may contribute to overall cancer cell survival, remain to be elucidated.
Cellular energy can be quantified in units of ATP, which is required to be at a considerably high concentration to ensure cell survival [10]. The primary function of adenosine monophosphate-activated protein kinase (AMPK) in the cell is to respond, much like the flip of a switch, to drops in ATP levels, sensed by a subsequent rise in AMP and direct binding to its β regulatory subunit. The heterotrimeric enzyme then undergoes a conformational change, followed by phosphorylation of its α subunit at Thr172 by upstream kinases, liver kinase B1 (LKB1), calcium/calmodulin-dependent protein kinase 2 (CaMKK), or transforming growth factor-beta-activated kinase 1 (Tak1) [11,12]. The consequence of AMPK activation is shutdown of processes that would further consume ATP energy, like the synthesis of fatty acids and cholesterol, and conversely the stimulation of energy-producing pathways like glucose uptake and fatty acid oxidation [13]. Therefore, AMPK regulation of cellular metabolism is necessary for both cell survival and cell growth inhibition, complicating its role as a potential anticancer drug target. Nevertheless, in the past 10 years, AMPK has received a lot of attention in the cancer research field, perhaps owing to the now widely accepted AMPK-activating ability of metformin, an FDA-approved agent and first-line treatment for type II diabetes [14]. Although metformin is currently being assessed in phase II and III cancer-related clinical trials, the benefit of AMPK activation in vitro and in vivo remains controversial in various cancer types, due to a need for more detailed mechanistic studies [15,16].
In the current study, we report that BCA2 has a negative regulatory function on the tumor-suppressing, cellular stress-sensing kinase, AMPK. We show that on the endogenous level, BCA2 inhibits both basal and inducible levels of AMPKα1 phosphorylation at Thr172 and therefore its activation. This inhibition was dependent on its E3 ligase-specific RING domain and was also affected by S132/S133 mutations in its predicted Akt phosphorylation site [17]. In addition, we demonstrate that chemical activation of AMPK by metformin or 5-amino-1-β-Dffff-ribofuranosyl-imidazole-4-carboxamide (AICAR) also increases BCA2 and pAkt protein levels, suggesting the presence of a possible cell survival feedback mechanism. Furthermore, the tumor cell growth inhibitory benefit of metformin was significantly improved once BCA2 was silenced [through small interfering RNA (siRNA)] or destabilized [through phosphatidylinositide 3 (PI3)/Akt kinase inhibition], compared to treatment with metformin alone, demonstrating that suppression of BCA2 function is necessary for enhancement of metformin's anticancer efficacy.
Materials and Methods
Cell Culture
Human breast cancer MDA MB 231, MDA MB 468, MCF7, and Hs578t cells and the human embryonic kidney fibroblast HEK293T cells were obtained from the American Type Culture Collection (Manassas, VA). MDAMB 231 and MDA MB 468 cells were cultured in Dulbecco's modified Eagle's medium (DMEM), and MCF7 and Hs578t cells in RPMI 1640 (Invitrogen, Carlsbad, CA) containing 10% FBS (Hyclone from Fisher Scientific, Pittsburgh, PA), 100 µg/ml streptomycin, and 100 units/ml penicillin (Invitrogen). HEK293T cells were also grown in DMEM containing 10% FBS but no antibiotics. Cells were passaged routinely and maintained at 37°C and 5% CO2.
Reagents
Anti-BCA2 antibody was purchased from LifeSpan BioSciences, Inc (Seattle, WA). FLAG (M2) and β-actin antibodies were obtained from Sigma-Aldrich Corp (St Louis, MO). Antibodies to phosphorylated AMPK (pAMPK; Thr172), acetyl-CoA carboxylase (ACC; Ser79), and pAKT (S473) as well as to total AMPK, ACC, and AKT were all purchased from Cell Signaling Technology (Beverly, MA). HRP-conjugated anti-rabbit IgG and anti-mouse IgG secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). All primary antibodies were stored at -20°C and secondary at 4°C. The AMPK inhibitor Compound C and DMSO were purchased from Sigma-Aldrich Corp. AMPK activators, metformin and AICAR, were obtained from Toronto Research Chemicals Inc (North York, Ontario) and Tocris Bioscience (Minneapolis, MN), respectively. The PI3 kinase inhibitor LY294002 was purchased from Cell Signaling Technology. The stocks of these chemical reagents were made up as follows: 20 mM Compound C dissolved in DMSO, 1 M metformin in sterile H2O, 75 mM AICAR in sterile H2O, and 10 mM LY294002 in DMSO. All drug stocks were stored at -20°C.
Expression Vectors and Constructs
The BCA2 amplicon was subcloned into the FLAG-tagged pCMV-tag2B vector as previously described [6,17]. Mutations in the BCA2 RING domain and predicated AKT phosphorylation site were engineered in each wild-type (wt) construct as also previously described [17]. The AMPKα1 catalytic subunit (PRKAA1) pCMV6-XL5 vector construct was purchased from the TrueClone Human Collection, OriGene Technologies Inc (Rockville, MD). Bacterial stocks of all vector constructs were generated in our laboratory. Once amplified and DNA isolated, plasmid Maxiprep stocks were stored at -20°C.
Transfection
Transfection using AMPKα and BCA2 wt and mutant constructs (created by site-directed mutagenesis [17]). Cells were plated into 100-mm dishes at 80% confluency and allowed to settle overnight. A fresh complete medium change, void of antibiotics, was given 2 hours before transfection. For transfection experiments using MDA MB 231 cells, 1 to 3 µg of FLAG-BCA2, wt, S132/S133 mutant (mt; serine 132 and 133 to alanine [8,17]), or RING (cysteine 228 and 231 to alanine [8,17]) mt vector DNA was transfected using the Lipofectamine LTX reagent according to the manufacturer's protocol (Invitrogen), with pCMV-tag2B empty vector used as a control. Following a 48-hour incubation period, cells were treated as indicated, harvested, and lysed for Western blot analysis. For co-transfection experiments, 2.5 µg of FLAG-BCA2 wt, S132/S133, or RING mt and 2.5 µg of AMPKα vector DNA were co-transfected into HEK293T cells using the FuGENE HD transfection reagent and protocol (Promega, Madison, WI), using pCMV-tag2B empty vector co-transfection with AMPKα as a control. For co-transfection experiments studying the concentration-dependent effect of BCA2 wt or mt on AMPKα, the AMPKα DNA concentration was held constant at 1 µg, while that of BCA2 ranged from 1 to 5 µg. After 48 hours, cells were treated as indicated, harvested, and lysed for Western blot analysis.
Transfection using BCA2-specific siRNA. The custom-designed siRNA duplexes were purchased from QIAGEN (Valencia, CA) [BCA2 siRNA duplex 2: sense, r(CGUCUGAAUAGAAUUAAUU) dTdT, antisense, r(AAUUAAUUCUAUUCAGACG)dGdG] and dissolved in siRNA suspension buffer to yield a stock of 20 µmol/l, stored at -20°C. Non-silencing control siRNA was used as a negative control and RNAiFect (QIAGEN) as the transfection reagent. Experiments were carried out in six-well plates following the manufacturer's protocol. siRNA transfection incubation period lasted 72 hours, after which cells were treated accordingly, harvested, lysed, and analyzed by Western blot analysis.
Western Blot Analysis
Cells were transfected, drug treated, or both, and then harvested. Cell lysates (30–40 µg) were mixed with 3x sodium dodecyl sulfate buffer, boiled for 5 minutes, and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 4% to 20% tris-glycine gradient gels (Invitrogen), and then transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA) and blocked in 5% milk powder in Tris-buffered saline tween 20 (0.2%; TBST) for 1 hour at room temperature. The primary antibodies were diluted in blocking buffer (phosphorylated antibodies at 1:500 and all remaining at 1:1000) and incubated overnight at 4°C. Membranes were washed in TBS-Tween 20 (0.2%) and incubated with species-specific secondary antibodies conjugated to HRP (1:5000) for 1 hour at room temperature. Signals were developed using the Immobilon Western Chemiluminescent HRP Substrate (Millipore) and the FOTO/Analyst Luminary/FX Systems Flexible chemiluminescent and fluorescent imagining workstation (FOTODYNE, Hartland, WI). Densitometry analysis was done using ImageJ software, and relative intensity was calculated as a percentage of the control.
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide Assay
Cells were grown under standard conditions as described above and seeded in 96-well plates (20,000 cells per well) and allowed to adhere for 24 hours. BCA2 siRNA (0.75 µg) or non-silencing scrambled control (0.75 µg) was added for a period of 72 hours after which metformin was added in concentrations ranging from 5 to 30 mM for 24 hours. Cell proliferation was determined by addition of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Invitrogen). The conversion of MTT to purple formazan by viable cells was measured using a Wallac Victor3 1420 multilabel plate counter (550 nm) and analyzed in Microsoft Excel. Growth curves were generated as percent of the control and statistical analysis was done as described below.
Colony Formation Assay
An agar concentration of 0.75% in DMEM, containing 10% FBS, 100 µg/ml streptomycin, and 100 units/ml penicillin, was dispensed at 200 µl/well of a 24-well plate to create a base layer and allowed to solidify. MDA MB 231 cells (200 µl) in 0.4% agar were added in the next layer (1000/well) and placed at 4°C for 5 minutes to solidify. Cells are incubated at 37°C and 5% CO2 overnight after which the respective drugs are added in a total volume of 200 µl/well and colonies are allowed to form over a period of 5 to 7 days. Once the colonies formed, iodonitrotetrazolium blue (2 mg/ml) was added at 100 µl/well and incubated overnight at 37°C and 5% CO2. Colonies of at least 30 µm in diameter were counted using the Oxford Optronix GelCount automated mammalian cell colony counter and CHARM algorithm software. Colony-forming ability was determined as a percent of those in the control.
RNA Analysis by Real-Time PCR
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN) and was reverse transcribed to cDNA (Two Step DNA kit; Invitrogen). The cDNA was amplified using primer pairs for BCA2: forward, 5′-GGGGTCACCAGACTCACACT-3′ and reverse, 3′-CAGGAAAAAGGGTGTGGAGA-5′. The loading control β-actin primers are given as follows: forward, 5′-GAGCGCGGCTACAGCTT-3′ and reverse, 5′-TCCTTAATGTCACGCACGATTT-3′.
BCA2 Promoter-Luciferase Assay
Cells were plated into 24-well plates and co-transfected with BCA2 promoter-luciferase vector (SwitchGear Genomics, Menlo Park, CA) and Renilla vector (Promega) as previously described [7]. After 24 hours, cells were treated with 20 mM metformin or 1 mM AICAR for 6 hours. Luciferase activity was measured using the Dual Luciferase Reporter Assay Kit (Promega), and promoter activity was calculated as relative luciferase units.
Immunoprecipitation
HEK293T cells were co-transfected with AMPKα and FLAG-tagged BCA2 (as above), and co-immunoprecipitation was performed using 700 µg of protein, FLAG antibody, and the Pierce Classic IP Kit (Thermo Scientific, Rockford IL) as per the manufacturer's protocol.
Statistical Analysis
Western blot densitometry analysis was performed using ImageJ analysis software (National Institutes of Health, Bethesda, MA). All phosphorylated proteins were normalized to corresponding total protein expression and BCA2 to β-actin. Fold change was calculated on the basis of the vehicle control/untreated lane, and error bars are means ± SEM for repeated experiments. Statistical analyses of the MTT and colony formation assays were carried out using an unpaired parametrical t test with Welch's correction to compare the means of six observations for each combination treatment to the single agent condition in both the colony formation and the MTT assays, followed by one-way analysis of variance to compare the combination treatments to single agent and then to the control. All statistical tests were two-tailed, and P < .05 was considered to be statistically significant. GraphPad Prism 6 Software was used.
Results
BCA2 Is an Endogenous Inhibitor of AMPKα Thr172 Phosphorylation and Therefore Activation
To study the potential relationship between BCA2- and AMPK-involved pathways, we first assessed the basal expression of each protein in a panel of four breast cancer cell lines (Figure 1A). We observed an inverse relationship of BCA2 to pAMPKα1 (Thr172): Those cell lines with higher levels of BCA2, like MDA MB 468 and Hs578t, have lower pAMPKα1 (Thr172) levels when compared to the other two examined, namely, MCF7 and MDA MB 231, which contain relatively lower amounts of BCA2 but higher levels of pAMPKα1 (Thr172; Figure 1A). It has previously been reported that BCA2 contains a predicted AKT phosphorylation site and that AKT-mediated phosphorylation at this site is responsible for stabilization of the BCA2 protein [17]. We detected levels of pAkt (S473) to be correlated positively to that of BCA2 (Figure 1A). These data provide grounds to further investigate the inverse relationship between BCA2 and pAMPK, allowing us to hypothesize that BCA2 may be an endogenous inhibitor of AMPK activation in breast cancer cells.
Figure 1.

BCA2 inhibits AMPKα1 phosphorylation and activation. (A) The assessment of basal protein expression levels in the four indicated human breast cancer cell lines. Exponentially growing cells were lysed for Western blot analysis with specific antibodies to BCA2, pAMPK (Thr172), total AMPKα1, pAKT (S473), total AKT, and β-actin (loading control). (B) Immunoblot of MDA MB 231 cells (grown in a six-well plate) transfected with BCA2 siRNA (x-axis, µg) for 72 hours. Scrambled, non-silencing siRNA and untreated cells were used as controls. Cell extracts were used for Western blots with specific antibodies to those in A as well as pACC (Ser79) and total ACC. Densitometry analysis is a representative of the mean ± SEM for three independent experiments. pAMPK was normalized to total AMPKα, pACC to total ACC, and BCA2 to β-actin. Fold change was calculated compared to the control. (C, D) MCF7 (C) and MDA MB 468 (D) cells were transfected with 5 µg of either BCA2 siRNA or scrambled control for assessment of the effect on pAMPK as done in B. (E) Immunoblot of HEK293T cells co-transfected with BCA2 wt and AMPKα1 (2.5 µg each). After a 48-hour transfection, cells were either treated with metformin (20 mM) or AICAR (1 mM) for 6 hours or left untreated. Cell extracts were used for Western blots with specific antibodies as previously stated. FLAG was used as a measure of transfection efficiency.
To provide direct support for our hypothesis, we used BCA2-specific siRNA to inhibit the expression of BCA2 in MDA MB 231 cells, followed by measurement of subsequent levels of endogenous pAMPKα1 (Thr172). Suppression of BCA2 occurred in an siRNA concentration-dependent manner (reaching 80% inhibition; Figure 1B); importantly, this was accompanied by a significant (11-fold) increase in pAMPKα1. Total AMPKα1 protein levels, however, remained relatively unchanged (Figure 1B). To further confirm that the increase of pAMPK was also active, the phosphorylation status of ACC, a direct downstream target of AMPK, was assessed [18]. Knockdown of BCA2 protein resulted in a concentration-dependent increase (up to seven-fold) in levels of phosphorylated ACC (pACC; S79), mirroring pAMPK, while levels of total ACC were unchanged compared to the control (Figure 1B). The non-silencing scrambled siRNA negative control had no effect on either BCA2 protein expression or AMPK signaling (Figure 1B, lane 2 vs lane 1). When this experiment was repeated in two other breast cancer cell lines, MCF7 and MDA MB 468 (Figure 1, C and D), inhibition (68% and 42%, respectively) of BCA2 expression by its siRNA again resulted in increased levels of pAMPKα1 (2.5- and 1.4-fold, respectively; Figure 1, C and D).
More evidence of BCA2 as an endogenous AMPK inhibitor is shown in Figure 1E, where the BCA2 gene was subcloned into a pCMV-tag2B vector construct and herein used to co-transfect HEK293T fibroblast cells together with a pCMV6-XL5 AMPK α1 subunit containing construct. HEK293T cells express low basal levels of AMPKα1 and BCA2 (Figure 1E, lane 1), making them a good model for transfection experiments. Co-transfection of the AMPKα1 subunit and the pCMV empty vector increased cellular levels of pAMPKα1 (Figure 1E, lane 2 vs lane 1), while co-transfection of AMPKα1 and BCA2 had little or no increase in the basal level of pAMPKα1 (Figure 1E, lane 3 vs lane 2 vs lane 1), confirming that BCA2 inhibits basal levels of AMPKα1 activation. To determine whether BCA2 could also inhibit induced AMPKα activation, the co-transfected cells were treated with AMPK activator metformin or AICAR. Metformin is an indirect activator of AMPK, while AICAR is an analog of AMP and can therefore mimic its cellular effect and directly bind its β regulatory subunit, activating AMPK in culture [19]. A robust induction of pAMPKα1 was observed in cells co-transfected with AMPKα and pCMV empty vector after treatment with metformin or AICAR (Figure 1E, lanes 4 and 6 vs lane 2). Importantly, this induction was almost completely inhibited by transfected BCA2 (Figure 1E, lane 5 vs lane 4 and lane 7 vs lane 6). Again, BCA2 transfection had little inhibitory effect on total AMPKα1 (Figure 1E). Therefore, these transfection studies reveal that BCA2 is an inhibitor of both basal and induced AMPK phosphorylation/activation. An increase in BCA2 levels was also observed in response to AMPK activators (Figure 1E, lanes 4 and 6 vs lane 2), suggesting that AMPK signaling may trigger BCA2 up-regulation as a feedback mechanism (see Figure 4).
Figure 4.
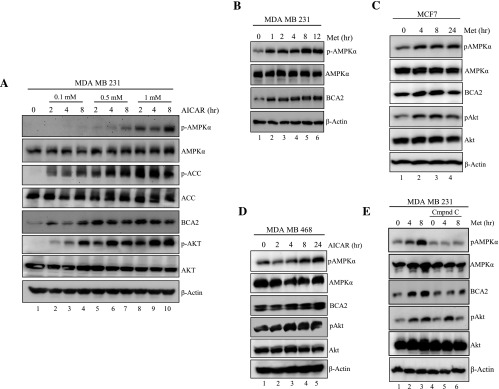
Activation of AMPK increases BCA2 protein levels in MDA MB 231 breast cancer cells. (A) Immunoblot of MDA MB 231 cells treated with AICAR at the indicated concentrations and time points. (B) Immunoblot of MDA MB 231 cells treated for up to 12 hours with 20 mM metformin. (C) Immunoblot of MCF7 and (D) of MDA MB 468 cells treated with 20 mM metformin for up to 24 hours. (E) MDA MB 231 cells were pretreated with 20 µm Compound C for 12 hours (lanes 4–6) or untreated (lanes 1–3), followed by treatment with 20 mM metformin for 4 or 8 hours (lanes 2, 3 and 5, 6). The prepared cell extracts in each experiment (representative of three repeats) were then used for Western blots with specific antibodies as indicated.
Requirement of the BCA2 RING Domain and S132/S133 Phosphorylation Sites for Inhibition of AMPK Activation
To gain insight into the molecular basis by which BCA2 suppresses AMPK activation, we determined whether its RING domain and S132/S133 phosphorylation sites, residues within its predicted Akt-binding domain, were required. The RING domain of BCA2 is critical for its E3 ligase function, and a cysteine mutation to alanine, at positions 228 and 231, renders the protein “ligase dead” and therefore ubiquitination negative [6], while AKT-mediated phosphorylation of BCA2 has been reported to increase its stability [8,17]. The serine 132 and 133 residues mutated to alanine would therefore increase its ubiquitination/autoubiquitination ability and subsequent degradation by the proteasome. It should be noted that these serines were predicted to be phosphorylated by Akt but may be susceptible to phosphorylation by other kinases in addition to Akt, which needs to be further characterized.
HEK293T cells were first co-transfected with the AMPKα1 subunit and a wt or an mt variant of BCA2, followed by examination of the effect on basal pAMPKα1 levels by Western blot analysis. Again, an increase in basal pAMPKα1 levels was seen when AMPKα1 was co-transfected with the pCMV empty vector control, which was inhibited in the presence of BCA2 wt (Figure 2A, lanes 1–3). However, co-transfection of the BCA2 RING mt failed to inhibit an increase in pAMPKα1 (Figure 2A, lane 4 vs lanes 3 and 2), indicating that the RING domain, and therefore the E3 ligase function of BCA2, is required for negative regulation of AMPK activation. The co-transfection of BCA2 S132/S133 mt still retained a partial inhibitory effect on pAMPKα1 levels in this experiment, compared to BCA2 wt and RING mt transfected cells (Figure 2A, lane 5 vs lanes 1–4), suggesting that this mt still confers enough stability (as shown by the level of FLAG) and therefore has partial inhibitory activity, perhaps due to its RING domain still being intact. Levels of FLAG and total AMPKα proteins were used as transfection efficiency controls and that of β-actin as the loading control (Figure 2A).
Figure 2.
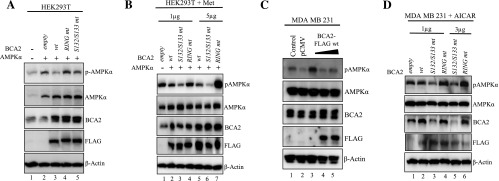
The requirement of the BCA2 RING domain and AKT phosphorylation sites for inhibition of AMPK. (A) Immunoblot of HEK293T cells (grown in 100-mm dishes) co-transfected with AMPKα1 and BCA2 wt, RING mt, or S132/S133 mt for the assessment of basal pAMPK levels. (B) Immunoblot of HEK293T cells co-transfected with a constant amount of AMPKα1 (1 µg) and increasing amounts of BCA2 wt or mt (1 or 5 µg) treated with metformin (20 mM) for 6 hours. (C) Immunoblot of MDA MB 231 cells transfected with 1, 3, and 5 µg of BCA2-FLAG wt to observe the effect on pAMPK. (D) MDA MB 231 breast cancer cells transfected with increasing concentrations of BCA2 wt and mt (1 or 3 µg) to detect changes in inducible (by AICAR) pAMPK levels. The prepared cell extracts in each experiment were used for Western blots with the specific antibodies as indicated. All experiments were repeated three times for consistency, with the included figure as a representative.
To further assess the requirement of the BCA2 RING domain and predicted AKT phosphorylation site, co-transfected HEK293T cells were treated with metformin (Figure 2B). While transfection of BCA2 wt (1 and 5 µg) inhibited the induction of pAMPKα1 by metformin (Figure 2B, lanes 2 and 5 vs lane 1), the BCA2 RING mt failed to do so (Figure 2B, lanes 4 and 7); in fact, a large increase in pAMPKα1 was seen in cells transfected with 5 µg of RING mt BCA2 (lane 7 vs lanes 4 and 1), confirming that the RING domain of BCA2 is critical for its ability to inhibit both basal and inducible levels of pAMPKα1, suggesting that the RING mt acts as a dominant negative inhibitor of endogenous BCA2 function. However, the transfection of BCA2 S132/S133 mt also inhibited AMPKα1 phosphorylation in a dose-dependent manner (Figure 2B, lanes 3 and 6 vs lane 1) and this inhibition was even greater than that seen by BCA2 wt (Figure 2B; lane 3 vs lane 2 and lane 6 vs lane 5) in this experiment. As the predicted Akt phosphorylation site residues, the results with this site mt were not entirely expected. While they may, in part, be explained by the intact RING domain of this construct, as seen under basal conditions in Figure 2A, the molecular mechanism for such a large inhibitory effect this mt produces on inducible AMPK remains unclear and will be further investigated in the near future.
We next progressed to confirm these results in human breast cancer cells. MDA MB 231 cells were transfected with increasing concentrations (1–5 µg/well) of BCA2 wt to observe the effect on basal pAMPKα1 levels. In Figure 2C, it can be seen that with increasing amounts of transfected BCA2 wt, basal AMPK phosphorylation is prevented, with some slight activation seen in the 1 µg lane (lane 3), but subsequent decrease with 3 and 5 µg of BCA2 wt (Figure 2C, lanes 4 and 5 vs lane 3). The same experiment was repeated in MDA MB 231 cells transfected with 1 and 3 µg of BCA2 wt, RING mt, S132/S133 mt, or pCMV empty vector control, followed by treatment of AICAR and measurement of the effect on induced pAMPKα1 (Figure 2D). The transfection of BCA2 wt had little inhibitory activity on inducible pAMPK levels (probably due to already high basal levels of endogenous BCA2 and AMPKα proteins; Figure 2D, lane 2 vs lane 1), while the transfected S132/S133 mt was again able to partly inhibit the induction of AMPK activation (Figure 2D, lanes 3 and 5 vs lane 1). Interestingly, the decrease in endogenous BCA2 protein in the presence of transfected S132/S133 mt compared to the RING mt was also observed (lanes 3 and 5 vs lanes 4 and 6). This may be explained by the still robust ubiquitination ability of this mt [17] and therefore targeted degradation of endogenous BCA2. Importantly, the BCA2 RING mt not only fully abolished the BCA2 inhibitory effect but also enhanced AICAR-mediated AMPK activation in a dose-dependent manner (Figure 2D, lanes 4 and 6 vs lanes 1 and 2). Taken together, our data demonstrate that BCA2 E3 ligase activity is essential for its inhibitory function on both basal and inducible AMPK activation.
Requirement of PI3/AKT Kinase Signaling for BCA2-Mediated AMPK Inhibition
To determine the role of AKT signaling on BCA2-mediated AMPK suppression, we used a chemical inhibitor of the PI3/AKT kinase signaling pathway, LY294002. Treatment of MDA MB 231 cells with LY294002 caused a dose-dependent inhibition (>90%) of pAkt at S473 associated with decreased levels of BCA2 protein (about 60%; Figure 3A). Importantly, this was also accompanied by a concentration-dependent increase in pAMPKα1 levels (up to four-fold; Figure 3A, lanes 2–5 vs lane 1). Moreover, in a kinetics experiment, 3- to 6-hour inhibition of pAkt (up to 90%) in MDA MB 231 cells occurred and was associated with BCA2 decrease (>75%) and an up to 2-fold increase in pAMPKα1 levels, which reached its highest point (almost 4.5-fold) at 24 hours (Figure 3B, lanes 2–5 vs lane 1). However, we again noticed an increase in BCA2 levels following AMPK activation and return of pAkt at 6 to 24 hours (Figure 3B), consistent with the idea that a potential feedback pathway is present (see Figure 4). Our data demonstrate that the dose- and time-dependent inhibition of AKT activity leads to a decrease in BCA2 levels and, subsequently, significantly increased levels of AMPK activation.
Figure 3.
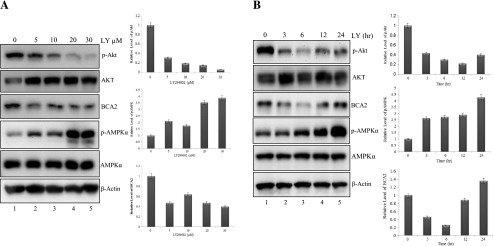
AKT inhibition decreases endogenous BCA2 protein levels, followed by increased AMPK activation. MDA MB 231 cells were treated with LY294002 in either a dosing study (A; 12-hour time point) or a kinetics experiment (B; 10 µm). The included figures are representatives of experiments done in triplicates, and the included densitometry is the mean ± SEM of phosphorylated proteins (normalized to total) and BCA2 to β-actin, compared to vehicle control (set to 1) for fold change expression. The prepared cell extracts in each experiment were used for Western blots with specific antibodies to pAKT (S473), BCA2, pAMPKα (Thr172), total AMPKα1, and β-actin as loading control. LY, LY294002.
Treatment of Breast Cancer Cells with an AMPK Activator Increases Endogenous BCA2 Protein Levels
In our previous experiments (Figures 1–3), we noted an increase in endogenous levels of BCA2 in response to the activation of AMPK. To test if AMPK signaling can, in turn, lead to up-regulation of the BCA2 protein, MDA MB 231 cells were treated with various concentrations of the direct AMPK activator, AICAR, for 2, 4, or 8 hours, followed by measurement of the effect on BCA2 by Western blot (Figure 4A). We observed a clear dose- and time-dependent increase in BCA2 protein levels, which corresponded to the induction of AMPK activation, as measured by specific phosphorylation of pAMPK and pACC (Figure 4A), demonstrating that BCA2 increase is a consequence of AMPK activation. Because AKT can phosphorylate and stabilize BCA2 [8,17], we determined whether AICAR treatment could also activate AKT. Indeed, levels of pAkt were increased in MDA MB 231 cells in an AICAR concentration- and time-dependent fashion, correlating well with that of the observed BCA2 protein increase (Figure 4A).
When MDA MB 231 cells were treated with metformin in a kinetics experiment, a time-dependent increase in BCA2 protein levels was again observed, in association with an induction of AMPK activation (Figure 4B). Furthermore, kinetics experiments using MCF7 cells treated with metformin (Figure 4C) or MDAMB 468 cells treated with AICAR (Figure 4D) also produced increased levels of BCA2 and pAkt proteins, associated with AMPK activation. Finally, the use of the chemical AMPK inhibitor, Compound C, not only suppressed activation of AMPKα1 but also inhibited the metformin-induced increase in BCA2 in MDA MB 231 cells (Figure 4E), suggesting the involvement of AMPK activity in a potential regulatory feedback mechanism with pAkt and BCA2 (see Figure 6).
Figure 6.

Proposed schematic. At the basal level, AKT works to stabilize BCA2 so that it is free to ubiquitinate protein substrates, including those controlling phosphorylation/dephosphorylation of AMPKα1 at Thr172, targeting them for degradation through the ubiquitin-proteasome pathway, resulting in inhibition of AMPK signaling in breast cancer cells. The activation of AMPK by metformin or AICAR, however, increases BCA2 protein levels, inciting a potential cancer cell survival involving PI3K/Akt. Therefore, the balance of AMPK and PI3K/AKT/BCA2 might control breast cancer cell sensitivity to metformin therapy and the tumor cell life-death switch.
To determine whether the metformin-mediated increase in BCA2 occurred on the transcriptional level, BCA2 mRNA levels and promoter activity were assessed in both co-transfected HEK293T and MDA MB 231 cells before and after metformin or AICAR treatment. From these experiments, we were able to conclude that AMPK activation increases levels of BCA2 protein only but not mRNA (see Figures W1 and W2).
Inhibition of BCA2 Significantly Enhances the Tumor Growth Inhibitory Effect of Metformin
If BCA2 is an endogenous inhibitor of AMPK phosphorylation/activation, it is logical to assess the growth inhibitory potential of metformin in combination with BCA2 inhibition. To do so, breast cancer MDA MB 231 (Figure 5A), MDA MB 486 (Figure 5B), and Hs578t (Figure 5C) cells were pretreated with BCA2 siRNA or negative control scrambled siRNA for 72 hours, followed by treatment with metformin at concentrations ranging from 1 to 30 mM for an additional 24 hours. Cell growth status was then analyzed by MTT assay. Again, BCA2 siRNA, but not control siRNA, inhibited the growth of MDA MB 231 breast cancer cells (59.2 vs 2.5%; Figure 5A). The combination of metformin and the control scrambled siRNA resulted in metformin dose-dependent growth inhibition, 5.5%, 31.0%, and 41.2% for 5, 15, and 30 mM, respectively (Figure 5A, bars 3–5). Importantly, the addition of metformin to BCA2 siRNA-transfected cells resulted in an even more significant reduction in growth, compared to metformin plus scrambled siRNA, reaching 82.8% (P < .001) at 5 mM, 86.0% (P < .001) at 15 mM, and 92.6% (P < .001) at 30 mM (Figure 5A, bars 7–9). MDA MB 468 and Hs578t cells (Figure 5, B and C) rendered also favorable and statistically significant results when combining BCA2 siRNA with metformin, compared to metformin alone. BCA2 siRNA alone was capable of inhibiting cell growth by 68.4% in MDA MB 468, followed by up to 89.7% inhibition with the addition of metformin (P < .001; Figure 5B). Hs578t, while not as sensitive to such treatment (31.1% inhibition with BCA2 siRNA), also reached up to 79% growth inhibition with the addition of metformin (P < .05; Figure 5C).
Figure 5.
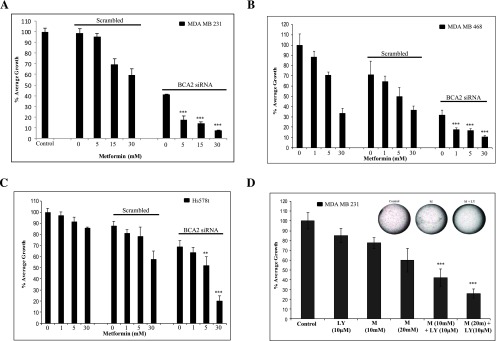
Inhibition of BCA2 enhances the antiproliferative effect of metformin. (A) MTT assay using MDA MB 231 cells transfected with BCA2 siRNA (0.75 µg per well of a 96-well plate) for 72 hours followed by treatment with 5, 15, and 30 mM metformin for 24 hours. Scrambled, non-silencing siRNA (0.75 µg) and untreated MDA MB 231 cells were used as controls. The included figure is a representative of three repeats, and the data are means ± SEM of those and based on an average of six observations for each condition in each experiment (two-tailed t test with Welch's correction comparing the means of combination treatment to metformin alone). (B, C) MDA MB 468 (B) and Hs578t (C) cells were used for reproducibility of those results shown in A and are as previously described for the indicated concentrations of metformin. (D) Soft agar colony formation assay using MDA MB 231 cells. Metformin (M) and LY294002 (LY) were added at concentrations shown for 6 days. Images (inserted) shown are representatives of six independent wells from the same treatment. The bar graph is a representative of repeated experiments, and the data are means ± SEM based on an average of six wells per condition per experiment (two-tailed t test with Welch's correction comparing means of the combination treatment to metformin alone). ***P < .001 and ** P < .05.
Similarly, a soft agar, anchorage-independent, colony formation assay was performed to further confirm the efficacy of metformin plus BCA2 inhibition using breast cancer MDA MB 231 cells, which were treated with either metformin alone, the PI3 kinase/AKT inhibitor LY294002 alone (which leads to BCA2 destabilization), or their combination (Figure 5D). The results show that the metformin and LY294002 combination is more effective than either agent alone, reaching 57.9% and 74.3% inhibition vs 22.5% and 40.2% with 10 and 20 mM metformin alone vs 14.8% with 10 µm LY294002 alone (bars 5 and 6 vs bars 2–4, P < .001; Figure 5D). Although Akt has been implicated in a number of cancer cell survival pathways and is therefore not specific to BCA2, previous published works, together with our positively correlated data herein, indicate it to be important for BCA2 stability and this experiment furthermore suggests that the disruption of the BCA2-mediated feedback regulatory loop may be a new avenue for improving metformin efficacy in breast cancer patients with aggressive breast tumors that overexpress BCA2.
Discussion
BCA2 overexpression in breast cancer cells and its growth-promoting activity have been well established, justifying it as a potentially attractive anticancer drug target [5–7,20]. Our current study first identifies an inverse relationship between endogenous BCA2 and pAMPKα1 (Thr172) expression levels in multiple human breast cancer cell lines (Figure 1A). Herein, we hypothesized that BCA2 is an endogenous inhibitor of AMPK activation and that BCA2 inhibition could therefore increase the efficacy of metformin (Figure 6). To test these hypotheses, we investigated if, and how, BCA2 is involved in regulating the tumor-suppressing AMPK signaling pathway. Toward this goal, we have identified BCA2 as an inhibitor of an upstream event controlling AMPK activation (Figure 6 and see discussion below). Furthermore, our data suggest the presence of a potential feedback mechanism where AMPK activation increases BCA2 protein expression (Figure 6). Although the existence of such a phenomenon adds to the controversy surrounding the use of the AMPK activator metformin for the treatment of breast cancer, it provides newfound awareness for the limitations that this type of therapy may have.
Posttranslational regulation of AMPK is just beginning to be understood. As the master regulator of cellular metabolism, this field of study has large implications in our understanding of cancer development, progression, and even occurrence and prevention, as recently nicely summarized in two review articles [21,22]. Evidence for ubiquitination of the catalytic α and regulatory β subunits of AMPK does exist. The E3 ligase, cidea, is the only identified E3 in this regard to be responsible for the ubiquitination of the AMPK β subunit in brown fat tissue, resulting in decreased AMPK activity due to proteasomal degradation [23], whereas the α subunit-specific E3 still remains to be discovered. Upstream of AMPK is a group of kinases including, but not limited to, LKB1, CaMKK, Tak1, MAP/microtubule affinity-regulating kinase 1 (MARK1), and AMPK related protein kinase 5, NUAK1, responsible for phosphorylation of the α subunit at the Thr172 site, and evidence of their individual ubiquitination, leading to decreased AMPK activity, also exists [24,25]. We have identified BCA2 as a negative regulator of AMPK by regulating an event(s) that controls the phosphorylation and dephosphorylation of the Thr172 site within the α1 subunit (Figure 1). Immunoprecipitation experiments have revealed that BCA2 does not bind the AMPK α1 subunit directly (see Figure W3), leading us to postulate that its negative effect is a result of BCA2 E3-mediated degradation of a target protein that is located upstream of AMPK and is responsible for regulating the (de)phosphorylation of the α1 subunit (Figure 6). On this account and in conjunction with our proteomics core, we have recently performed titanium dioxide (TiO2) phosphopeptide enrichment using MDA MB 231 cells treated with BCA2-specific siRNA or control siRNA, followed by mass spectrometry to identify potential leads that would have a place in the regulatory mechanism outlined within this work. We have identified several potential BCA2 target proteins that might directly regulate AMPKα phosphorylation and dephosphorylation. The complete analysis of these results is currently under way. Furthermore, the data in Figure 2 suggest that the BCA2 target protein is also a substrate of its E3 ligase activity since the RING domain is required for its inhibitory effect. Consistent with this argument, if this were a direct effect on the α1 subunit, we would expect the RING mt to have a similar effect to the wt, as this would be the substrate-binding ubiquitination site. The no change in total AMPKα observed is also supportive in this regard (Figure 2).
Phosphorylation is possibly the most common occurring posttranslational protein modification, often resulting in changes in protein conformation and stability, and BCA2 is no different in this regard. The fact that the transfected BCA2 S132/S133 mt also produced an inhibitory effect on basal pAMPKα1 in HEK293T cells (Figure 2A) points to the importance of an intact RING domain for its inhibitory activity. Furthermore, the even greater inhibitory effect of this mt protein seen in the presence of metformin/AICAR (Figure 2, B and D) also suggests the need for a more detailed characterization of these serine mutations made within the predicted Akt phosphorylation site. It is possible that other kinases may be in competition for phosphorylation of BCA2 at these sites and delineating the function of each residue under such conditions that induce AMPK activation as in Figure 2, B and D, is beyond the scope of this work and will be determined in future work. We also observed that the level of endogenous BCA2 in breast cancer cells transfected with S132/S133 mt after AICAR treatment was lower than those with BCA2 wt and RING mt (Figure 2D). This could also be due to the ubiquitination ability of this mt [17], which would target endogenous BCA2 protein for degradation. The detailed mechanisms behind this need to be investigated.
Therefore, our BCA2 mt variant analysis data draw at least one main conclusion: An event upstream of AMPK, regulating its (de)phosphorylation and activation, is likely the substrate of BCA2 E3 ligase activity (Figures 2A and 6). The importance of AKT in suppressing AMPK activation through BCA2 was confirmed in Figure 3 where the use of a PI3/AKT kinase inhibitor produced a correlative effect among pAkt inhibition, BCA2 protein decrease, and AMPK activation. These data suggest that BCA2-mediated inhibition of AMPK activation on the basal level is AKT-dependent.
Interest in metformin for the treatment of cancer, particularly breast cancer, sprouted as a result of the 2005 report on reduced cancer risk in diabetic patients to daily metformin intake, compared to diabetic populations on other therapies [26]. Since then, thousands of papers assessing metformin's anticancer properties both in vitro and in vivo have erupted. Phase II and III clinical trials are also under way. The use of metformin has been experimented with in a wide variety of cancer types, but that is not to say without much controversy. Generally speaking, it is agreeable that metformin works through indirect activation of AMPK. The mechanisms leading up this event are unclear, but it is believed to be due to metformin's ability to inhibit the mitochondrial respiratory complex I, thereby disrupting the ATP/AMP ratio and activating AMPK [27–29]. However to our knowledge, no direct data that show this binding taking place are available. Nevertheless, sustained activation of AMPK, and therefore limited ATP production, is expected to produce a state of energy homeostasis halting further energy consuming processes and creating an unfavorable environment for neoplastic cells. While many laboratories report these results, which have been recently reviewed [30], several other groups have also shown that AMPK activation in certain tumor types may contribute to cancer cell survival through different mechanisms [31–36], of which the downstream survival effectors have not yet been defined.
Our data show that the use of metformin and AICAR not only activates AMPK signaling but also produces a subsequent increase in BCA2 protein levels (Figure 4). This increase in BCA2 was further shown to be specific to AMPK activation, as it does not occur in the presence of the AMPK inhibitor, Compound C (Figure 4E). Given what is already known about BCA2 and its dependence on such a classic growth-promoting signal as AKT, it is logical to suggest that the observed BCA2 increase in response to AMPK activation by metformin or AICAR is related to a potential cell survival mechanism, a to-be-determined feedback loop ignited by AKT, and may incite cancer cell growth (Figure 6). This argument was supported by increased levels of pAKT and BCA2 proteins in AICAR- or metformin-treated breast cancer cells (Figure 4). Directly opposing these results are Zakikhani et al., who report inhibition of pAkt levels in response to metformin [36]. However, it is important to note the difference in experimental conditions between the work of Zakikhani et al. and the current study, which may affect what results are observed. Zakikhani et al. performed metformin treatment in serum-starved conditions for 72 hours, whereas the experiments done herein use complete media containing 10% serum. In addition, for the purpose of our molecular studies, AMPK activation at shorter time points is sufficient to observe the increase in pAkt and BCA2. Furthermore, literature in support of AMPK-activating pAkt results can be seen in various cancer types [37–40]. The reason for these discrepancies is still unclear; however, published reports in agreement with both observations are in existence. To confirm the regulatory role of BCA2 on AMPK activation, we treated three breast cancer cell lines with metformin in the absence or presence of BCA2 inhibition. We show that through the inhibition of BCA2, either directly by siRNA or indirectly by PI3/AKT kinase inhibition, the desirable anticancer effect of metformin is significantly enhanced (Figure 5).
While retrospective epidemiological data link metformin intake to reduced overall cancer occurrence in diabetics by 30% and some preliminary data testing its use in patients with breast cancer are available, it is yet to be proven that metformin improves the overall current clinical outcome for breast cancer victims, and furthermore, in the laboratory setting, the mechanisms underlying its reported preclinical anticancer activity are still unclear [26,41–44]. Most of the best available therapies are beneficial to fixed tumor types, for example, anti-estrogens for hormone-responsive breast cancers [45,46] or Herceptin for Her2-positive cases [47]. Taking into account the mixed reports in the literature on the effect of AMPK activation in a cancer context, it is important to consider that metformin use may also have beneficial or adverse effects depending on the cancer type and background, including the overexpression of BCA2. Therefore, our data convey that the screening of breast tumors, particularly those with a more aggressive phenotype, for BCA2 may not only provide a strong predictor of which patients may respond to metformin-based therapies but also suggest that the combination of metformin and a BCA2 inhibitor may be a more effective anticancer strategy than metformin alone in which the desirable reprogrammed cellular energy homeostatic state, characteristic of AMPK activation, could be maintained.
Supplementary Material
Acknowledgments
The authors acknowledge Yutaka Amemiya for providing the BCA2 (wt and mt) vector constructs, Di Chen for his assistance in the initial phase of this project, and the Translational Core at Karmanos Cancer Institute for the use of the FOTO/Analyst imaging workstation and Optronix GelCount colony counter.
Footnotes
This work was partially supported by National Cancer Institute R01CA127258 (to A.M.B. and Q.P.D.), National Institutes of Health Center grant P30 CA022453 (to Karmanos Cancer Institute), and the DeRoy Testamentary Foundation (PhD fellowship to D.B.).
This work is dedicated to the memory of Professor Angelika M. Burger who initiated the work on BCA2 here at Wayne State University. Dr Burger was exceptionally dedicated to science, focusing on the identification of novel molecular drug targets for the treatment of breast cancer and primarily ubiquitin E3 ligases. As a gifted researcher and colleague, as well as an outstanding mentor whose work was cut short by a battle with cancer in May of 2011, this article is a token of our appreciation for her efforts and devotion.
This article refers to supplementary materials, which are designated by Figures W1 to W3 and are available online at www.neoplasia.com.
References
- 1.Ciechanover A, Orian A, Schwartz AL. The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J Cell Biochem Suppl. 2000;34:40–51. doi: 10.1002/(sici)1097-4644(2000)77:34+<40::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP. From bortezomib to other inhibitors of the proteasome and beyond. Curr Pharm Des. 2013;19:4025–4038. doi: 10.2174/1381612811319220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen D, Dou QP. The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Curr Protein Pept Sci. 2010;11:459–470. doi: 10.2174/138920310791824057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nalepa G, Wade Harper J. Therapeutic anti-cancer targets upstream of the proteasome. Cancer Treat Rev. 2003;29(suppl 1):49–57. doi: 10.1016/s0305-7372(03)00083-5. [DOI] [PubMed] [Google Scholar]
- 5.Burger A, Amemiya Y, Kitching R, Seth AK. Novel RING E3 ubiquitin ligases in breast cancer. Neoplasia. 2006;8:689–695. doi: 10.1593/neo.06469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger AM, Gao Y, Amemiya Y, Kahn HJ, Kitching R, Yang Y, Sun P, Narod SA, Hanna WM, Seth AK. A novel RING-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005;65:10401–10412. doi: 10.1158/0008-5472.CAN-05-2103. [DOI] [PubMed] [Google Scholar]
- 7.Kona FR, Stark K, Bisoski L, Buac D, Cui Q, Dou QP. Transcriptional activation of breast cancer-associated gene 2 by estrogen receptor. Breast Cancer Res Treat. 2012;135:495–503. doi: 10.1007/s10549-012-2107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacopulos S, Amemiya Y, Yang W, Zubovits J, Burger A, Yaffe M, Seth AK. Effects of partner proteins on BCA2 RING ligase activity. BMC Cancer. 2012;12:63. doi: 10.1186/1471-2407-12-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyakawa K, Ryo A, Murakami T, Ohba K, Yamaoka S, Fukuda M, Guatelli J, Yamamoto N. BCA2/Rabring7 promotes tetherin-dependent HIV-1 restriction. PLoS Pathog. 2009;5:e1000700. doi: 10.1371/journal.ppat.1000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson DL, Lehninger AL, Cox MM. Lehninger Principles of Biochemistry. New York: W.H. Freeman; 2008. [Google Scholar]
- 11.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 12.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carling D, Hardie DG. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta. 1989;1012:81–86. doi: 10.1016/0167-4889(89)90014-1. [DOI] [PubMed] [Google Scholar]
- 14.American Diabetes Association, author. Standards of medical care in diabetes—2009. Diabetes Care. 2009);32(suppl 1):S13–S61. doi: 10.2337/dc09-S013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2:778–790. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 16.Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol. 2012;48:R31–R43. doi: 10.1530/JME-12-0007. [DOI] [PubMed] [Google Scholar]
- 17.Amemiya Y, Azmi P, Seth A. Autoubiquitination of BCA2 RING E3 ligase regulates its own stability and affects cell migration. Mol Cancer Res. 2008;6:1385–1396. doi: 10.1158/1541-7786.MCR-08-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies SP, Sim AT, Hardie DG. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem. 1990;187:183–190. doi: 10.1111/j.1432-1033.1990.tb15293.x. [DOI] [PubMed] [Google Scholar]
- 19.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 20.Burger AM, Kona F, Amemiya Y, Gao Y, Bacopulos S, Seth AK. Role of the BCA2 ubiquitin E3 ligase in hormone responsive breast cancer. Open Cancer J. 2010;3:116–123. doi: 10.2174/1874079001003010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- 22.Zungu M, Schisler JC, Essop MF, McCudden C, Patterson C, Willis MS. Regulation of AMPK by the ubiquitin proteasome system. Am J Pathol. 2011;178:4–11. doi: 10.1016/j.ajpath.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P. Downregulation of AMP-activated protein kinase by Cideamediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Control of AMPK-related kinases by USP9X and atypical Lys29/Lys33-linked polyubiquitin chains. Biochem J. 2008;411:249–260. doi: 10.1042/BJ20080067. [DOI] [PubMed] [Google Scholar]
- 25.Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. 2008;65:3737–3755. doi: 10.1007/s00018-008-8244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Logie L, Harthill J, Patel K, Bacon S, Hamilton DL, Macrae K, McDougall G, Wang HH, Xue L, Jiang H, et al. Cellular responses to the metal-binding properties of metformin. Diabetes. 2012;61:1423–1433. doi: 10.2337/db11-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 29.Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes. 2008;57:1414–1418. doi: 10.2337/db07-1552. [DOI] [PubMed] [Google Scholar]
- 30.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park HU, Suy S, Danner M, Dailey V, Zhang Y, Li H, Hyduke DR, Collins BT, Gagnon G, Kallakury B, et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther. 2009;8:733–741. doi: 10.1158/1535-7163.MCT-08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle. 2009;8:2385–2398. doi: 10.4161/cc.8.15.9082. [DOI] [PubMed] [Google Scholar]
- 34.Ríos M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, Señarís R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013;73:2628–2638. doi: 10.1158/0008-5472.CAN-12-0861. [DOI] [PubMed] [Google Scholar]
- 35.Phoenix KN, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERα negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat. 2009;113:101–111. doi: 10.1007/s10549-008-9916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;123:271–279. doi: 10.1007/s10549-010-0763-9. [DOI] [PubMed] [Google Scholar]
- 37.Kuznetsov JN, Leclerc GJ, Leclerc GM, Barredo JC. AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Mol Cancer Ther. 2011;10:437–447. doi: 10.1158/1535-7163.MCT-10-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sengupta TK, Leclerc GM, Hsieh-Kinser TT, Leclerc GJ, Singh I, Barredo JC. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol Cancer. 2007;6:46. doi: 10.1186/1476-4598-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leclerc GM, Leclerc GJ, Fu G, Barredo JC. AMPK-induced activation of Akt by AICAR is mediated by IGF-1R dependent and independent mechanisms in acute lymphoblastic leukemia. J Mol Signal. 2010;5:15. doi: 10.1186/1750-2187-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tao R, Gong J, Luo X, Zang M, Guo W, Wen R, Luo Z. AMPK exerts dual regulatory effects on the PI3K pathway. J Mol Signal. 2010;5:1. doi: 10.1186/1750-2187-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 42.Goodwin PJ, Pritchard KI, Ennis M, Clemons M, Graham M, Fantus IG. Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer. 2008;8:501–505. doi: 10.3816/CBC.2008.n.060. [DOI] [PubMed] [Google Scholar]
- 43.Campagnoli C, Pasanisi P, Abbà C, Ambroggio S, Biglia N, Brucato T, Colombero R, Danese S, Donadio M, Venturelli E, et al. Effect of different doses of metformin on serum testosterone and insulin in non-diabetic women with breast cancer: a randomized study. Clin Breast Cancer. 2012;12:175–182. doi: 10.1016/j.clbc.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 44.Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, Jellema G, Deharo S, Hardie DG, Pusztai L, et al. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat. 2011;128:783–794. doi: 10.1007/s10549-011-1612-1. [DOI] [PubMed] [Google Scholar]
- 45.Early Breast Cancer Trialists' Collaborative Group, author. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 46.Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 47.Dent S, Oyan B, Honig A, Mano M, Howell S. HER2-targeted therapy in breast cancer: a systematic review of neoadjuvant trials. Cancer Treat Rev. 2013;39:622–631. doi: 10.1016/j.ctrv.2013.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.