

A crystal structure of the L7Ae protein bound to the near-consensus k-turn Kt-7 shows that the protein binds in the exterior major groove using a helix and a hydrophobic loop. The interactions show how the protein is selective for the structure of the k-turn. The RNA structure in the complex is almost identical to that of protein-free RNA, consistent with a conformational selection model of binding.
Keywords: RNA structure, RNA-protein recognition, k-turn motif, X-ray crystallography
Abstract
L7Ae is a member of a protein family that binds kink-turns (k-turns) in many functional RNA species. We have solved the X-ray crystal structure of the near-consensus sequence Kt-7 of Haloarcula marismortui bound by Archaeoglobus fulgidus L7Ae at 2.3-Å resolution. We also present a structure of Kt-7 in the absence of bound protein at 2.2-Å resolution. As a result, we can describe a general mode of recognition of k-turn structure by the L7Ae family proteins. The protein makes interactions in the widened major groove on the outer face of the k-turn. Two regions of the protein are involved. One is an α-helix that enters the major groove of the NC helix, making both nonspecific backbone interactions and specific interactions with the guanine nucleobases of the conserved G•A pairs. A hydrophobic loop makes close contact with the L1 and L2 bases, and a glutamate side chain hydrogen bonds with L1. Taken together, these interactions are highly selective for the structure of the k-turn and suggest how conformational selection of the folded k-turn occurs.
INTRODUCTION
The L7Ae family of proteins (Koonin et al. 1994; Watkins et al. 2000) include the ribosomal proteins L7Ae and L30e, yeast snu31p, human 15.5-kDa protein (Nottrott et al. 1999), and bacterial homologs such as YbxF (Baird et al. 2012). These proteins bind to kink-turns (k-turns) in duplex RNA, stabilizing their folded conformation. These interactions are found in most types of functional RNA. In the ribosome, they mediate tertiary interactions and stabilize the structure. In box C/D snoRNPs, the binding of an L7Ae family protein to k-turn structures is the first step of assembly (Kuhn et al. 2002; Watkins et al. 2002) that then serves to recruit additional proteins to generate the functional apparatus for the fibrillarin-catalyzed site-specific methylation of RNA. L7Ae is also a critical component of the box H/ACA snoRNP (Rozhdestvensky et al. 2003) and U3 snoRNP (Marmier-Gourrier et al. 2003). In mammals, the 15.5-kDa protein binds to a k-turn in the U4 snRNA during the spliceosome cycle (Nottrott et al. 1999; Vidovic et al. 2000). L7Ae has recently been shown to be a subunit of archaeal RNaseP (Cho et al. 2010) required for tRNA maturation. Thus, L7Ae-family proteins are rather general k-turn binding proteins that are important in ribosome structure, spliceosome assembly, and the site-specific modification of RNA.
The standard k-turn comprises a 3-nucleotide (nt) bulge followed on its 3′ side by G•A and A•G pairs, and usually one further non-Watson-Crick pair (Fig. 1A). Although substitutions can be tolerated in most positions in varying degrees in some sequence environments, k-turns that are close to the consensus sequence appear to adopt the most stable folded conformation. Kt-7 of the Haloarcula marismortui 50S ribosomal subunit 23S rRNA is very close to being a consensus k-turn, and its interaction with Archaeoglobus fulgidus L7Ae protein has been extensively studied. It was shown that binding of L7Ae strongly shifted the conformational population to the folded state (Turner et al. 2005), with an apparent binding affinity of 10 pM (Turner and Lilley 2008). Recently in single-molecule experiments, we detected no folding intermediates down to a timescale of 8 msec, consistent with a conformational selection mechanism (Wang et al. 2012). A crystal structure at 2.7-Å resolution was determined for L7Ae bound to the box C/D k-turn (Moore et al. 2004) and for L7Ae bound to Kt-15 in the ribosome (Ban et al. 2000). However, Kt-15 is a complex k-turn whose structure does not map linearly onto the sequence.
FIGURE 1.
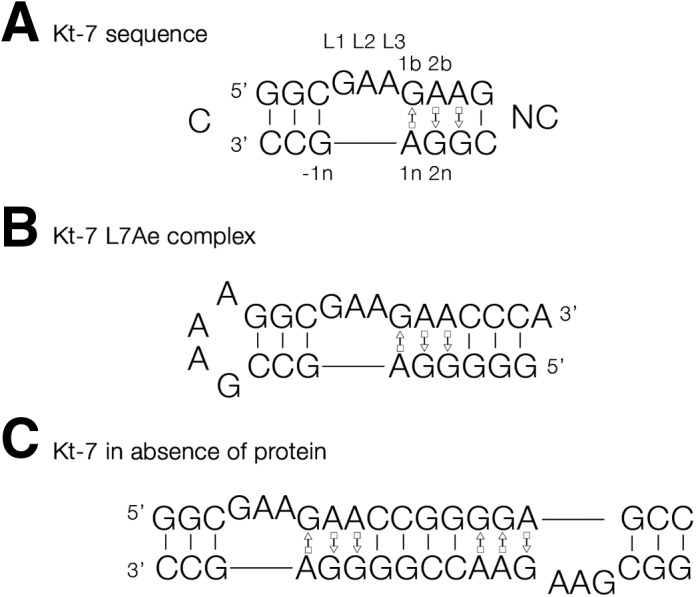
Sequence of Kt-7 and the RNA species used for crystallization. (A) The sequence of H. marismortui Kt-7, with key nucleotides labeled using our standard nomenclature (Liu and Lilley 2007). (B) A stem–loop RNA containing a single Kt-7 sequence. This was used for the crystallization of the complex of A. fulgidus L7Ae bound to Kt-7. (C) A duplex RNA containing two Kt-7 sequences with twofold rotational symmetry. This was crystallized in the absence of protein.
Kt-7 exhibits an interesting structural plasticity in terms of the hydrogen bonding in the core (Daldrop and Lilley 2013). Like most k-turns, the folded structure of Kt-7 is stabilized by two near-universal cross-strand hydrogen bonds, accepted by the nucleobases of the two conserved adenines. L1 O2′ donates its proton to N1 of the A1n, whereas the -1n O2′ donates its proton to either N3 or N1 of A2b. Almost all the known k-turn structures divide into one of two classes, depending on whether the acceptor is N3 or N1. We have previously shown that when the Kt-7 sequence is inserted into the SAM-I riboswitch, it is a member of the N3 class (Daldrop and Lilley 2013); whereas in the ribosome, it is an N1 class structure. This conformational switch results in a significant change of the rotational setting of the NC helix of the k-turn, with consequences for any tertiary interaction made by that helix. No atomic resolution structure has been determined to date for Kt-7 bound to a L7Ae family protein, so its structural class under these conditions is unknown. Nor is the structure of Kt-7 known in the absence of bound proteins or tertiary constraints.
We therefore decided to determine the crystal structure of the A. fulgidus L7Ae protein bound to the archetypal k-turn Kt-7 from H. marismortui, given the wealth of biophysical data that has been collected for this interaction. We have succeeded in obtaining a crystal structure of the complex at 2.3 Å. This is the highest resolution obtained to date for the complex of L7Ae with a standard k-turn, with excellent electron density that well defines the protein side chains in their interaction with the k-turn RNA. From this we present a general model for the recognition of k-turn structure by the L7Ae family of proteins.
RESULTS
Crystallization of a complex of Kt-7 with L7Ae
Well-diffracting crystals were obtained from an equimolar mixture of L7Ae with a 26-nt RNA oligonucleotide, where the C helix was closed by a GAAA tetraloop (Fig. 1B). The structure was determined by molecular replacement and subsequently refined to 2.3-Å resolution. In addition, we also succeeded in obtaining crystals of a 19-nt RNA containing the Kt-7 sequence in the absence of protein. The RNA formed a self-complementary duplex with two k-turns related by twofold symmetry (Fig. 1C). The structure was determined by molecular replacement and refined to 2.2-Å resolution. Crystallographic statistics are presented in Table 1.
TABLE 1.
Details of data collection and refinement statistics for the crystallographic data as deposited in the PDB
The interaction of L7Ae with Kt-7
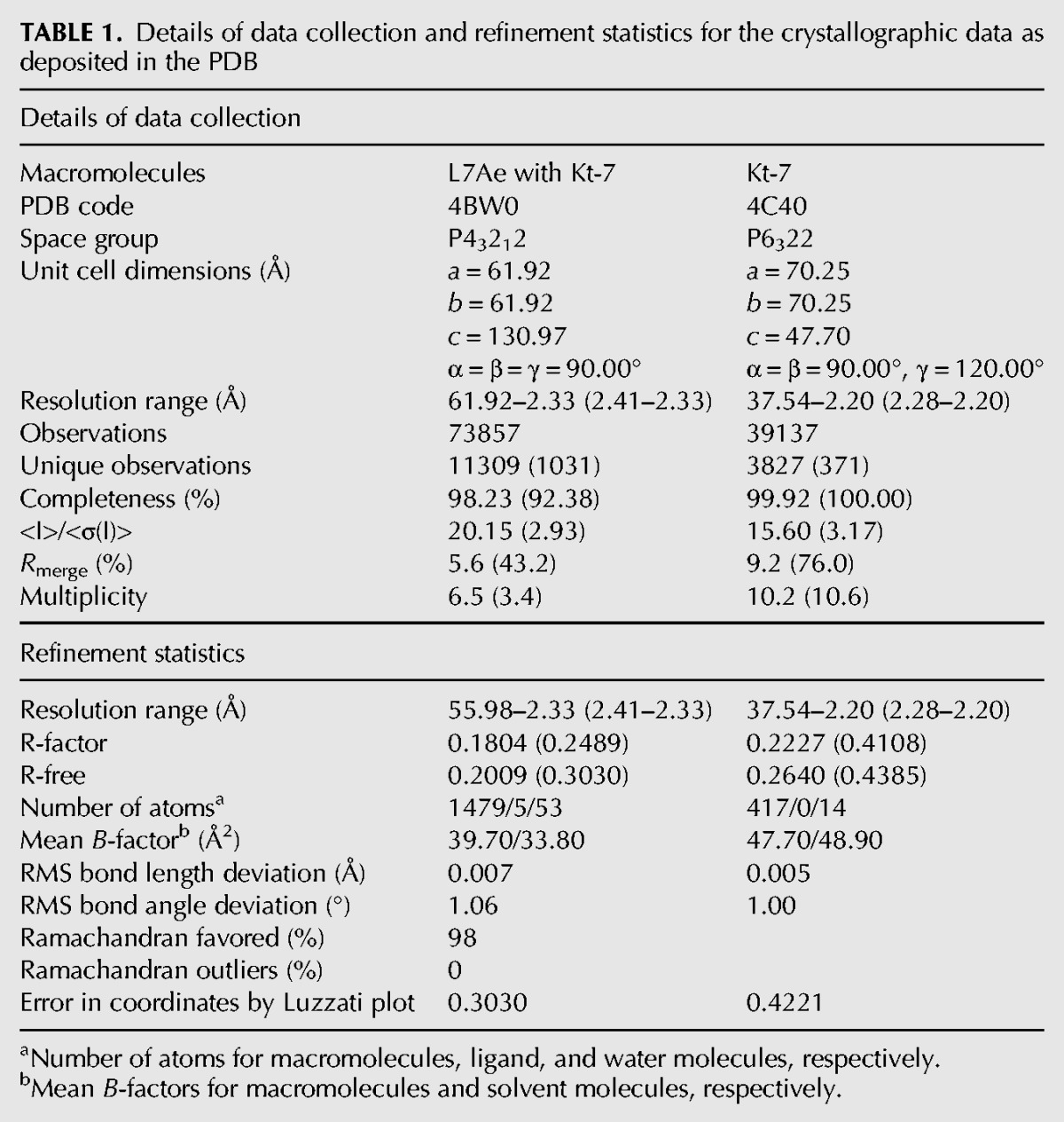
L7Ae adopts its standard fold in the complex (Fig. 2), comprising a four-strand mixed β-sheet flanked on both sides by α-helical sections. In overall terms, the structure is similar to that of the complex with the box C/D k-turn (Moore et al. 2004), with an RMSD of 0.46 Å. The protein binds in the major groove on the outer face of the Kt-7 k-turn, and two sections form the principle contacts with the RNA. The definition of the electron density of the new structure is excellent, and we can position most amino acid side chains with good confidence and thus identify important contacts with the k-turn. To exclude the possibility of model bias, we have calculated a composite omit map of the structure, and all the electron density shown is taken from that.
FIGURE 2.
The structure of the complex of L7Ae bound to Kt-7. Stereoscopic pairs (A,D,E) are shown as parallel-eye stereoscopic pairs. Electron density is taken from the composite omit map and contoured at 2σ. (A) An overall view of the complex. The basic β-strand:turn:helix and hydrophobic loop sections are colored blue, with hydrogen bonds between amino acid side chains and the RNA highlighted in red. The nucleotides of the k-turn are colored in our conventional scheme (loop purple; G•A pairs green; NC helix gray; C helix yellow), with the NC and C helices indicated. (B,C) Nonstereoscopic images of the structures of the two recognition elements of the protein, i.e., the α-helix (B) and the hydrophobic loop (C). (D,E) The interaction between the α-helix (D) and the hydrophobic loop (E) of the k-turn RNA. The electron density map is shown for the protein. Hydrogen bonds between amino acid side chains and the RNA are highlighted in red. Note that the N-terminal end of the α-helix is directed toward O6 of G1b.
One major set of contacts is made by the second α-helix from the N terminus, comprising residues 32–41, together with the preceding short β-strand (residues 27–30) that is part of the four-strand β-sheet (Fig. 2B,D). The helix is directed into the major groove of the NC helix, with its N-terminal end directed toward the guanine nucleobases of the two G•A pairs of the k-turn. The β-strand:turn:helix element is highly basic, and side chains make a number of contacts with the RNA. At the C-terminal end of the α-helix, basic side chains from successive turns are hydrogen bonded to backbone phosphate proS nonbridging oxygen atoms on the nonbulged strand of the NC helix. Arginine R41 is bonded to the 3n/4n phosphate (N–O distance = 3.0 Å) and lysine K37 to the 4n/5n phosphate (N–O distance = 2.7 Å). The side chains of lysine residues K27 and K29 are directed toward the backbone of the same nonbulged strand; these are too distant to form hydrogen bonds but could contribute to the electrostatic stabilization of the complex. At the N-terminal end of the α-helix, two successive amino acid side chains make specific hydrogen-bonded contacts to the G1b and G2n nucleobases. Glutamate E34 accepts a proton from N1 of G1b (N–O distance = 3.0 Å), and glutamine N33 donates a proton to O6 of G2n (N–O distance = 2.6 Å). These two interactions specifically recognize the position of the conserved guanine nucleobases of the k-turn in the major groove. Additionally, the positive pole of the helix dipole is directed toward the G1b O6 atom, providing significant electrostatic stabilization.
The other major protein–RNA contact occurs at the loop comprising residues 86–93 (Fig. 2C,E). This has the sequence VGIEVPC. This very hydrophobic loop makes close van der Waals contact with the L2 and L1 nucleobases of the k-turn loop. The hydrocarbon side chains of isoleucine I88 and valine V90 are on the lower face, with I88 directly facing the L2 nucleobase in its syn conformation. Altogether the contact buries 732 Å2. The single charged amino acid of the loop, glutamate E89 is hydrogen bonded to N1 of GL1 (N–O distance = 2.9 Å); the position of this side chain is very well defined by the electron density.
The structure of Kt-7 in the complex
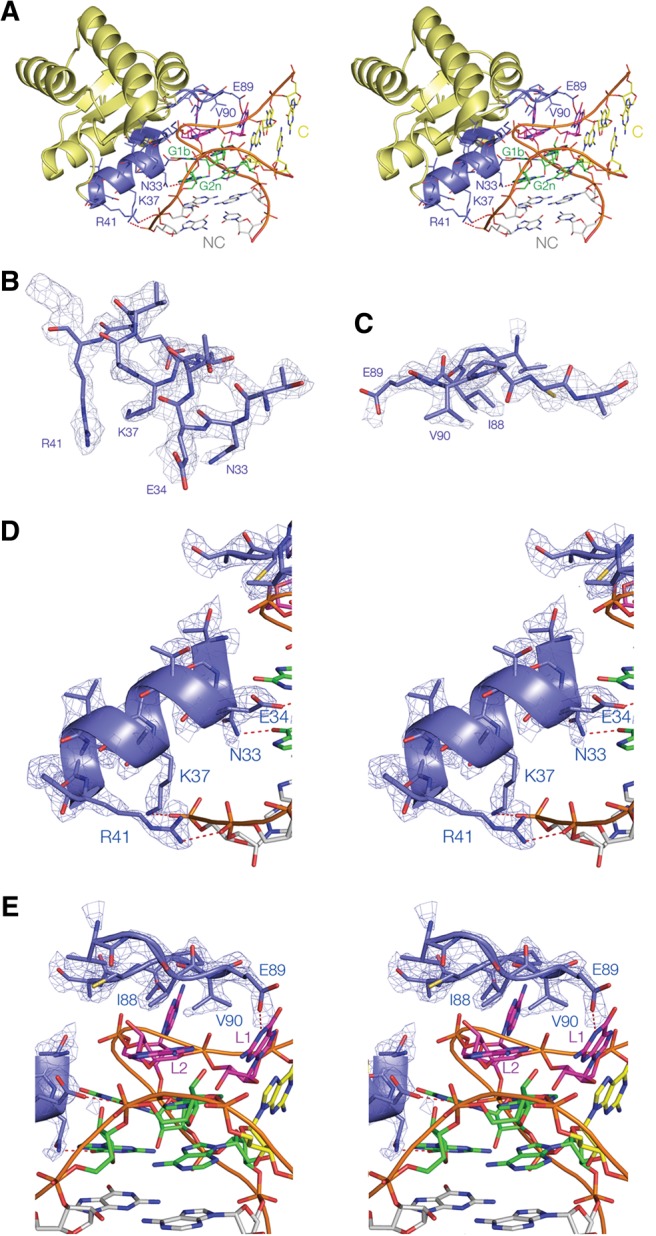
Within the complex with L7Ae, Kt-7 adopts a standard N3-class k-turn structure (Fig. 3A,B). The C helix is fully base-paired, and the loop nucleotides adopt their normal positions, i.e., the GL1 and AL2 nucleobases are stacked onto the C and NC helices, respectively, and AL3 is directed away from the RNA. AL2 has a syn glycosyl bond conformation, positioning it to stack on A1n. The G•A pairs at the 1b•1n and 2n•2b positions are conventional trans G (sugar)•A (Hoogsteen) pairs, followed by another in the 3n•3b position, after which Watson-Crick pairing resumes. Kt-7 has the standard cross-strand A-minor hydrogen bonds (Liu and Lilley 2007; Daldrop and Lilley 2013), from L1 O2′ to A1n N1 (O–N = 2.6 Å) and from G-1n O2′ to A2b N3 (O–N = 2.7 Å). Thus, the structure falls into the N3 class of k-turn structures (Daldrop and Lilley 2013). The near-universal hydrogen bond from AL3 O2′ to P1/P2 O proS (O–O = 2.3 Å) closes the loop of the k-turn. All the hydrogen bonds identified have acceptable angular geometry.
FIGURE 3.
The structure of Kt-7 in the complex with L7Ae and in the absence of bound protein. These are shown as parallel-eye stereoscopic pairs. (A) The complete k-turn structure, viewed from the side of the bulged strand, with the electron density shown contoured at 2σ is taken from the composite omit map. (B,C) The structures of Kt-7 in the complex (B) and protein-free (C), both viewed from the nonbulged strand side with the C-helix directed rightward.
The structure of Kt-7 in isolation
We have also solved the structure of Kt-7 as a duplex RNA in the absence of bound L7Ae, at a resolution of 2.2 Å (Fig. 3C). The structure of the k-turn is closely similar to that in the L7Ae-bound complex. The two structures superimpose with an RMSD of 0.83 Å (Fig. 4). All the standard hydrogen bonds are present, including the cross-strand A-minor interactions (Liu and Lilley 2007; Daldrop and Lilley 2013) and the hydrogen bond from AL3 O2′ to P1/P2 O proS (O–O = 2.6 Å). G-1n O2′ donates its proton to A2b N3 (O–N = 2.5 Å), and thus the structure is a member of the N3 class of k-turn structures (Daldrop and Lilley 2013).
FIGURE 4.
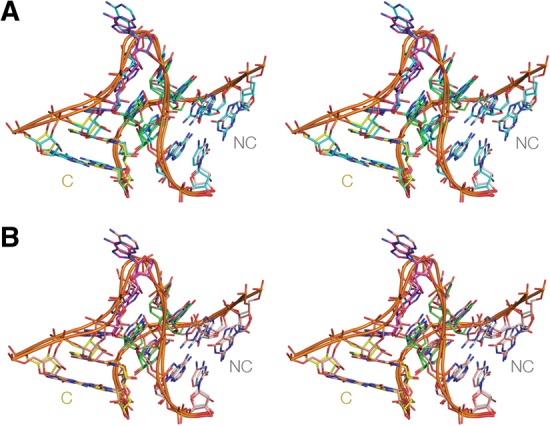
Superposition of crystal structures of Kt-7. The structure of Kt-7 from the complex with L7Ae (PDB:4BW0) is shown in our standard coloring. The structures are viewed from the side of the bulged strand. The images are shown in parallel-eye stereo. (A) Superimposition with the structure of Kt-7 as a protein-free duplex colored cyan (PDB:4C40). The RMSD between the two structures is 0.83 Å. (B) Superimposition with the structure of Kt-7 inserted into the SAM-I riboswitch (Daldrop and Lilley 2013) colored orange (PDB:4B5R). The RMSD between the two structures is 1.13 Å. Note that all three Kt-7 structures are in the N3 conformation.
Variability of adenine 2n base-pairing and its A-minor interaction with -1n O2′
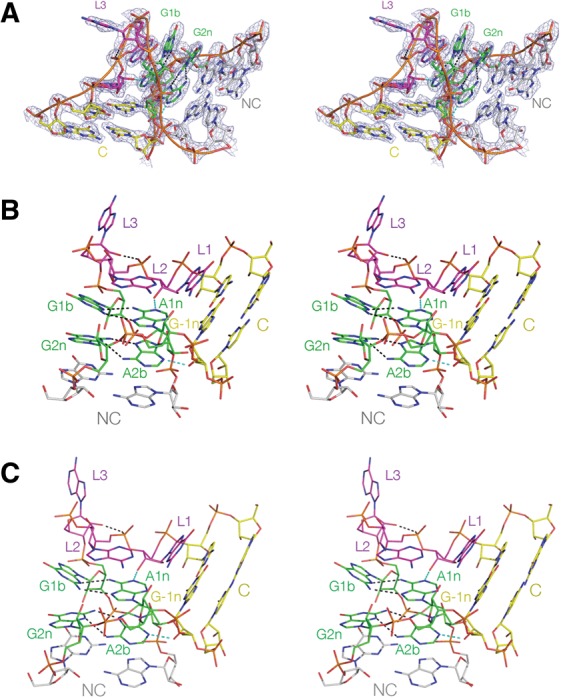
The structure of Kt-7 falls in the N3 class in both the complex with L7Ae and as a protein-free duplex. The angular position of the A2b nucleobase thus permits both hydrogen bonds to form with G2n, with a normal bond from A2b N6 to G2n N3 (N–N = 3.1 Å for the complex and 3.0 Å for the isolated k-turn). The consequent rotational setting of the helical axis results in a long distance between A3b O2′ and C-2n O2′ (O–O = 6.8 and 6.9 Å for the complex and free k-turn, respectively). The structures of the G2n•A2b–G-1n interactions are compared with the corresponding regions of Kt-7 in the SAM-I riboswitch (Daldrop and Lilley 2013) and the ribosome (Ban et al. 2000) in Figure 5. Kt-7 adopts the N3 structure in the L7Ae complex, free duplex, and the SAM-I riboswitch, whereas in the ribosome, it forms the N1 structure, with a consequent loss of the A2b N6 to G2n N3 hydrogen bond. The structure of the free Kt-7 in particular suggests that the N3 structure is intrinsically more stable than the N1 structure, perhaps because of a more stable G2n•A2b pair. In the ribosome Kt-7 is bound by L24, and the terminal loop of the C helix makes a loop–loop interaction with the adjacent stem–loop, connected via a three-way helical junction. Given the change in axial rotation between the N3 and N1 structures (Daldrop and Lilley 2013) (which is also the reason for the marked lengthening of the A3b–C-2n O2′ to O2′ distance), it is probable that the orientational requirements of the tertiary interactions stabilize the N1 conformation in the ribosome. This is a further demonstration of the conformational plasticity of the k-turn and its ability to accommodate the structural requirements of its environment.
FIGURE 5.

Comparison of the G2n•A2b–A-1n interaction in four Kt-7 structures. (A) Bound by L7Ae protein (this work, with electron density from the composite omit map); (B) as free RNA (this work, with electron density from the composite omit map); (C) engineered into the SAM-I riboswitch (Daldrop and Lilley 2013); and (D) in the H. marismortui ribosomal 50S subunit 23S rRNA (Ban et al. 2000). Structures A–C fall into the N3 class, with G-1n O2′ donating a proton to A2b N3 and two hydrogen bonds connecting A2b and G2n, whereas the ribosomal structure (D) is N1 class where G-1n O2′ donates a proton to A2b N1. The resulting rotation of the A2b nucleobase results in an A2b N6 to G2n N3 N–N length of 4.3 Å, i.e., too long to be hydrogen bonded.
DISCUSSION
We have presented a structure of L7Ae bound to a standard k-turn at the highest resolution available to date, such that the electron density for all the protein side chains can be observed with confidence. In light of this and comparison with other related structures available (Vidovic et al. 2000; Moore et al. 2004), we can describe the molecular recognition of a k-turn by the L7Ae-related proteins in general terms.
These proteins bind exclusively in the major groove of the k-turn. This would not be possible in regular duplex RNA due to the deep, narrow characteristics of the major groove in double-stranded RNA. However the structure of the k-turn splays open the groove, forming a continuous ribbon-like surface around the outer face of the structure, where it is very accessible.
The binding interface of L7Ae-related proteins comprises two elements; first is a highly basic β-strand:turn:α-helix element; and second, a short loop of hydrophobic residues (plus a glutamate at one end) that makes a 180° turn.
The three-turn α-helix enters the major groove of the NC helix so as to direct its N-terminal end toward the major groove edges of the guanine nucleobases of the G•A pairs. This resembles the entry of the recognition helices of helix-turn-helix elements interacting with DNA.
The C terminus of the α-helix lies close to the nonbulge strand of the NC RNA helix and contributes nonspecific interactions to the binding. The arginine in the third turn (R41 in A. fulgidus L7Ae) hydrogen bonds to the 3n/4n phosphate. This interaction is near-universal, proposed in the complexes of the same species L7Ae with box C/D and the human 15.5-kDa protein with U4 snRNA. The lysine at the second turn (K37 in A. fulgidus L7Ae) is more variable in its interactions but is bonded to the 4n/5n phosphate in the Kt-7 complex. Two of the basic side chains at the N-terminal end of the β-sheet section are also directed toward the NC helix nonbulge strand, not close enough for hydrogen bonding, but these will contribute to nonspecific electrostatic stabilization.
The N-terminal end of the α-helix is involved in specific recognition of the core of the k-turn. The glutamate side chain from the first turn (E34 in A. fulgidus L7Ae) is hydrogen bonded to N1 of G1b. This interaction is also observed in the interactions of L7Ae with box C/D and the 15.5-kDa protein with U4 snRNA and is likely to be universal. The adjacent asparagine residue (N33 in A. fulgidus L7Ae) is hydrogen bonded to O6 of G2n, also observed in the box C/D interaction. Interestingly, in a similar complex of L7Ae with Telohenia solenopsae Kt-23, where the 2n position is adenine (not guanine as in a standard k-turn), N33 is hydrogen bonded to N6 (L Huang and DMJ Lilley, unpubl.). In the L7Ae–Kt-7 complex, as well as the other structures considered here, the O6 atom of G1b is located almost on the axis of the α-helix at the N-terminal end. Guanine O6 has a significant partial negative charge, and this will be electrostatically stabilized by the positive pole of the helix dipole. This appears to be another universal interaction in the L7Ae–k-turn complexes. Lastly, the C-terminal lysine of the β-strand is directed toward N7 of the guanine at the L2 position. However, this is too long to be hydrogen bonded in the Kt-7 complex, and a similar situation probably exists in the box C/D and 15.5-U4 snRNA complexes.
The hydrophobic loop covers the nucleobases in the L2 and L1 position, making good van der Waals contact and burying a surface area of 732 Å2. The syn conformation of L2 places it maximally in contact with the loop. Isoleucine I88 in particular is located directly over GL2 in the Kt-7 complex. A very similar interaction is observed in the box C/D and 15.5-U4 snRNA complexes. The glutamate of the loop (E89 in A. fulgidus L7Ae) is hydrogen bonded to N1 of the L1 guanine, thus making a specific contact. This is also observed in the box C/D complex, but is not found in the complex between the 15.5-kDa protein with U4 snRNA, where the L1 nucleotide is cytosine. However, the glutamate side chain is probably flexible enough to make contact with bases other than guanine.
The combined effect of these elements is a highly specific molecular recognition of the structure of the k-turn in double-stranded RNA. By placing the α-helix in the major groove of the NC arm and capping the L2 nucleobase it effectively “measures” the angle between these two elements in the major groove. This then juxtaposes the N terminus of the α-helix with the major groove side of the guanine bases of the two conserved G•A, where side chains make specific hydrogen bonds, and G1b O6 is placed to maximize the interaction with the positive pole of the helix dipole.
The L7Ae proteins make no significant interaction with the C helix. Thus, these proteins should recognize a k-loop structure (where the C helix is replaced by a terminal loop) equally well as a k-turn. For example, the L7Ae-family proteins bind the k-loops of the box H/ACA RNA in the assembly of the snoRNP required for pseudouridylation (Rozhdestvensky et al. 2003; Hamma and Ferré-D'Amaré 2004; Li and Ye 2006; Youssef et al. 2007).
The folding of Kt-7 that occurs on the binding of L7Ae might in principle occur by conformational selection, or alternatively by a more active induced fit process (Fig. 6). We have studied the binding of Kt-7 to L7Ae in single molecule experiments and failed to detect any intermediate folded state down to a timescale of 8 msec (Wang et al. 2012). This is consistent with a conformational selection process, although we cannot exclude a protein-induced conformational change in the RNA on a faster timescale. However, a prerequisite of the conformational selection model is that the folded k-turn is in equilibrium with the unfolded RNA (as shown previously by time-resolved FRET experiments), and the conformation of the folded RNA in the free and protein-bound states are closely similar. Comparison of the two structures presented here show that this is the case with an all-atom RMSD of 0.83 Å. Thus, no conformational adjustment of the RNA structure should be required on binding to L7Ae.
FIGURE 6.

Scheme showing two alternative pathways of k-turn folding upon binding to L7Ae protein. The Kt-7 RNA is in equilibrium between the extended and folded conformation in free solution, biased toward the extended state at low ionic concentration. In the conformational selection mechanism (the vertical equilibrium shown right), the L7Ae selectively binds to the folded form and thus draws the equilibrium toward this state. Alternatively, the protein could bind the extended state and mechanically force the RNA to adopt the k-turn structure in an induced fit process (left). The conformational selection model requires that L7Ae structure is complementary to that of the folded k-turn, and the structure of the k-turn in the absence of protein is closely similar to that of the bound RNA. The structures determined here (shown far right) show that these requirements are met.
YbxF is a bacterial homolog of L7Ae protein (Sojka et al. 2007). Interestingly, this protein binds k-turns but not k-loops. A high-resolution structure of the complex with the SAM-I riboswitch k-turn has been solved by Ferré D'Amaré and coworkers (Baird et al. 2012). The structure of YbxF is similar to L7Ae; and the general orientation of the complex is similar, with the α-helix (although not preceded by a basic β-strand) entering the NC helix major groove and directed toward O6 of G1b and the hydrophobic loop stacked over L2 and L1. Yet there is a striking difference in that none of the interactions with the side chains of the α-helix is observed. It may be relevant that the binding affinity of YbxF to the k-turn (Kd = 270 nM) (Baird et al. 2012) is orders of magnitude weaker than that of L7Ae to Kt-7 (Kd = 10 pM) (Turner and Lilley 2008).
In the ribosome, k-turns are recognized by a number of different proteins that selectively bind to their cognate targets. We have also found that S17 and L24 stabilize the kinked structure of their cognate k-turns on binding in a similar manner to L7Ae (L Huang and DMJ Lilley, unpubl.). The structures of these proteins have little in common, and they bind to different faces of the k-turn. For example, in the small ribosomal subunit, the binding domain of S17 comprises largely β-sheet that binds to the C helix of Kt-11. The protein makes no contact with the 1b or 2n nucleobases, but a long α-helix projects over the loop region with side chains stacked on the loop nucleobases. S11 places an antiparallel β-sheet into the splayed out minor groove of the NC helix. Evidently, the k-turn presents a number of distinctive structural features that can be recognized by a variety of different binding proteins.
However, L7Ae is the most extensive family of k-turn binding proteins. Binding of L7Ae family proteins to k-turns and k-loops is required in many aspects of RNA function in the cell, including translation (L7Ae and L30e in the ribosome), site-specific RNA modification (L7Ae binding a k-turn in the formation of box C/D and H/ACA complexes), and spliceosome assembly (15.5-kDa protein binding U4 snRNA). These interactions are therefore very important in many aspects of RNA function.
MATERIALS AND METHODS
RNA synthesis
Ribooligonucleotides were synthesized using t-BDMS phosphoramidite chemistry (Beaucage and Caruthers 1981), as described in Wilson et al. (2001). Oligoribonucleotides were deprotected in 25% ethanol/ammonia solution for 3 h at 20°C and evaporated to dryness. Oligoribonucleotides were redissolved in 100 µL DMSO to which was added 125 µL 1 M triethylamine trihydrofluoride (TEA, 3HF) (Sigma-Aldrich) and incubated for 2.5 h at 65°C to remove t-BDMS protecting groups. All oligonucleotides were purified by gel electrophoresis in polyacrylamide in the presence of 7 M urea. The full-length RNA product was visualized by UV shadowing. The band was excised and electroeluted using an Elutrap (Whatman) into 45 mM Tris.borate (pH 8.5), 5 mM EDTA buffer for 8 h at 200 V at 4°C. The RNA was precipitated with ethanol, washed once with 70% ethanol, and suspended in water. The concentration of RNA was determined by measuring the absorbance at 260 nm, using an extinction coefficient of 366.1 mM−1cm−1 and 264.3 mM−1cm−1.
Expression and purification of A. fulgidus L7Ae
A gene encoding full-length A. fulgidus L7Ae was cloned into a modified pET-Duet1 plasmid (Novagen) (Huang et al. 2011) using the HindIII and EcoRI sites. The L7Ae gene was fused upstream of a hexahistidine-encoding sequence with a PreScission-cleavable linker. The hexahistidine-L7Ae fusion protein was expressed in Escherichia coli BL21-Gold (DE3) pLysS cells (Stratagene) induced with 0.2 mM IPTG for 12 h at 20°C. Harvested cells were resuspended in 20 mM Tris.HCl, pH (8.0), 500 mM NaCl, 10 mM imidazole, 1 mM phenylmethylsulfonyl fluoride (buffer A) and lysed by sonication. The protein suspension was heated for 20 min at 85°C in the presence of 10 mM MgCl2 to denature endogenous protein, and this was removed by centrifugation at 18,000 rpm for 30 min at 4°C. L7Ae was loaded onto a HisTrap column (GE Healthcare), washed with 25 mM imidazole in buffer A, and the protein was eluted with 500 mM imidazole in buffer A. The His6 tag was cleaved from the L7Ae fragments by PreScission protease in 20 mM Hepes-Na (pH 7.6), 100 mM NaCl, 0.5 mM EDTA for 16 h at 4–8°C. L7Ae was loaded onto a heparin column (GE Healthcare) and eluted at 250 mM in a 50–2000 mM gradient of NaCl in 20 mM Hepes.Na (pH 7.6). The protein was further purified using a Superdex 200 gel filtration column in a buffer containing 5 mM Tris.HCl (pH 8.0), 100 mM NaCl. The protein concentration was measured by absorbance at 280 nm using a molar extinction coefficient of 5240 M−1 cm−1 for L7Ae. This value was calculated on the basis of amino acid composition. The protein was concentrated to 20 mg/mL in buffer containing 5 mM Tris.HCl (pH 8.0), 100 mM NaCl, and stored at −20°C as aliquots.
Crystallization, structure determination, and refinement
Kt-7 complexed with L7Ae
A number of RNA constructs containing the H. marismortui Kt-7 sequence were used in crystallization trials of the L7Ae–HmKt7 complex. Two main designs were investigated: One comprised a set of 25–28 nt RNA oligonucleotides, each generating a k-turn where the C helix was closed by a GAAA tetraloop (Fig. 1B); and the other comprised 17–25 nt RNA oligonucleotides, containing two self-complementary copies of the Kt-7 sequence related by a twofold axis (Fig. 1C).
A mixture of 0.25 mM RNA and 0.25 mM L7Ae in 5 mM Tris.HCl (pH 8.0) and 100 mM NaCl was heated for 1 min to 85°C. The solution was slowly cooled to 20°C; MgCl2 was added to a final concentration of 10 mM. Crystals were grown by vapor diffusion using drops prepared by mixing 1.0 μL of the RNA–protein complex with 1 μL of a reservoir solution comprising 100 mM Hepes.Na (pH 7.0), 2.3 M ammonium sulfate at 20°C. Crystals (approximate dimensions 500 × 50 × 50 μm3) of space group P43212 appeared after 3 d. Crystals were transferred to a solution containing 100 mM Hepes.Na (pH 7.0), 2.0 M ammonium sulfate, and 30% glycerol for ∼5 sec. The crystals were flash frozen by mounting in nylon loops and plunging into liquid nitrogen. The crystals were characterized in the laboratory with a MicroMax HF007 copper rotating anode X-ray generator equipped with an ACTOR sample changer system and a Saturn 944HG+ CCD detector (Rigaku). Suitable crystals were stored and subsequently used to measure full data sets on beamline I03 of the Diamond Light Source (Harwell, UK). The crystals had unit cell dimensions a = 61.9 Å, b = 61.9 Å, and c = 131.0 Å. From crystal density considerations (Mathews 1968), one RNA–protein complex was expected to be present in the asymmetric unit.
The structure was determined by molecular replacement. In the first stage, the A. fulgidus 7Ae structure taken from PDB file 1RLG was used as the search model, followed by the H. marismortui Kt-7 structure taken from PDB file 4B5R, using the program PHASER (McCoy et al. 2007). The resulting electron density map revealed the remaining GAAA tetraloop and the N-terminal region of L7Ae, and a model was built using the difference map. Composite omit maps were calculated using PHENIX (Adams et al. 2010).
Kt-7 as free RNA
Three types of design were explored in attempts to crystallize Kt-7 RNA in the absence of protein. These were (1) the same 25–28 nt RNA with a GAAA tetraloop closing the C-helix as above (Fig. 1B); (2) 30–36 nt RNA in which the C-helix was a stem–loop closed by a U1A protein binding loop (data not shown); and (3) the 17–25 nt RNA oligonucleotides with two inverted copies of the Kt-7 described above (Fig. 1C).
A solution of 1 mM RNA in 5 mM Tris.HCl (pH 8.0) and 100 mM NaCl was heated for 1 min to 95°C. The solution was slow cooled to 20°C; MgCl2 was added to a final concentration of 10 mM. The hanging-drop vapor diffusion method was used for crystallization. A volume of 1.0 μL of prefolded RNA was mixed 1:1 with well solution comprising 3.5 M Na formate, 0.1 M Na acetate (pH 4.0) at 20°C. Crystals (approximate dimensions 100 × 10 × 10 μm3) with space group P6322 appeared after 5 d. Crystals were briefly washed in well solution supplemented with 30% glycerol. The crystals were flash frozen by mounting in nylon loops and plunging into liquid nitrogen. A 2.45 Å resolution data set was collected on a tunable microfocus beamline I24 of the Diamond Light Source (Harwell, UK). Later, a bigger crystal (approximate dimensions 200 × 30 × 30 μm3) was obtained from which a 2.2-Å resolution data set was collected on beamline ID23-1 at the European Synchrotron Radiation Facility (Grenoble, France). The crystals had unit cell dimensions a = 70.2 Å, b = 70.2 Å, and c = 47.7 Å. The molecular weight of a subunit is 6.3 kDa, and the asymmetric unit consists of one subunit with a VM value of 2.7 Å3 Da−1 and solvent content of ∼69% (Mathews 1968). The structure was determined by molecular replacement. H. marismortui Kt-7 (PDB 4bw0) was used as the search model using the program PHASER (McCoy et al. 2007). The remaining nucleotides were added to the model based on inspection of electron density difference maps.
Both structural models were built in Coot (Emsley et al. 2010) and refined with Refmac5 (Murshudov et al. 2011) from the CCP4 suite of programs (Winn et al. 2011) and Phenix (Adams et al. 2010). Ramachandran analysis shows that 100% of amino acid residues are in the most favored and additionally allowed regions. Model geometry and the fit to electron-density maps were monitored with MOLPROBITY (Chen et al. 2010) and the validation tools in COOT.
DATA DEPOSITION
Atomic coordinates and structure factor amplitudes have been deposited with the PDB with accession codes 4BW0 and 4C40.
ACKNOWLEDGMENTS
We thank our colleagues Drs. Jia Wang and Paul Fyfe for discussion, Scott McPhee for RNA synthesis, and the staff of I24 and I03 beam stations at Diamond Light Source UK for their assistance with data collection. Cancer Research UK and the Wellcome Trust provided financial support.
Footnotes
Freely available online through the RNA Open Access option.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallog 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Zhang J, Hamma T, Ferré-D'Amaré AR. 2012. YbxF and YlxQ are bacterial homologs of L7Ae, and bind K-turns but not K-loops. RNA 18: 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289: 905–920. [DOI] [PubMed] [Google Scholar]
- Beaucage SL, Caruthers MH. 1981. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett 22: 1859–1862. [Google Scholar]
- Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst D 66: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho IM, Lai LB, Susanti D, Mukhopadhyay B, Gopalan V. 2010. Ribosomal protein L7Ae is a subunit of archaeal RNase P. Proc Natl Acad Sci 107: 14573–14578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daldrop P, Lilley DMJ. 2013. The plasticity of a structural motif in RNA: Structural polymorphism of a kink turn as a function of its environment. RNA 19: 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Cryst D 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamma T, Ferré-D'Amaré AR. 2004. Structure of protein L7Ae bound to a K-turn derived from an archaeal box H/ACA sRNA at 1.8 Å resolution. Structure 12: 893–903. [DOI] [PubMed] [Google Scholar]
- Huang L, Yin P, Zhu X, Zhang Y, Ye K. 2011. Crystal structure and centromere binding of the plasmid segregation protein ParB from pCXC100. Nucleic Acids Res 39: 2954–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Bork P, Sander C. 1994. A novel RNA-binding motif in omnipotent suppressors of translation termination, ribosomal proteins and a ribosome modification enzyme? Nucleic Acids Res 22: 2166–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn JF, Tran EJ, Maxwell ES. 2002. Archaeal ribosomal protein L7 is a functional homolog of the eukaryotic 15.5kD/Snu13p snoRNP core protein. Nucleic Acids Res 30: 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ye K. 2006. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature 443: 302–307. [DOI] [PubMed] [Google Scholar]
- Liu J, Lilley DMJ. 2007. The role of specific 2′-hydroxyl groups in the stabilization of the folded conformation of kink-turn RNA. RNA 13: 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmier-Gourrier N, Cléry A, Senty-Ségault V, Charpentier B, Schlotter F, Leclerc F, Fournier R, Branlant C. 2003. A structural, phylogenetic, and functional study of 15.5-kD/Snu13 protein binding on U3 small nucleolar RNA. RNA 9: 821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews BW. 1968. Solvent content of protein crystals. J Mol Biol 33: 491–497. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Zhang Y, Fenley MO, Li H. 2004. Molecular basis of box C/D RNA-protein interactions; Cocrystal structure of archaeal L7Ae and a box C/D RNA. Structure 12: 807–818. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst D 67: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottrott S, Hartmuth K, Fabrizio P, Urlaub H, Vidovic I, Ficner R, Lührmann R. 1999. Functional interaction of a novel 15.5kD [U4/U6.U5] tri-snRNP protein with the 5′ stem–loop of U4 snRNA. EMBO J 18: 6119–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhdestvensky TS, Tang TH, Tchirkova IV, Brosius J, Bachellerie J-P, Hüttenhofer A. 2003. Binding of L7Ae protein to the K-turn of archaeal snoRNAs: A shared RNA binding motif for C/D and H/ACA box snoRNAs in Archaea. Nucleic Acids Res 31: 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojka L, Fučík V, Krásný L, Barvík I, Jonák J. 2007. YbxF, a protein associated with exponential-phase ribosomes in Bacillus subtilis. J Bacteriol 189: 4809–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B, Lilley DM. 2008. The importance of G·A hydrogen bonding in the metal ion- and protein-induced folding of a kink turn RNA. J Mol Biol 381: 431–442. [DOI] [PubMed] [Google Scholar]
- Turner B, Melcher SE, Wilson TJ, Norman DG, Lilley DMJ. 2005. Induced fit of RNA on binding the L7Ae protein to the kink-turn motif. RNA 11: 1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidovic I, Nottrott S, Hartmuth K, Lührmann R, Ficner R. 2000. Crystal structure of the spliceosomal 15.5 kD protein bound to a U4 snRNA fragment. Mol Cell 6: 1331–1342. [DOI] [PubMed] [Google Scholar]
- Wang J, Fessl T, Schroeder KT, Ouellet J, Liu Y, Freeman AD, Lilley DM. 2012. Single-molecule observation of the induction of k-turn RNA structure on binding L7Ae protein. Biophys J 103: 2541–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins NJ, Ségault V, Charpentier B, Nottrott S, Fabrizio P, Bachi A, Wilm M, Rosbash M, Branlant C, Lührmann R. 2000. A common core RNP structure shared between the small nucleolar box C/D RNPs and the spliceosomal U4 snRNP. Cell 103: 457–466. [DOI] [PubMed] [Google Scholar]
- Watkins NJ, Dickmanns A, Lührmann R. 2002. Conserved stem II of the box C/D motif is essential for nucleolar localization and is required, along with the 15.5K protein, for the hierarchical assembly of the box C/D snoRNP. Mol Cell Biol 22: 8342–8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TJ, Zhao Z-Y, Maxwell K, Kontogiannis L, Lilley DMJ. 2001. Importance of specific nucleotides in the folding of the natural form of the hairpin ribozyme. Biochemistry 40: 2291–2302. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. 2011. Overview of the CCP4 suite and current developments. Acta Cryst D 67: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef OA, Terns RM, Terns MP. 2007. Dynamic interactions within sub-complexes of the H/ACA pseudouridylation guide RNP. Nucleic Acids Res 35: 6196–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]