

Abstract
This paper reports an overview on the chemistry of the indole alkaloid goniomitine focusing, mainly, on the methods of synthesis related to this natural product and analogs.
1. Introduction
The indole alkaloids belong to the class of natural substances displaying biological activities as well as a broad structural diversity. In view of these important properties, these products are target of study in the areas of isolation, identification, and synthesis [1–5]. Goniomitine (1) (Figure 1) is an indole alkaloid that was isolated and identified by Randriambola et al. [6] and Hashimoto and Husson [7]. The unique structure and biological activity of goniomitine have attracted the attention of several groups. This review describes the isolation, biogenesis hypothesis, chemical transformations, and syntheses of this alkaloid and analogs.
Figure 1.

Natural (−)- and unnatural (+)-goniomitine (1).
2. Isolation of Goniomitine
In the course of studies of the alkaloids of the genus Gonioma, Randriambola et al. [6] isolated, from the root bark of Gonioma malagasy, a crystalline compound named goniomitine with melting point of 150°C (ether-methanol), [α]D 20 −80° (c 0.9 in CHCl3), and molecular formula C19H26N2O (HRMS, M+∙ 298.2080, calculated for 298.2045). The structure of goniomitine was initially proposed as indicated in Figure 1, with 20S, 21R configuration, based on its NMR spectra. Its absolute structure was deduced through the correlation with other alkaloids from Aspidosperma found in the same plant from where goniomitine had been isolated. The relative structure of goniomitine (1) was soon after confirmed by Takano et al. [8] through the total enantioselective synthesis of the natural form of this alkaloid. It could be evidenced that the absolute structure of the compound 1 is enantiomeric to the one that had been initially proposed for 20S, 21R configuration.
3. Biogenesis of Goniomitine
Randriambola et al. [6] proposed that goniomitine (1) may be derived from the Aspidosperma skeleton of vincadifformine (2) by the successive steps depicted in Scheme 1.
Scheme 1.

Biogenetic hypothesis of transformation of vincadifformine (2) into goniomitine (1): (a) oxidative fission of the C-5, N-4 bond; (b) decarboxylation; (c) retro-Mannich reaction; (d) nucleophilic attack of the indole nitrogen on the iminium moiety.
4. Chemical Transformations and Syntheses of Goniomitine and Analogs
4.1. Chemical Transformations of Goniomitine
For the occasion of the structural determination of goniomitine (1) [6], this compound was transformed into the N-acetyl derivative 5 upon treatment with Ac2O in MeOH and into the N,O-diacetyl derivative 6 upon treatment with Ac2O in pyridine (Scheme 2). The formation of the acetylated compounds 5 and 6 confirmed the presence of the groups OH and NH in the structure of 1.
Scheme 2.

Chemical transformations of goniomitine (1) into the acetyl derivatives 5 and 6.
4.2. Synthesis of the Goniomitine Analog (+/−)-12
In order to ascertain unambiguously the unprecedented structure of the alkaloid goniomitine (1), Hashimoto and Husson [7] synthesized the goniomitine analog (+/−)-12 by the sequence of reactions depicted in Scheme 3.
Scheme 3.

Reagents and conditions: (a) (i) n-BuLi (2.2 equiv), hexane (reflux, 6 h) and (ii) methyl 3-(3-pyridyl)propanoate, THF (−78 to 15°C); (b) MeMgI (10 equiv), ethylene oxide (10 equiv), Et2O (1 h), reflux (2 h); (c) MeI, CH2Cl2 (reflux, 2 h); (d) H2, PtO2, MeOH (3 h); (e) H2, PtO2, NaOMe, MeOH (3 h).
4.3. Total Synthesis of (−)-Goniomitine by Takano
The first enantiocontrolled total synthesis of natural (−)-goniomitine (1) was published in 1991 by Takano et al. [8], who established the absolute stereochemistry of this alkaloid. This total synthesis, depicted in Scheme 4, starts with the chiral cyclopentadienone synthon (−)-13.
Scheme 4.

Reagents and conditions: (a) Zn (5.0 equiv), AcOH-EtOH (1 : 3), reflux (4 h); (b) EtI (2.0 equiv), t-BuOK (1.2 equiv), THF (−70 to −30°C, 15 min); (c) allyl bromide (2.0 equiv), t-BuOK (1.2 equiv), THF (−30°C, 5 min); (d) o-dichlorobenzene (reflux, 24 h); (e) LiAlH4 (1.0 equiv), CuI (0.5 equiv), HMPA-THF (1 : 4), −75°C (15 min); (f) propane-1,3-diyldithiotosylate (1.5 equiv), t-BuOK (3.0 equiv), t-BuOH-THF (1 : 4), 0°C; (g) KOH (5.0 equiv), t-BuOH (70°C, 12 h); (h) CH2N2, Et2O; (i) MeI (1.0 equiv), CaCO3 (5.0 equiv), 10% aq. MeCN (reflux, 1 h); (j) Ph3P (4.0 equiv), CBr4 (2.0 equiv), Et3N (3.0 equiv), CH2Cl2 (0°C, 5 min); (k) LDA (3.0 equiv), THF (−78°C, 10 min); (l) compound 20 (1.1 equiv), PdCl2(PPh3)2 (2%), CuI (5%), Et3N (reflux, 30 min); (m) NaOEt (10 equiv), Et3N (5%), EtOH (reflux, 3 h); (n) (i) dicyclohexylborane (1.5 equiv), THF (0°C, 30 min), (ii) 10% NaOH (1.0 equiv), 30% H2O2 (3.0 equiv), 0°C (30 min); (o) phthalimide (1.3 equiv), Ph3P (1.3 equiv), (i-PrO2CN)2 (1.3 equiv), THF (0°C, 10 min); (p) NH2NH2·H2O (4.0 equiv), EtOH (reflux, 2 h); (q) [Me2N=CH2]Cl (1.5 equiv), CH2Cl2 (r.t., 30 min); (r) MeI, MeOH (r.t., 10 min); (s) NaCN (1.3 equiv), DMF (100°C, 10 min); (t) POCl3 (6.0 equiv), toluene (reflux, 2 h); (u) NaBH4, MeOH, 0°C; (v) DIBAL (1.5 equiv), CH2Cl2 (−75°C, 10 min); (x) dil. H2SO4; (y) NaBH4; (z) 30% HCl-MeOH (1 : 10), reflux (30 min).
4.4. The First Biomimetic Approach to the Skeleton of Goniomitine from an Aspidosperma Alkaloid
The results from the study of biomimetic transformation of an Aspidosperma alkaloid (2) into the substances 39-40, with the skeleton of goniomitine (1), were published in 1995 by Lewin et al. [9]. The sequences of reactions for the discovery of a new biomimetic in vitro rearrangement are depicted in Scheme 5. Scheme 6 displays the proposed mechanism [9] for the transformation of compound 36 into the alkaloids 39 and 40.
Scheme 5.

Reagents and conditions: (a) m-CPBA (1.1 equiv), CH2Cl2 (r.t., 3 h); (b) 0.2 mol L−1 NaOH-MeOH (r.t., 5 min); (c) NaI (3.0 equiv), AcOH (r.t., 1.5 h); (d) 11 mol L−1 HCl (105°C, 10 min); (e) TFA (16 equiv), CH2Cl2 (r.t., 20 min); (f) TFA (r.t., 4 h); (g) TFA (16 equiv), CH2Cl2 (r.t., 15 h); (h) TFA (12.5 equiv), CH2Cl2 (r.t., 45 h).
Scheme 6.

4.5. Semisynthesis of (+)-(16S,20S,21R)-16-Hydroxymethyl-goniomitine from (−)-Vincadifformine
In continuation to the studies of chemical transformations of vincadifformine (2) into alkaloids analogs to goniomitine (1), Lewin and Schaeffer [10] published in 1995 the semisynthesis of (+)-16-hydroxymethyl-goniomitine (45). This alkaloid was obtained as a result of the attempts to synthesize (+)-goniomitine (1) from the compound 40, previously obtained from (−)-vincadifformine (2) (Scheme 5) [9]. In Scheme 7 are depicted the sequences of reactions that led to the synthesis of compound 45 as well as other alkaloids with tetracyclic skeleton of goniomitine (1).
Scheme 7.

Reagents and conditions: (a) NaBH3CN, AcOH (r.t., 1.5 h); (b) NaOH-MeOH (120°C, 1 h); (c) LiAlH4 (excess), THF (reflux, 3 h); (d) H2 (1 atm), 10% Pd-C, MeOH (r.t., 5 h); (e) TiCl3-H2O, MeOH (r.t., 20 h); (f) 30% HCl-MeOH (120°C, 1.5 h).
4.6. Synthesis of the Goniomitine Analogs 52–55 by Cycloaddition Reactions
In the year 1996, Gürtler et al. [11] published the synthesis of the goniomitine analogs 52–55 by [4 + 2] cycloaddition reactions between 2-vinylindoles and substituted cyclic enamines, via anodic oxidation (Scheme 8).
Scheme 8.

Reagents and conditions: (a) vinylindole 47 (1.0 equiv), enamine 49 (2.37 equiv), CH3CN, LiClO4 (0.1 mol L−1), electrolysis (480 mV versus Ag/AgNO3, current (20 to 2 mA), 200 min); (b) vinylindole 48 (1.0 equiv), enamine 49 (6.17 equiv), CH3CN, LiClO4 (0.1 mol L−1), electrolysis (480 mV versus Ag/AgNO3, current (20 to 2 mA), 200 min); (c) vinylindole 47 (1.0 equiv), enamine 50 (1.4 equiv), CH3CN, LiClO4 (0.1 mol L−1), electrolysis (480 mV versus Ag/AgNO3, current (20 to 2 mA), 40 min); (d) vinylindole 47 (1.0 equiv), enamine 51 (2.1 equiv), CH3CN, LiClO4 (0.1 mol L−1), electrolysis (480 mV versus Ag/AgNO3, current (20 to 2 mA), 200 min).
4.7. Proposal of Synthesis of Goniomitine by Alves
In the year 2000, Alves [12] presented his qualification exam of doctorate about a plan of synthesis of the indole alkaloid goniomitine (1). The convergent strategies and synthetic routes for the synthesis of this alkaloid, idealized on that occasion, are described in the supplementary material of this review, available online at http://dx.doi.org/10.1155/2013/292396.
4.8. Syntheses of Cytotoxic Bisindole Alkaloids
In the year 2000, Lewin et al. [13] published an article about a slight modification of the Borch reductive amination method (delayed addition of NaBH3CN) [14, 15], applied to compound 40, analog of the natural alkaloid goniomitine (1). As a result of this reaction, a series of new cytotoxic bisindole alkaloids was prepared, as depicted in Scheme 9.
Scheme 9.

Reagents and conditions: (a) compound 56 (5.0 equiv), NaBH3CN (immediate addition), MeOH (r.t., 16 h); (b) compound 56 (5.0 equiv), NaBH3CN (delayed addition, 20 min), MeOH (r.t., 16 h); (c) TiCl3-H2O (6.0 equiv), MeOH (r.t., 20 h); (d) Ac2O, Py (r.t., 48 h); (e) Ac2O, Py (r.t., 3 h); (f) CH2O, NaBH3CN, AcOH (r.t., 2 h); (g) LiAlH4, THF (reflux, 3 h).
In continuation to the studies of synthesis of cytotoxic bisindole alkaloids, Raoul et al. [16] published, in the year 2001, an article with a novel series of these alkaloids prepared by reductive amination of the compound 40 with various anilines, using the modified Borch amination conditions described in Scheme 9 (delayed addition (20 min) of NaBH3CN) [15]. The influence of substitution of the starting aniline on the reaction and on cytotoxicity of produced dimers is discussed in the paper.
4.9. Total Synthesis of (+/−)-Goniomitine by Pagenkopf
In the year 2008, Morales and Pagenkopf [17] published the total synthesis of racemic (+/−)-goniomitine (1), accomplished in 17 linear steps with 5.2% overall yield starting from commercially available δ-valerolactam (65). Their synthetic approach includes the application of a formal [3+2] cycloaddition between the highly functionalized nitrile 68 and the activated cyclopropane 69 to prepare the indole nucleus (Scheme 10).
Scheme 10.
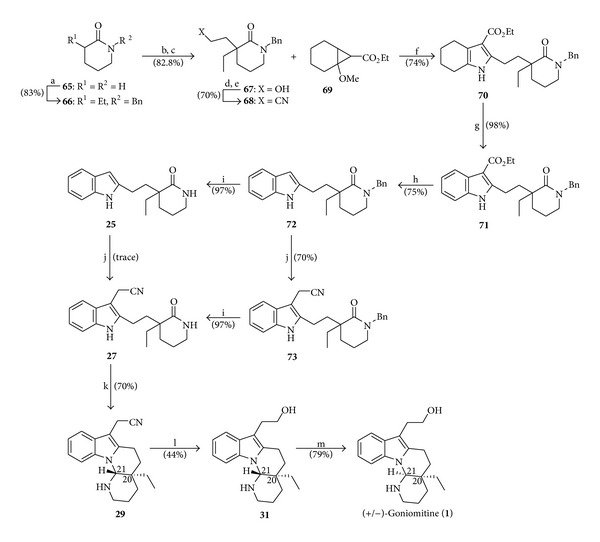
Reagents and conditions: (a) (i) n-BuLi (2.0 equiv), THF (−78°C), (ii) EtI (1.0 equiv), −78°C (1 h), (iii) BnBr (1.0 equiv), r.t. (overnight); (b) (i) LDA (1.0 equiv), THF (−78°C, 15 min), (ii) BrCH2CH2OTHP (1.1 equiv), r.t. (overnight); (c) TsOH (0.1 equiv), MeOH (ice-brine bath, 4 h); (d) Et3N (2.1 equiv), MsCl (1.0 equiv), CH2Cl2 (0°C to r.t., 3 h); (e) NaCN (2.0 equiv), MeCN, 120°C (μw, 8 h, 900 rpm stirring); (f) Nitrile 68 (1.0 equiv), cyclopropane 69 (2.9 equiv), TMSOTf (1.0 equiv), EtNO2 (−30°C, 24 h); (g) 5% Pd-C (0.03 equiv), mesitylene (reflux, 24 h); (h) NaOH (10 equiv), EtOH-H2O (1 : 1), 150°C (μw, 3 h, 900 rpm stirring); (i) Na (5.0 equiv), liq. NH3 (0.042 mol L−1), THF (−78°C, 10 min); (j) (i) [Me2N=CH2]Cl (1.5 equiv), CH2Cl2 (r.t., 15 min), (ii) MeI (40 equiv), MeOH (r.t., 10 min), (iii) NaCN (1.3 equiv), DMF (100°C, 10 min); (k) (i) POCl3 (6.0 equiv), toluene (reflux, 2 h), (ii) NaBH4 (2.0 equiv), MeOH (0°C, 30 min); (l) (i) DIBAL (1.5 equiv), CH2Cl2 (−78°C, 10 min), (ii) 0.75 mol L−1 H2SO4, (iii) NaBH4 (2.2 equiv), EtOH (0°C, 30 min); (m) TsOH (cat.), Et3N-MeOH (3 : 5, v/v), reflux (30 min).
4.10. Total Synthesis of (+/−)-Goniomitine by Waser
De Simone et al. [18] published the synthesis of racemic goniomitine (1) with the first study of its bioactivity, revealing significant cytotoxicity against several cancer cell lines [18, 19]. The strategy of this synthesis is based on cyclization of aminocyclopropanes [20], applied to cyclopropyl ketone 83 to lead to compound 84 with tetracyclic skeleton of goniomitine (Scheme 11).
Scheme 11.
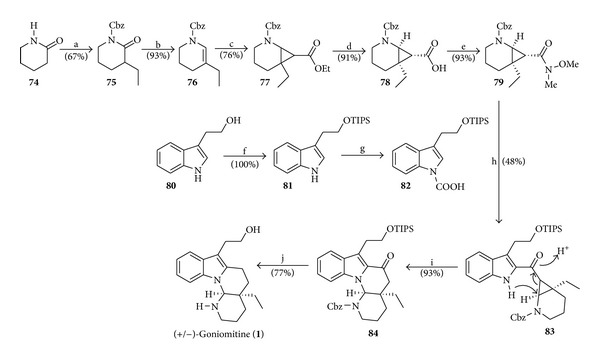
Reagents and conditions: (a) (i) n-BuLi (2.2 equiv), THF (0°C, 30 min), (ii) EtI (1.5 equiv), 0°C (20 min), (iii) benzyl chloroformate (1.05 equiv), 0°C (20 min); (b) (i) NaBH4 (1.05 equiv), MeOH (0°C, 15 min), (ii) conc. H2SO4, Et2O (r.t., 1 h); (c) N2CH2COOEt (4.0 equiv), (CuOTf)2·C7H8 (0.02 equiv), CH2Cl2 (18 h); (d) (i) BF3·OEt2 (0.15 equiv), CH2Cl2 (−20 to 0°C), (ii) NaOH (9.0 equiv), H2O-THF-EtOH (1 : 1 : 3), 0°C to 60°C (2 h); (e) (i) DMTMM (1.5 equiv), THF (r.t., 60 min), (ii) MeNHOMe.HCl (1.0 equiv), NMM (2.0 equiv), r.t. (36 h); (f) TIPSCl (1.05 equiv), imidazole (2.1 equiv), DMF (r.t., 1 h); (g) (i) n-BuLi (1.2 equiv), Et2O (0°C then reflux, 2 h), (ii) CO2 (0°C, 30 min), (iii) H3O+ (pH 2); (h) (i) t-BuLi (3.0 equiv), compound 82 (1.5 equiv), TMEDA (2.0 equiv), THF (−78°C, 3 h), (ii) amide 79 (1.0 equiv), THF (0°C, 20 min); (i) TsOH (0.2 equiv), CH2Cl2 (r.t., 10 min); (j) (i) NaBH4, MeOH (0°C to r.t., 3 h), (ii) Ac2O, Py (r.t., overnight), (iii) H2, Pd-C (0.1 equiv), EtOH, (iv) TBAF (4.4 equiv), THF (r.t., 30 min).
4.11. Total Syntheses of (+/−)-, (−)-, and (+)-Goniomitine by Mukay
In the year 2011, Mizutani et al. [21] published the syntheses of both racemic and optically active goniomitine, whose principal steps are the preparation of the indole skeleton by their own developed procedure [22] and alkene cross-metathesis. The synthesis of racemic (+/−)-goniomitine (1) was performed, as a preliminary study, by the sequence of reactions depicted in Scheme 12.
Scheme 12.

Reagents and conditions: (a) (i) compound 85 (1.3 equiv), n-BuLi (1.2 equiv), THF (−78°C, 1.5 h), (ii) compound 86 (1.0 equiv), THF (r.t., 22 h); (b) MsCl (1.6 equiv), Et3N (2.0 equiv), CH2Cl2 (0°C to r.t., 20 min); (c) (i) LDA (2.4 equiv), n-Bu3SnH (2.4 equiv), THF (−78°C, 1 h), (ii) CuBr·SMe2 (2.7 equiv), −78°C (40 min), (iii) mesylate 88 (1.0 equiv), THF (−78°C, 1 h); (d) (i) 2-I-PhNHBoc (1.28 equiv), compound 89 (1.0 equiv), TBAC (3.29 equiv), TFP (0.25 equiv), Pd2(dba)3 (0.03 equiv), CuI (0.11 equiv), DMF (r.t., 2 h); (e) (i) o-NO2PhSeCN (1.54 equiv), n-Bu3P (1.55 equiv), THF (r.t., 5 h), (ii) 30% aq. H2O2 (1.48 mol L−1), THF (0°C (20 min), r.t. (17 h)); (f) compound 90 (1.0 equiv), lactam 91 (9.44 equiv), Hoveyda-Grubbs-II cat. (0.3 equiv), neat (140°C, 3 h); (g) H2, 5% Pd-C (0.1 equiv), AcOEt (r.t., 23 h); (h) DIBAL (3.4 equiv), THF (−78°C to r.t.); (i) H2, 20% Pd (OH)2, AcOH-EtOH (5 : 2), r.t. (2 h); (j) TBAF (3.3 equiv), THF (r.t., 14 h).
The convergent total synthesis of the natural (−)-goniomitine (1) [21] was completed by the sequence of reactions depicted in Scheme 13.
Scheme 13.

Reagents and conditions: (a) (i) O3, MeOH (−78°C, 15 min) and (ii) NaBH4 (1.5 equiv), −78°C to r.t. (2 h); (b) (i) o-NO2PhSeCN (3.9 equiv), n-Bu3P (6.0 equiv), THF (r.t., 3 h) and (ii) 30% aq. H2O2, THF (0°C to r.t., 9 h); (c) indole 90 (1.0 equiv), lactam 97 (3.5 equiv), Hoveyda-Grubbs-II cat. (0.31 equiv), xylene (140°C, 3 h); (d) H2, 5% Pd-C (0.1 equiv), AcOEt (r.t., 27 h); (e) (i) NaH (17.8 equiv), Et2O (0°C, 30 min) and (ii) DIBAL (1.07 equiv), 0°C to r.t. (10 min), repeat three-times; (f) H2, 20% Pd (OH)2, n-PrOH/1,4-dioxane (1 : 1), r.t. (11 h); (g) TBAF (3.3 equiv), THF (r.t., 14 h).
Using the synthetic route described in Scheme 13, but starting from the enantiomer of the lactam 97 (ent- 97) Mizutani et al. [21] synthesized the unnatural (+)-goniomitine (ent-1). With the racemic, natural, and unnatural goniomitine in hand, the authors [21] executed the preliminary bioactive assays, which revealed that natural (−)-goniomitine has stronger antiproliferative activity in Mock and MDCK/MDR1 cells than its enantiomer.
4.12. Total Synthesis of (+/−)-Goniomitine by Bach
In the year 2012, Jiao et al. [23] published the total synthesis of racemic goniomitine (1), using the strategy of C-2 alkylation of indoles catalyzed by palladium via a norbornene-mediated C–H activation [24]. The steps for the synthesis of (+/−)-goniomitine (1), by this strategy, are depicted in Scheme 14.
Scheme 14.

Reagents and conditions: (a) (i) NaI (1.5 equiv), TMSCl (1.5 equiv), MeCN (r.t., 30 min), (ii) lactone 101 (1.0 equiv), MeCN (r.t., 16 h), (iii) TMSCl (0.5 equiv), EtOH (r.t., 71 h); (b) compound 102 (1.0 equiv), norbornene (2.01 equiv), K2CO3 (4.01 equiv), iodide 103 (4.01 equiv), PdCl2 (0.1 equiv), DMF-DMSO (9 : 1), H2O (0.5 mol L−1), air (60°C, 26 h); (c) (i) indole 104 (1.0 equiv), LiHMDS (3.0 equiv), THF (−78°C to r.t.), (ii) CH2=CHCH2Br (3.0 equiv) (−78°C (40 min), r.t. (30 min)); (d) (i) lactam 105 (1.0 equiv), 9-BBN (1.39 equiv), (0°C (15 min), r.t. (1 h)), (ii) aq. NaOH (1 mol L−1), 35% aq. H2O2 (0.18 mol L−1), 0°C (30 min); (e) alcohol 106 (1.0 equiv), PPh3 (2.08 equiv), DPPA (2.94 equiv), DIAD (2.8 equiv), 0°C to r.t. (3.5 h); (f) azide 107 (1.0 equiv), LiAlH4 (4.01 equiv), THF (0°C to r.t., 2 h); (g) AcOH-THF-H2O (3 : 1 : 1, v/v), 40°C (24 h).
4.13. Synthesis of (+)- and (−)-Goniomitine by Lewin
In the year 2013, Lewin et al. [25] have published the first biomimetic semisynthesis of goniomitine (1), in nine steps with 11% overall yield, starting from vincadifformine (2). Natural (−)- and unnatural (+)-goniomitine were prepared from (+)- and (−)-vincadifformine, respectively. The steps for the synthesis of unnatural (+)-goniomitine (1) are depicted in Scheme 15.
Scheme 15.

Reagents and conditions: (a) compound 40 (1.0 equiv), NaBH3CN (5.73 equiv), AcOH (r.t., 2.5 h); (b) TiCl3 (3.1 equiv), MeOH (r.t., 20 h); (c) compound 109 (1.0 equiv), HCO2NH4 (5.71 equiv), 10% Pd-C (0.33 equiv), MeOH (reflux, 45 min); (d) 4 mol L−1 HCl (100°C, 1 h).
Lewin et al. [25] have synthesized the natural (−)-goniomitine (1), starting from (+)-vincadifformine (ent-2), using the same conditions described in Scheme 15. The evaluation of the antiproliferative effect of (+)- and (−)-goniomitine (1), undertaken on five human cancer cell lines, has demonstrated that unnatural (+)-goniomitine is more potent than its enantiomer (−)-goniomitine [25], in opposition to Mizutani et al.'s results on a canine kidney cell line (MDCK II) [21].
4.14. Synthesis of (+/−)-Goniomitine by Zhu
In the year 2013, Xu et al. [26] have published a seven-step total synthesis of (+/−)-goniomitine (1) through two key steps: (i) a novel palladium-catalyzed decarboxylative coupling reaction between the potassium nitrophenyl acetate 118 and the vinyl triflate 115 for a rapid production of the functionalized cyclopentene 119; (ii) a late-stage construction of the whole tetracyclic scaffold of goniomitine (1) from the functionalized cyclopentene 120 by a one-pot integrated oxidation/reduction/cyclization (IORC) sequence (Scheme 16).
Scheme 16.

Reagents and conditions: (a) (i) compound 113 (2.0 equiv), CH3MgCl (2.0 equiv), THF (−78°C to r.t.), (ii) Mg (2.2 equiv), reflux (3 h), (iii) compound 112 (1.0 equiv), THF (reflux, 2 h), (iv) 2 mol L−1 HCl (0°C, 3 h); (b) TBSCl (1.1 equiv), imidazole (1.5 equiv), DMF (r.t, 3 h); (c) (i) CuBr·Me2S (2.0 equiv), EtMgBr (4.0 equiv), THF (−78 to −40°C, 40 min), (ii) compound 114 (1.0 equiv), THF (−40°C, 3 h), (iii) Comins' reagent (2.0 equiv), THF (r.t., 24 h); (d) compound 116 (1.0 equiv), ICH2CH2OBn (1.2 equiv), Cs2CO3 (1.3 equiv), DMF (60°C, overnight); (e) compound 117 (1.0 equiv), 10% aq. KOH, MeOH-THF (5 : 1), r.t. (5-6 h); (f) t-BuOK (1.0 equiv), EtOH (r.t., 1 h); (g) (i) compound 118 (1.2 equiv), [PdCl(allyl)]2 (5 mol%), X-Phos (15 mol%), triflate 115 (1.0 equiv), diglyme (100°C, 2 h), (ii) TBAF (4.0 equiv), THF (r.t., 4 h); (h) compound 119 (1.0 equiv), Ph3P (2.1 equiv), DPPA (2.9 equiv), DIAD (2.8 equiv), THF (0°C to r.t., 3.5 h); (i) (i) compound 120 (1.0 equiv), NaHCO3 (5.0 equiv), MeOH, O3 (−78°C, 20–30 seg), (ii) Me2S (50 equiv), −78°C to r.t. (24 h), (iii) Zn (70 equiv), CaCl2 (20 equiv), MeOH (reflux, 2 h); (j) compound 121 (1.0 equiv), sodium naphthalenide (6.0 equiv), THF (−20°C, 15 min).
5. Conclusions
In summary, it may be concluded that this brief survey on the chemistry of goniomitine has covered the literature relative to this alkaloid and analogs from 1987 to the first semester of the year 2013. Taking into account the results published in this period, a considerable progress on the synthesis of this alkaloid has been verified in the last years (2008–2013) with the publications of five racemic and two enantiomeric syntheses. It is also important to emphasize the recent pioneering works on the bioactive assays performed with the racemic mixtures as well as both enantiomers of goniomitine. In spite of these progresses, the development of new efficient enantioselective synthetic strategies for this indole alkaloid, with low operational costs, is still a target to be reached.
Supplementary Material
This supplementary material displays the proposals of synthesis of the indole alkaloids (+/-)-goniomitine, the natural (-)-goniomitine, and the unnatural (+)-goniomitine. The idealized strategies and synthetic routes for the preparation of these alkaloids and stereisomers are described.
Abbreviations
- Ac:
Acetyl
- 9-BBN:
9-Borabicyclo[3.3.1] nonane
- Boc:
tert-Butoxycarbonyl
- Bn:
Benzyl
- n-Bu:
n-Butyl
- t-Bu:
tert-Butyl
- Bz:
Benzoyl
- DIAD:
Diisopropyl azodicarboxylate
- DIBAL:
Diisobutylaluminum hydride
- DMF:
N,N-Dimethylformamide
- DMSO:
Dimethyl sulfoxide
- DMTMM:
2,4-Dimethoxy-6-(4-methylmorpholin-4-ium-4-yl) chloride
- DPPA:
Diphenylphosphoryl azide
- Et:
Ethyl
- HMPA:
Hexamethylphosphoramide
- LDA:
Lithium diisopropylamide
- LiHMDS:
Lithium bis(trimethylsilyl)amide
- m-CPBA:
meta-Chloroperbenzoic acid
- Me:
Methyl
- Ms:
Mesyl (methanesulfonyl)
- NMM:
N-methylmorpholine
- Pd2(dba)3:
Tris(dibenzylideneacetone)dipalladium(0)
- i-Pr:
iso-Propyl
- n-Pr:
n-Propyl
- Ph:
Phenyl
- Py:
Pyridine
- TBAC:
Tetrabutylammonium chloride
- TBAF:
Tetrabutylammonium fluoride
- TBDPS:
tert-Butyldiphenylsilyl
- TBS:
tert-Butyldimethylsilyl
- Tf:
Trifluoromethanesulfonyl
- TFA:
Trifluoroacetic acid
- TFP:
Tetrafluorophenyl
- THF:
Tetrahydrofuran
- THP:
Tetrahydropyranyl
- TIPSCl:
Triisopropylsilyl chloride (chlorotriisopropylsilane)
- TMEDA:
N, N, N′, N′-Tetramethylethylenediamine
- TMSCl:
Trimethylsilylchloride
- TMSOTf:
Trimethylsilyl trifluoromethanesulfonate
- Ts:
Tosyl (p-toluenesulfonyl)
- X-Phos:
2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl.
References
- 1.Saxton JE. Recent progress in the chemistry of the monoterpenoid indole alkaloids. Natural Product Reports. 1997;14(6):559–590. doi: 10.1039/np9951200385. [DOI] [PubMed] [Google Scholar]
- 2.El-Sayed M, Verpoorte R. Catharanthus terpenoid indole alkaloids: biosynthesis and regulation. Phytochemistry Reviews. 2007;6(2-3):277–305. [Google Scholar]
- 3.Frederich M, Tits M, Angenot L. Potential antimalarial activity of indole alkaloids. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2008;102(1):11–19. doi: 10.1016/j.trstmh.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Ziegler J, Facchini PJ. Alkaloid biosynthesis: metabolism and trafficking. Annual Review of Plant Biology. 2008;59:735–769. doi: 10.1146/annurev.arplant.59.032607.092730. [DOI] [PubMed] [Google Scholar]
- 5.Hájícek J. Recent developments in syntheses of the post-secodine indole alkaloids. Part III: rearranged alkaloid types. Collection of Czechoslovak Chemical Communications. 2011;76(12):2023–2083. [Google Scholar]
- 6.Randriambola L, Quirion J-C, Kan-Fan C, Husson H-P. Structure of goniomitine, a new type of indole alkaloid. Tetrahedron Letters. 1987;28(19):2123–2126. [Google Scholar]
- 7.Hashimoto C, Husson H-P. Synthetic and structural studies in the goniomitine alkaloid series: a new reductive cyclization reaction in the indole field. Tetrahedron Letters. 1988;29(36):4563–4566. [Google Scholar]
- 8.Takano S, Sato T, Inomata K, Ogasawara K. The enantiocontrolled total synthesis of natural (-)-goniomitine. Journal of the Chemical Society, Chemical Communications. 1991;(7):462–464. [Google Scholar]
- 9.Lewin G, Schaeffer C, Lambert PH. New rearrangement of an Aspidosperma alkaloid: the first biomimetic entry in the goniomitine skeleton. The Journal of Organic Chemistry. 1995;60(11):3282–3287. [Google Scholar]
- 10.Lewin G, Schaeffer C. Semisynthesis of (+)-(16S, 20S, 21R)-16-hydroxymethyl-goniomitine from (−) vincadifformine. Natural Product Letters. 1995;7(3):227–234. [Google Scholar]
- 11.Gürtler CF, Steckhan E, Blechert S. [4 + 2]-cycloaddition reactions between β-acceptor-substituted enamines and 2-vinylindole radical cations acting as hetero-dienes. The Journal of Organic Chemistry. 1996;61(12):4136–4143. doi: 10.1021/jo951551o. [DOI] [PubMed] [Google Scholar]
- 12.Alves JCF. Proposal of enantioselective synthesis of the indole alkaloid (−)-goniomitine and stereoisomers, [Qualification Exam of Doctorate] Rio de Janeiro, Brazil: NPPN, Universidade Federal do Rio de Janeiro; 2000. [Google Scholar]
- 13.Lewin G, Schaeffer C, Hocquemiller R, et al. Access to new cytotoxic bisindole alkaloids by a modified Borch reductive amination process. Heterocycles. 2000;53(11):2353–2356. [Google Scholar]
- 14.Borch RF, Bernstein MD, Durst HD. The cyanohydridoborate anion as a selective reducing agent. Journal of the American Chemical Society. 1971;93(12):2897–2904. [Google Scholar]
- 15.Lewin G, Schaeffer C. One-pot access to 2,3-disubstituted 1,2,3,4-tetrahydroquinolines by reductive amination of aldehydes with sodium cyanoborohydride. Heterocycles. 1998;48(1):171–174. [Google Scholar]
- 16.Raoul M, Schaeffer C, Léonce S, et al. Synthesis of a novel series of cytotoxic bisindole alkaloids. Bioorganic & Medicinal Chemistry Letters. 2001;11(1):79–81. doi: 10.1016/s0960-894x(00)00600-4. [DOI] [PubMed] [Google Scholar]
- 17.Morales CL, Pagenkopf BL. Total synthesis of (±)-goniomitine via a formal nitrile/donor-acceptor cyclopropane [3 + 2] cyclization. Organic Letters. 2008;10(2):157–159. doi: 10.1021/ol702376j. [DOI] [PubMed] [Google Scholar]
- 18.de Simone F, Gertsch J, Waser J. Catalytic selective cyclizations of aminocyclopropanes: formal synthesis of aspidospermidine and total synthesis of goniomitine. Angewandte Chemie International Edition. 2010;49(33):5767–5770. doi: 10.1002/anie.201001853. [DOI] [PubMed] [Google Scholar]
- 19.De Simone F, Gertsch J, Waser J. Corrigendum: catalytic selective cyclizations of aminocyclopropanes: formal synthesis of aspidospermidine and total synthesis of goniomitine. Angewandte Chemie International Edition. 2011;50(18):p. 4038. doi: 10.1002/anie.201001853. [DOI] [PubMed] [Google Scholar]
- 20.De Simone F, Waser J. Cyclization of aminocyclopropanes in indole alkaloids synthesis. Synlett. 2011;(5):589–593. doi: 10.2533/chimia.2012.233.P00710ST [DOI] [PubMed] [Google Scholar]
- 21.Mizutani M, Inagaki F, Nakanishi T, Yanagihara C, Tamai I, Mukai C. Total syntheses of (−)- and (+)-goniomitine. Organic Letters. 2011;13(7):1796–1799. doi: 10.1021/ol200320z. [DOI] [PubMed] [Google Scholar]
- 22.Mukai C, Takahashi Y. A new entry to the synthesis of 2,3-disubstituted indoles. Organic Letters. 2005;7(26):5793–5796. doi: 10.1021/ol052179u. [DOI] [PubMed] [Google Scholar]
- 23.Jiao L, Herdtweck E, Bach T. Pd(II)-catalysed regioselective 2-alkylation of indoles via a norbornene-mediated C-H activation: mechanism and applications. Journal of the American Chemical Society. 2012;134(35):14563–14572. doi: 10.1021/ja3058138. [DOI] [PubMed] [Google Scholar]
- 24.Jiao L, Bach T. Palladium-catalyzed direct 2-alkylation of indoles by norbornene-mediated regioselective cascade C-H activation. Journal of the American Chemical Society. 2011;133(33):12990–12993. doi: 10.1021/ja2055066. [DOI] [PubMed] [Google Scholar]
- 25.Lewin G, Bernadat G, Aubert G, Cresteil T. Semisynthesis of (+)- and (−)-goniomitine from (−)- and (+)-vincadifformine. Tetrahedron. 2013;69(5):1622–1627. [Google Scholar]
- 26.Xu Z, Wang Q, Zhu J. Palladium-catalyzed decarboxylative vinylation of potassium nitrophenyl acetate: application to the total synthesis of (+/−)-goniomitine. Angewandte Chemie International Edition. 2013;52(11):3272–3276. doi: 10.1002/anie.201209970. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This supplementary material displays the proposals of synthesis of the indole alkaloids (+/-)-goniomitine, the natural (-)-goniomitine, and the unnatural (+)-goniomitine. The idealized strategies and synthetic routes for the preparation of these alkaloids and stereisomers are described.