

In this paper, the authors describe a universal stem–loop primer-based RT-PCR protocol for TaqMan probes which is more cost-efficient and can be used to detect small noncoding RNAs. This new method is efficient for the detection of synthetic siRNAs, double-stranded miRNA mimics, single-stranded synthetic miRNAs, and in vitro transcribed miRNA precursors, as well as for the detection of endogenous bacterial small RNAs.
Keywords: noncoding RNA detection, TaqMan PCR, stem–loop primer, real-time PCR
Abstract
Several methods for the detection of RNA have been developed over time. For small RNA detection, a stem–loop reverse primer-based protocol relying on TaqMan RT-PCR has been described. This protocol requires an individual specific TaqMan probe for each target RNA and, hence, is highly cost-intensive for experiments with small sample sizes or large numbers of different samples. We describe a universal TaqMan-based probe protocol which can be used to detect any target sequence and demonstrate its applicability for the detection of endogenous as well as artificial eukaryotic and bacterial small RNAs. While the specific and the universal probe-based protocol showed the same sensitivity, the absolute sensitivity of detection was found to be more than 100-fold lower for both than previously reported. In subsequent experiments, we found previously unknown limitations intrinsic to the method affecting its feasibility in determination of mature template RISC incorporation as well as in multiplexing. Both protocols were equally specific in discriminating between correct and incorrect small RNA targets or between mature miRNA and its unprocessed RNA precursor, indicating the stem–loop RT-primer, but not the TaqMan probe, triggers target specificity. The presented universal TaqMan-based RT-PCR protocol represents a cost-efficient method for the detection of small RNAs.
INTRODUCTION
To detect RNAs, a wide range of methods have been developed including Northern blotting, cloning, and RT-PCR, with real-time RT-PCR being considered as the gold standard for RNA quantification (O'Connor and Glynn 2010). Very short RNAs, such as microRNAs (miRNAs), however, are frequently not captured by these techniques. In particular, microRNAs are not long enough to provide sufficient complementarity for conventional linear RT-PCR forward and reverse primers. In 2005, a TaqMan RT-PCR protocol was reported which uses stem–loop RT primers to specifically trap the 3′ end of small RNAs (Fig. 1; Chen et al. 2005), reducing the amount of complementarity required. The authors attribute the observed higher specificity and sensitivity of the stem–loop RT primers as compared with linear primers with base stacking and spatial constraint of the stem–loop structure. By necessity, each amplicon requires a specific 3′ stem–loop primer and linear forward primer for first- and second-strand cDNA synthesis, respectively. Typically, the TaqMan probe spans the junction of the 3′ stem–loop primer and the 3′ end of the target amplicon, thus requiring, in addition, an individual TaqMan probe for each target to be detected. Such an experimental design renders the method expensive and not suitable for small sample sizes or high-throughput applications.
FIGURE 1.
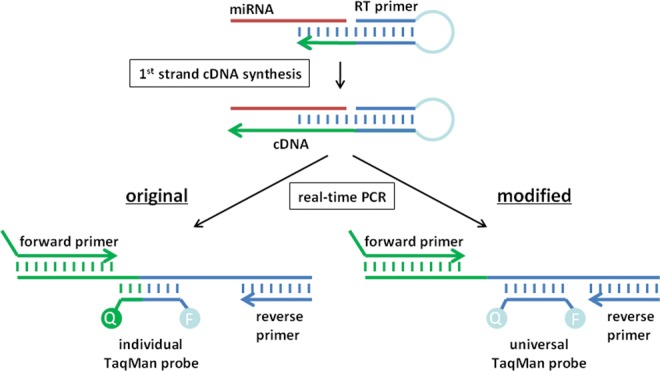
Schematic diagram depicting the principle of stem–loop primer-based small RNA detection. The original design requires one TaqMan probe for each target RNA (lower left). The universal design uses a universal TaqMan-based probe which does not overlap with the target binding domain of the stem–loop primer and, hence, can be used to detect any target sequence (lower right).
We considered that the TaqMan method could work equally well with a TaqMan-based probe that was completely complementary to the part of the stem–loop adjacent to the target overhang. As a consequence, a single universal TaqMan-based probe is now suitable for the detection of any target RNA, which significantly reduces the costs of this method, rendering extensive applications feasible. We compared the conventional and universal probe in terms of sensitivity and investigated the specificity for RNA 3′ ends. Our data confirm that both protocols are equally sensitive and specific in detecting small RNAs.
RESULTS
Experimental design and parameter testing
Our method conforms with the standard approach for stem–loop RT-PCR amplification (using a stem–loop reverse primer for first-strand cDNA synthesis and a linear primer for second-strand synthesis) but differs in the detection of the PCR amplicon (Fig. 1). Before comparing our universal with conventional TaqMan probes, we optimized the synthesis steps in order to maximize the effective range of our assay and, thus, the detection of any differences in probe sensitivity. Specifically, we investigated the effects of (1) the length of the target binding domain of the stem–loop primer (the 3′ overhang region), (2) the nature of the template (single- or double-stranded, mature or pre-miRNA), and (3) the reaction volumes (for reverse transcription and second strand synthesis). The forward primers were designed to have melting temperatures of 60°C –65°C and to closely align with the 5′ end of the target sequence. If necessary we increased the melting temperatures by extending the 5′ end of the forward primers with a random sequence. At the same time, an overlap with the target's 3′ end was avoided to prevent the forward primer from competing with the target for the hairpin primer.
The sensitivity of the assay was significantly affected by the length of the target binding site of the stem–loop primer. Four stem–loop primers with 5-, 6-, 8-, and 11-nt complementarity to the 3′ end of the antisense strand of the GFP22 siRNA were tested. The highest sensitivity was observed for the 5- and 6-nt target binding sites, both of which still produced a signal clearly above background and in a linear amplification range for 103 template molecules (Fig. 2A).
FIGURE 2.

Impact of length of target binding overhang, strand accessibility, and reaction volumes on the sensitivity of the modified protocol. The target used is the antisense strand of the GFP22 siRNA duplex. (A) Amplification using stem–loop primers with different lengths of the target binding overhang. (B) Detection of single-stranded vs. double-stranded target RNA using the stem–loop primer with a 6-nt target binding overhang. Technical triplicate, mean, and range of values (A,B). (C) Sensitivity of the method depending on the reverse transcription and PCR reaction volumes. (NTC) No template controls; (XTC) control template mature human miR146.
We next determined whether the template structure affected detection. The antisense strand of the GFP22 siRNA was detectable with equal sensitivity regardless of whether or not it was employed as single-stranded RNA or as part of the siRNA duplex, indicating that the duplex structure of the siRNA did not interfere with the detection (Fig. 2B).
Finally, we investigated the effect of reaction volume of the synthesis steps on detection sensitivity. The sensitivity of detection was clearly influenced by the volumes of the reverse transcription and subsequent PCR reaction and found to be highest (670 molecules per PCR reaction or 60 molecules per μL reverse transcription) in the lowest investigated RT reaction volume of 7.5 μL (Fig. 2C).
Universal and specific TaqMan-based probes are equally sensitive, though to a lower extent than originally reported
Having optimized the synthesis steps of the RT-PCR reactions, we next initiated a set of experiments to directly compare the performance of conventional and our universal TaqMan-based probe designs. To create a universal TaqMan-based probe, which can be used for the detection of any small RNA, we completely shifted it toward the conserved sequence of the stem–loop primer and eliminated any overlap with the target sequence (Fig. 1). The original TaqMan probe design was reported to detect as few as seven molecules of the lin-4 miRNA per reaction (Chen et al. 2005). To exclude errors in our reverse engineering and to consider all intricacies of the original design, we compared our universal design with the conventional design from a custom kit for the GFP22 siRNA target sequence. Both the conventional and our universal probe detected two independent lots of the GFP22 siRNA with the same sensitivity (Fig. 3A). However, only with the conventional custom kit was a strong background signal detected in the no-template control. For further validation, we compared our universal probe with two conventional ones targeting the mature human miRNAs hsa-miR-133b and hsa-miR-146a, either as mature miRNA single strands or as double-stranded siRNA-like miRNA mimics (Fig. 3B–E). Both the conventional probes and the universal probe gauged these two miRNAs with equal sensitivities of 103 to 104 template molecules which, however, were significantly lower than previously reported. This could be confirmed by another group (A Mantei and A Scheffold, pers. comm.; Mantei et al. 2008). Notably, the sensitivity of detection was very similar for the double-stranded and the single-stranded templates (Figs. 2B, 3B–E). A slightly higher background signal was detected in the NTCs with the conventional miR-133b and miR-146a detection kits. We could not achieve the previously reported sensitivity even when varying the lengths of hairpin primer overhangs (Fig. 2A). Indeed, a decrease of the reaction volumes for reverse transcription (RT) and PCR below the recommended volumes increased the sensitivity but not to the originally reported levels (Fig. 2C).
FIGURE 3.

Comparison of the sensitivity of the conventional probe (Com) and the universal probe (Mod) using serially diluted single- and double-stranded RNA. (A) Detection of the GFP22 siRNA antisense strand derived from two double-stranded siRNA batches using both protocols; mean of duplicates with standard deviation. Detection of mature miR-133b as part of a double-stranded miRNA mimic (B) or as RNA single-strand (C). Detection of mature miR-146a as part of a double-stranded miRNA mimic (D) or as RNA single-strand (E). Error bars represent value range from mean of two independent experiments, each performed in duplicate. (NTC) No template control; (NEC) no reverse transcriptase control; (NPC) nonforward primer control.
Universal and specific protocols exhibit equal specificity for small RNA detection
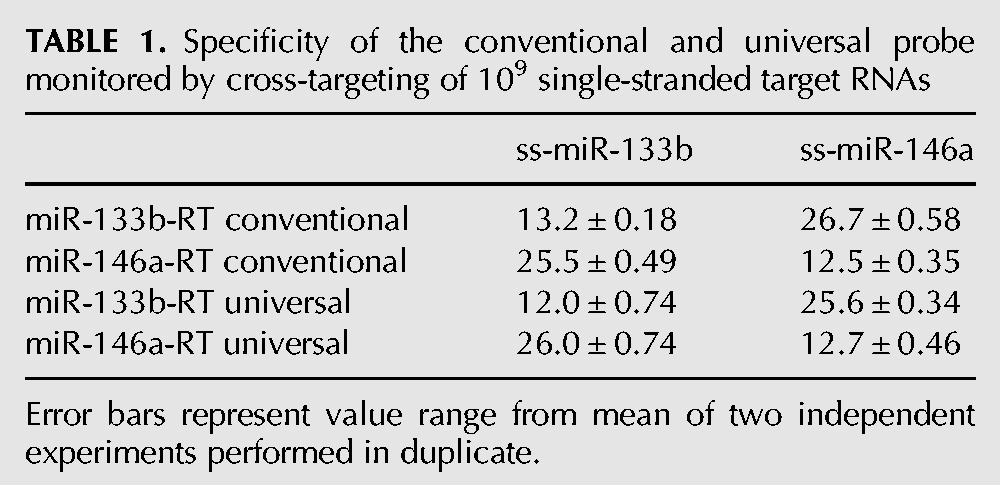
We next wished to compare the conventional and universal probe performance in assays quantifying precursor and mature microRNAs, whose endogenous levels can frequently differ by an order of magnitude or more. The stem–loop primer-based RT-PCR protocol has been claimed to efficiently discriminate between mature miRNAs and their unprocessed precursor structures. Stem–loop primers designed to target mature miRNAs produced Ct values that were at least 11 cycles higher with the corresponding miRNA precursor templates (Chen et al. 2005). We investigated how accurately our universal probe can discriminate mature miRNAs from their unprocessed precursors, which lack the correct 3′ ends to be captured by the stem–loop primer, as well as from other mature miRNA species. Precursor RNAs of miR-133b and miR-146a were transcribed in vitro from linear DNA templates as described. We then performed serial RNA dilutions and used the stem–loop primers designed to target the 3′ ends of the mature miRNAs to detect both the mature miRNAs and the corresponding RNA precursors, using the conventional and universal probes. Figure 4 shows the discrimination of mature miRNAs from their corresponding precursors. For the highest molecular concentration tested, i.e., 109 molecules per reaction, we observed a seven to 12 or nine to 11 cycle difference with the conventional probes or the universal probe, respectively. Our data suggest that if mature miRNAs are presented at equal concentrations as their precursors, the latter will contribute ∼1 to 0.1% to the overall signal. In addition, a cross-over experiment was performed to compare the specificity for perfectly matched or completely mismatched mature miRNA targets. We used the miR-133b RT primer to reverse-transcribe miR-146a and vice versa. The sensitivity of stem–loop RT primer-based RT-PCR was evaluated for 109 template/nontemplate molecules per reaction. In all cases, we observed a 13–14 cycle difference, indicating that both methods can efficiently and equally discriminate between the correct mature miRNA template and a false-template control sequence (Table 1). However, we still detected a clear signal with the false-template controls that was seven to eight cycles higher than in the nontemplate controls. This implies that, for detection of low-abundant miRNAs, highly-abundant off-target species can contribute to a false-positive signal. Summing up, both probe designs were found to be equally specific in discriminating between correct and incorrect small RNA targets or between a mature miRNA and its unprocessed RNA precursor.
FIGURE 4.

Comparison of the specificity of the conventional probe (Com) and the universal probe (Mod) using serially diluted single-stranded mature miRNAs and the corresponding miRNA precursors. Detection of miR-133b sequences using the conventional probe (A) or the universal probe (C). Detection of miR-146a sequences using the conventional probe (B) or the universal probe (D). (E) Ct values detected in A–D for 109 template molecules. Error bars represent value range from mean of one experiment performed in duplicate. (NTC) No template control.
TABLE 1.
Specificity of the conventional and universal probe monitored by cross-targeting of 109 single-stranded target RNAs
The universal probe detects prokaryotic endogenous and artificially delivered small RNAs as well as endogenous human miRNAs
Detection of small noncoding RNAs in prokaryotes represents a field of increasing interest. These RNAs typically range from 50 to 400 nt in length (Waters and Storz 2009), a size which can be difficult to detect using conventional RT-PCR protocols. We investigated whether the universal probe-based protocol is suitable to detect small noncoding RNAs isolated from bacteria. Bacterial noncoding RNAs range in transcript length from 70 to 500 nt and in some cases are processed by RNases to become significantly shorter (Mikulik 2003). After electroporation of the synthetic GFP22 siRNA into Listeria monocytogenes and purification of total RNA, a strong signal was detected directly after electroporation compared with unpulsed samples, with degradation of template detectable after 4 h. The nontemplate control and a false-template control siRNA did not give any signal (Fig. 5A). In a second experiment, we aimed at detecting two endogenous noncoding RNAs in Escherichia coli, LstR-1 and CsrB. LstR-1 is a 140-nt-long negative regulator of the SOS operon (Vogel et al. 2004) which is expressed during the logarithmic growth phase, while CsrB is predominantly expressed during growth in amino acid-depleted media (Jonas and Melefors 2009) and measures 359 nt. E. coli DH5α was grown in rich LB media until the logarithmic growth phase was reached and expression of LstR-1 and CsrB was determined. As expected, we detected a profound signal for LstR-1, while the CsrB signal was weaker (Fig. 5B). Another major application of stem–loop primer-based TaqMan RT-PCR is the detection of endogenous miRNAs in eukaryotic, including human, cells. We, therefore, applied the conventional and our universal design to the detection of endogenous miR-133b and miR-146a in the human adherent fibroblastic HEK293T, as well as suspension Jurkat T, cell lines. Both methods were equally sensitive in detecting both miRNAs in total RNA isolates (Fig. 5C,D).
FIGURE 5.

Detection of artificial and endogenous small noncoding RNAs in prokaryotes and eukaryotes with the universal probe. (A) Confirmation of delivery of siRNA after electroporation of Listeria monocytogenes compared with buffer and control siRNA. Relative values normalized to the Listeria 6PBG housekeeping gene and compared with unpulsed samples (2−(ΔΔCT) method). Errors represent standard deviation from ΔΔCT, one experiment in duplicate. (B) Detection of two small noncoding RNAs CsrB and Lst-R isolated from Escherichia coli grown in log phase in rich media. (NTC) No template control. (C,D) Detection of endogenous miR-133b and miR-146a using the universal design (Com) and universal modified (Mod) protocol in total RNA from human cell lines HEK293T and Jurkat. Error bars represent value range from mean of one experiment performed in duplicate.
Stem–loop primer-based RT protocols exhibit limited specificity for RNA 3′ ends
Stem–loop primer-based RT protocols are considered suitable for multiplex PCR. That is because the short target binding sites of the stem–loop primers are assumed to efficiently discriminate between perfect matching and imperfect binding of 3′ end targets, i.e., they can discriminate between intact and partially degraded, extended, or mutated 3′ ends. In an earlier publication, the authors investigated the specificity of the method to discriminate not only between mature miRNAs and their precursors but also among the closely related members of the let-7 miRNA family (Chen et al. 2005). Compared with a perfectly matching target, single mismatched targets were about 27- to 1000-fold less effective targets relating to Ct differences of five to 10. Such a degree of specificity may not consistently allow reliable quantification of closely related differentially expressed miRNAs, but it should be sufficient to monitor exact RNA 3′-end processing. In general, 3′ RACE is used to define RNA 3′ ends. We determined whether the 3′-end specificity of stem–loop primer-based RT-PCR is sufficiently high to discriminate between perfect and imperfect matching of RNA 3′ ends. Therefore, we designed sets of overlapping stem–loop primers with single-stranded 3′ ends of 6 nt in length and attempted to detect the exact 3′ end of an siRNA resembling a hairpin sequence at the end of the tuf mRNA of Mycobacterium tuberculosis. The tuf elongation factor is a highly conserved gene in Mycobacteria. It is up-regulated under anaerobic conditions in M. tuberculosis (Starck et al. 2004), a condition thought to occur during bacterial dormancy in vivo (Voskuil et al. 2004). The tuf gene has been extensively studied; its 3′ end is well defined from cloning of the mRNA analog of Mycobacterium leprae (Dhandayuthapani et al. 1994). We designed several stem–loop primers that harbored a perfectly matching 6-nt target binding site, which is considered to yield the highest target sensitivity and specificity, as well as other primers designed to either leave a gap toward (primers 1–6) or produce an overlap with (primer 8) the 3′ end of the siRNAs (Fig. 6A). Gaps weaken base stacking energies, while overlaps are associated with steric hindrance. Hence, both of the imperfect 3′-matching primer sets are considered suboptimal for target amplification. However, perfect matching as well as single-nucleotide gap-forming or overlapping stem–loop primers exhibited similar detection sensitivities. Only primers forming larger gaps with the target resulted in clearly decreased signal strengths (Fig. 6B).
FIGURE 6.

Specificity and sensitivity of different stem–loop primers with overlapping binding sites for a distinct RNA 3′ end and impact of forward primer position. The universal probe was applied to detect a chemically synthesized siRNA. (A) Positioning of stem–loop primers relative to the 3′ end of the siRNA. (B) Threshold cycles (CT) in PCR with template (siRNA) and without (NTC) for each stem–loop primer. CT was defined as 40 if not detectable. Error bars represent value range from mean of two independent experiments. (C) Impact of forward primer design CT, siRNA mimicking forward primer with 105 siRNA target molecules with (gray bars) or without template RNA (black bars) and conventional forward primer without template RNA (diagonal pattern).
DISCUSSION
There is an increasing demand in research and medical diagnostics for sensitive and specific methods that are suitable for detecting and quantifying small RNAs. Many of the established techniques, such as Northern blotting or conventional RT-PCR, are of insufficient sensitivity, require large samples sizes, are not high-throughput compatible, or harbor difficulties in the detection of very short targets. Unlike deep sequencing, RT-PCR represents a cost-efficient and rapid technique for RNA detection. In 2005, a TaqMan probe-based RT-PCR protocol was described that uses stem–loop RT primers to specifically capture 3′ ends of small RNA targets (Chen et al. 2005). This protocol is suitable for sensitive detection and quantification of small RNA, but the requirement for an individual TaqMan probe for each target RNA renders the method expensive for medium- or high-throughput applications. Nevertheless, the method has found application in many laboratories, especially for miRNA profiling.
In stem–loop RT-PCR, the TaqMan probe binds to the sequence of the stem–loop primer. Thus, in terms of sensitivity and specificity, it makes no difference whether the TaqMan probe overlaps with the miRNA binding site partially, as in the conventional protocol, or whether it exclusively binds to the invariable part of the stem–loop primer. This hypothesis was confirmed by our experiments, which show that a universal TaqMan-based probe can be designed by shifting its target site toward the conserved part of the stem–loop primer. The universal probe is suitable for detection of small RNAs derived from chemical synthesis or isolated from bacteria or human cells with the same sensitivity as conventional individual probes.
The specificity of a method relates to its ability to identify negative results with a low rate of false positives. Here, specificity is reflected by the ability of the test to discriminate between the sample and the nontemplate control, as well as between the correct target and incorrect or off-targets. We found both protocols to be equally specific in discriminating between correct and incorrect small RNA targets or between a mature miRNA and its unprocessed RNA precursor, confirming the hypothesis that the stem–loop RT-primer, but not the TaqMan probe, triggers target specificity. As expected, the discrimination between a mature miRNA and its precursor was found to be less pronounced compared with the discrimination between a correct and an incorrect mature miRNA target. These observations indicate that the structure of the precursor actively suppresses binding of the stem–loop primer, albeit not as low as levels observed with nonmatching targets.
Our results indicate previously unknown limitations of the stem–loop primer-based small RNA detection, discussed in the following.
Independent of the used protocol (TaqMan probe design), the sensitivity of the method was lower than originally reported. We have no plausible explanation for this finding because we used the same buffers and equipment, and others have confirmed the results with more targets. However, since we investigated several targets, the observed lower sensitivity seems to be independent of the target sequence.
Stem–loop primer-based TaqMan assays are frequently used to quantify mature miRNAs in samples that also contain related miRNAs with very similar but not identical 3′ ends. While the previous publication indicated that a single nucleotide mismatch can be discriminated with the stem–loop primer design, our results indicate that the protocol fails to discriminate perfectly matching 3′ ends from ends leaving single–nucleotide gaps or overhangs with the stem–loop primers. This limited 3′-end specificity may become challenging if closely related miRNAs, shRNAs, or siRNAs without mismatches have to be quantified or multiplexing is attempted with stem–loop primers that share the same identity at the +1 nt or −1 nt position. In addition, stem–loop RT-PCR appears unsuitable for RNA 3′-end mapping. However, degenerate stem–loop primers harboring constant and randomized (degenerate) positions within the target binding domain might be used to simultaneously detect related RNA species with similar but not identical 3′ends.
Our data indicate a low specificity of stem–loop RT-PCR in the case of distinct complementarities between the forward primer and the target binding site of the stem–loop primer. This becomes most evident if the forward primer 3′ end exactly resembles the 3′ end of the target as reflected by a strong signal in the nontemplate controls (Fig. 6C). We can exclude that competition between the forward primer and the target for the target binding site accounts for this effect since we performed a two-step RT-PCR protocol in which the forward primer was added only after the reverse transcription was complete. However, the forward primer could still be able to mimic the target sequence and prime the extension of remaining hairpin primer in the subsequent PCR reaction, thereby triggering a false-positive signal in the negative control. Surprisingly, this process seems to occur even under the hot-start PCR conditions we used, indicating that base stacking is sufficiently strong to stabilize the hexa-nucleotide duplex for hairpin-primer extension at the given concentrations and temperature. As a consequence, any overlap between the forward primer and the hairpin primer should be avoided or reduced to a minimum. The high signals in the nontemplate controls versus the nonprimer control indicate that the conventional probes may not necessarily consider this point (Figs. 3A, 6C). High false-positive background signals may strongly impair the sensitivity of a TaqMan PCR. Thus, it appears to be essential to include nontemplate controls for each pair of forward and hairpin primer in order to detect false-positive signals.
Notably, even without heat denaturation of the samples, the detection was equally sensitive for single-stranded and double-stranded small RNA templates. That might be explained by strand displacement activities of the MMuLV reverse transcriptase used in our assays that were reported previously, at least for DNA-DNA and RNA-DNA hybrids (Kelleher and Champoux 1998); although the method can distinguish between pre-miRNA and mature miRNA, the stem–loop primers likely are not able to discriminate single-stranded and double-stranded stages of processed mature miRNA, i.e., the stages from Dicer cleavage, passenger strand cleavage, to RISC loading complex (RLC)- and RNA-induced silencing complex (RISC)-associated RNA. Since the availability of Argonauts as the active component of RISC is known as the limiting factor in RNAi (Grimm et al. 2010; Winter and Diederichs 2011), the method must be applied with care in cases of overloading the RNAi pathway with miRNA, small hairpin (sh)RNA, or siRNA. Under these conditions, the results will not necessarily correlate with the RNAi effector quantity.
The terminology “TaqMan” is used by several vendors though the underlying chemistry is always probe hydrolysis causing a fluorescent signal. Other hydrolysis probes with alternative fluorophores should work as well, but a fluorophore-based quencher like TAMRA should probably be avoided. The minor groove binder (MGB)-stabilized probes we investigated are proprietary to Applied Biosystems. However, other companies have developed modifications that are supposed to act in the same manner and thus should give similar results. Non-MGB stabilized probes are usually a bit longer, but we would not expect any problem in using longer probes and longer hairpin primers in this detection system.
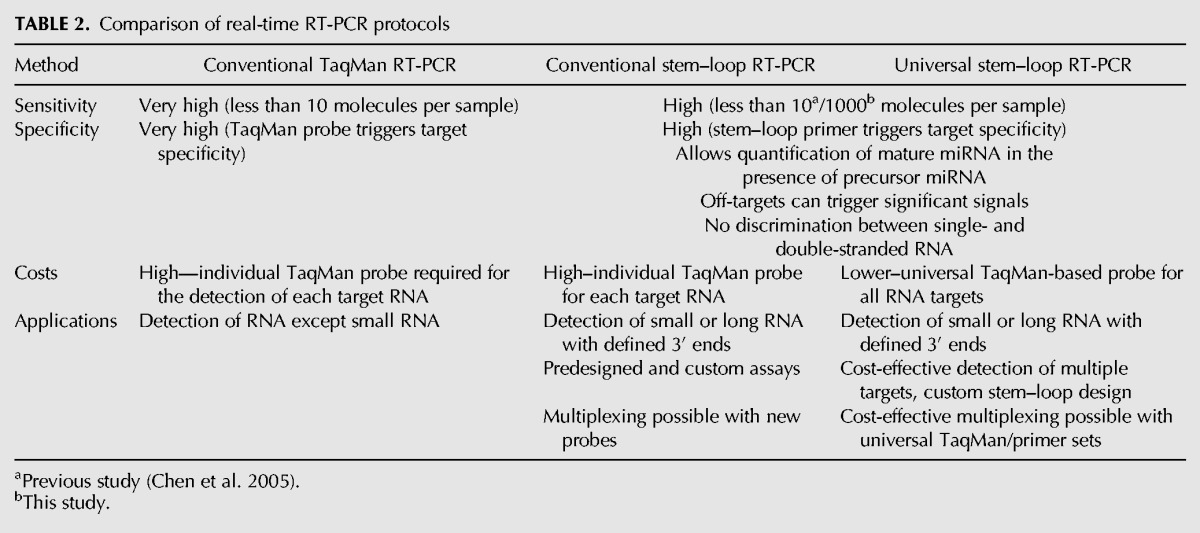
In conclusion, we report previously unknown limitations of a widely used protocol that are intrinsic to the method itself. However, a careful consideration of our results allows the robust application of stem–loop primer-based RT-PCR. The use of a universal TaqMan-based probe affects neither the sensitivity nor the specificity of the method but drastically reduces the costs for detection or quantification of multiple targets. Overall, there are several advantages and disadvantages between the different TaqMan RT-PCR protocols (Table 2) depending on the application. The standard TaqMan protocol does not allow detection of small RNAs and otherwise combines a very high sensitivity with relatively high cost. The conventional stem–loop TaqMan protocol is feasible for detection of both small RNAs as well as RNAs with defined unique 3′ ends while retaining a high-cost factor but seemingly lower sensitivity. As for this protocol, a new primer and probe set has to be obtained for each target; the costs for single-tube multiplexing do not differ from the basic price. The universal stem–loop TaqMan-based protocol, however, exhibits the same sensitivity and specificity as the conventional design but with a significant cost reduction especially when several targets or larger sample numbers are to be detected. The absolute price of the latter increases for single-tube multiplexing as additional stem–loop/probe sets have to be obtained. Relatively, this design still remains more cost-efficient since universal sequences can be used as well.
TABLE 2.
Comparison of real-time RT-PCR protocols
MATERIALS AND METHODS
Electro-competence and electroporation of Listeria monocytogenes
L. monocytogenes EGD strain was grown in 300 mL LB broth at 37°C until optical density at 600 nm reached a value between 0.18 and 0.25. PenicillinG was added at a final concentration of 3 μg/mL and the culture incubated for an additional 2 h. Afterward, the bacteria were collected by centrifugation at 4°C and 6000g for 10 min and the pellet resuspended in 35 mL ice cold HS buffer (1 mM HEPES, pH 7.0 PAA, 0.5 M sucrose [Sigma-Aldrich]). The centrifugation step was repeated, the pellet resuspended in 10 mL ice cold HS buffer, followed by another centrifugation step and resuspension of the pellet in 2.5 mL ice cold HS buffer. Aliquots of 100 μL bacteria suspension were electroporated. For electroporation, 100 pmol siRNA in 1 μL siRNA suspension buffer (Qiagen) were adjusted to 25 μL with water and incubated for 5–10 min at room temperature. After addition of 100 μL competent bacteria, electroporation was performed at 1 kV, 400 Ohm, 25 μFD with time constants around 5 msec.
mRNA purification
TRIzol (Invitrogen) was used according to the manufacturer's protocol for purification of mRNA from E. coli and human tissue culture cell lines HEK293T and Jurkat. For purification of L. monocytogenes mRNA, the protocol was modified with all steps performed at 4°C and material precooled on ice as follows: 4 h after electroporation, 3 mL of bacteria culture were collected by centrifugation at 3800g for 7 min and the pellet resuspended in 1 mL TRIzol. The suspension was transferred into 2-mL FastPrep Tubes Lysis Matrix B (Qbiogene) and cell walls ruptured by shaking at setting 6.5 for 20 sec in a Fast-Prep shaker (Qbiogene). This was followed by a subsequent incubation for 3–5 min on ice before the shaking process was repeated. Afterward, the tubes were centrifuged for 10 min at 18,000g and the supernatant carefully transferred into a new tube. In the following, the standard Trizol protocol was applied according to the manufacturer's recommendations except for a 20-min precipitation in 0.7 volumes of isopropanol, 0.2 volume of 5 M ammonium acetate, and 1/60 volumes of glycogen (Ambion) at −80°C. The RNA was pelleted at 18,000g for 15 min, and RNA quality was assessed using a Bioanalyzer (Applied Biosystems).
Reverse transcription and real-time PCR
Double-stranded miRNA mimics (Ambion), siRNA (Qiagen), single-stranded synthetic miRNAs (Dharmacon), and in vitro-transcribed miRNA precursors were serially diluted and served as templates for reverse transcription reactions. In reverse transcription reactions, 50 nM stem–loop RT primer, 1× RT buffer, 0.25 mM of each dNTP, 25 units MultiScribe reverse transcriptase (Applied Biosystems), and 3.8 units RNaseOUT (Invitrogen) were mixed with 2.5 μL containing varying amount of the RNA templates. The 7.5-μL reverse transcription mixture was incubated in an Applied Biosystems GeneAmp 9700 thermo cycler for 30 min at 16°C, 30 min at 42°C, 5 min at 85°C, and kept at 4°C. Then, 1.33 μL of the 7.5-μL reverse transcription reaction were used as templates for the real-time PCR amplification. The 1.33-μL cDNA was mixed with 1× TaqMan Universal PCR Master Mix (Applied Biosystems), 0.8 μM universal TaqMan-based probe (Applied Biosystems, FAM-MGB design), 1.5 μM forward primer, and 0.7 μM reverse primer. The real-time PCR was performed using the 7900HT Fast Real-Time PCR system from Applied Biosystems. All real-time PCR reactions were run in duplicate. When performing reverse transcription and real-time PCR using commercial kits, the recommendations from the manufacturer were followed.
RNA targets, primers, and probes
Target RNAs—GFP22 siRNA (Qiagen): sense 5′-GCAAGCUGACCCUGAAGUUCAU-3′, antisense 5′-GAACUUCAGGGUCAGCUUGCCG-3′; control siRNA (Dharmacon; seq. 6-5 in Patzel et al. 2005): sense 5′-GGAGCACAUUUGCAGUGAAdTdT-3′, antisense 5′-UUCACUGCAAAUGUGCUCCdGdU-3′; tuf-derived siRNA: sense 5′-UCAUGUUCUUUUGCGUCUGC-3′, antisense 5′-AGACGCAAAAGAACAUGAUG-3′; mature miR-133b double-stranded mimics (Applied Biosystems/Ambion PM10029); mature miR-146a double-stranded mimics (Applied Biosystems/Ambion PM13059); mature single-stranded miR-133b: 5′-UUUGGUCCCCUUCAACCAGCUA-3′ (Thermo Scientific/Dharmacon); mature single-stranded miR-146a: 5′-UGAGAACUGAAUUCCAUGGGUU-3′ (Thermo Scientific/Dharmacon). Full-length miRNA precursor genes including the T7 promoter sequence and a BamH1 restriction site at the 5′ end and the SP6 promoter sequence as well as a HindIII restriction site at the 3′ end were synthesized by gene synthesis (GeneArt, Applied Biosystems).
The complete synthesized sequences are as follows:
pre-miR-133b: 5′-CTAATACGACTCACTATAGGGATCGGATCC CCTCAGAAGAAAGATGCCCCCTGCTCTGGCTGGTCAAACGGAACCAAGTCCGTCTTCCTGAGAGGTTTGGTCCCCTTCAACCAGCTACAGCAGGGCTGGCAATGCCCAGTCCTTGGAGAAAGCTTGTATTCCTATAGTGTCACCTAAAT-3′;
pre-miR-146a: 5′-CTAATACGACTCACTATAGGGATCGGATCC CCGATGTGTATCCTCAGCTTTGAGAACTGAATTCCATGGGTTGTGTCAGTGTCAGACCTCTGAAATTCAGTTCTTCAGCTGGGATATCTCTGTCATCGTAAGCTTGTATTCCTATAGTGTCACCTAAAT-3′.
For RNA in vitro transcription, the precursor genes were PCR-amplified from the pMA-T synthesis vector using the following primers:
MA-T-133b-Fw: 5′-AGCTCCTAATACGACTCACTATAGGGATC-3′;
MA-T-133b-Rv: 5′-GCTCCAGGTACCATTTAGGTGACACT-3′;
MA-T-146a-Fw: 5′-GCCACTAATACGACTCACTATAGGGATC-3′;
MA-T-146a-Rv: 5′-GTGCTCCATTAATTAATATTTAGGTGACAC-3′.
The PCR fragments were digested by HindIII, and the purified DNA was used as templates. In vitro transcription was performed using T7 RNA polymerase and related reagents from Fermentas following the manufacturer's instructions. RNA transcripts were purified by TRIzol extraction (Invitrogen) and analyzed by 10% denaturing polyacrylamide gel electrophoresis. RT-PCR primers and probe were Universal stem–loop protocol reverse primer: 5′-GTGCAGGGTCCGAGGT-3′; Universal stem–loop protocol TaqMan- based probe: 5′-FAM-TCGCACTGGATACG–MGBNFQ-3′; Universal stem–loop primer sequence: 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAC…-3′; target-specific stem–loop 3′-overhangs (GFP22): 5 bp 5′-…CGGCA-3′, 6 bp 5′-…CGGCAA-3′, 8 bp 5′-…CGGCAAGC-3′, 11 bp 5′…CGGCAAGCTGA-3′; GFP22 forward primer: 5′-GCCGAACTTCAGGGTCAGCTT-3′. MiR-133b-specific stem–loop 3′-overhang: 5′…TAGCTG-3′. MiR-113b-specific forward primer: 5′-GCCCTTTGGTCCCCTTCAAC-3′; miR-146a-specific stem–loop 3′-overhang: 5′-…AACCCA-3′; miR-146a-specific forward primer: 5′-GCCCTGAGAACTGAATTCCATG-3′. Commercial TaqMan microRNA Assays (Applied Biosystems) were used for hsa-miR-133b (#002247) and hsa-miR-146a (#000468) detection. Tuf siRNA-specific stem–loop 3′-overhangs: #1 5′-…TTTTCGCG-3′, #2 5′-…TTTTCG-3′, #3 5′-…CGAGCAGA-3′, #4 5′-…CGAGCA-3′, #5 5′-…AGCAGACG-3′, #6 5′-…AGCAGA-3′, #7 5′-…GCAGAC-3′, #8 5′-…CAGACG-3′, #9 5′-…ACGCAA-3′; Tuf siRNA-specific conventional forward primer: 5′-CCAGCGCCCATCATGTTCTT-3′, internal forward primer: 5′- TCATGTTCTTTTGCGTCTGC-3′. E. coli lstr-1-specific 3′-overhang: 5′-…GTTGAC-3′; lstr-1-specific forward primer 5′-CCGCAAAGCACACTGTATTATGT-3′; CsrB- specific 3′-overhang: 5′-…AATAAA-3′; CsrB-specific forward primer 5′-GCTGTGAATACAGTGCTCCCTTTT-3′. Standard RT- PCR primer for housekeeping gene 6PBG in L. monocytogenes: forward 5′-CCATCCGATGTTTTGCTAGCTAA-3′, reverse 5′-TGAACGTCGCCGCAGAA-3′, 6PBG TaqMan-based Probe: 5′-FAM-CAGACGGAAGCAATGCACAGCCA–MGBNFQ-3′ (Applied Biosystems). Melting temperatures were calculated using Primer 3 output with Breslauer's enthalpy and entropy from the online Oligo Melting Tool (http://bioinformatics.weizmann.ac.il/blocks-bin/oligomelt.pl).
ACKNOWLEDGMENTS
We thank Sandra Leitner and Isabell Dietrich for technical assistance with electroporation of L. monocytogenes and reverse transcription/RT-PCR. We also thank Juan P. Patron for providing microRNA kits and mature microRNAs and Lisa Scherer and John J. Rossi for discussion of the manuscript. This work was supported by Network “RNA Technologies,” funded by BMBF, Senate of Berlin and Chiron Corp. (grant numbers 0313066C-11/SenBB3066C-1 to V.P. and S.H.E.K.); the NUS-Cambridge Start-up grant of the National University of Singapore (grant number R-182-000-163-646 to V.P.); the National Medical Research Council of Singapore (grant number NMRC/NIG/1058/2011 to V.P.); and the Academic Research Fund of the Ministry of Education of Singapore (grant number R-182-000-200-112 to V.P.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33: e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhandayuthapani S, Banu MJ, Kashiwabara Y. 1994. Cloning and sequence determination of the gene coding for the elongation factor Tu of Mycobacterium leprae. J Biochem 115: 664–669. [DOI] [PubMed] [Google Scholar]
- Grimm D, Wang L, Lee JS, Schurmann N, Gu S, Borner K, Storm TA, Kay MA. 2010. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J Clin Invest 120: 3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas K, Melefors O. 2009. The Escherichia coli CsrB and CsrC small RNAs are strongly induced during growth in nutrient-poor medium. FEMS Microbiol Lett 297: 80–86. [DOI] [PubMed] [Google Scholar]
- Kelleher CD, Champoux JJ. 1998. Characterization of RNA strand displacement synthesis by Moloney murine leukemia virus reverse transcriptase. J Biol Chem 273: 9976–9986. [DOI] [PubMed] [Google Scholar]
- Mantei A, Rutz S, Janke M, Kirchhoff D, Jung U, Patzel V, Vogel U, Rudel T, Andreou I, Weber M, et al. 2008. siRNA stabilization prolongs gene knockdown in primary T lymphocytes. Eur J Immunol 38: 2616–2625. [DOI] [PubMed] [Google Scholar]
- Mikulik K. 2003. Structure and functional properties of prokaryotic small noncoding RNAs. Folia Microbiol (Praha) 48: 443–468. [DOI] [PubMed] [Google Scholar]
- O'Connor L, Glynn B. 2010. Recent advances in the development of nucleic acid diagnostics. Expert Rev Med Devices 7: 529–539. [DOI] [PubMed] [Google Scholar]
- Patzel V, Rutz S, Dietrich I, Koberle C, Scheffold A, Kaufmann SH. 2005. Design of siRNAs producing unstructured guide-RNAs results in improved RNA interference efficiency. Nat Biotechnol 23: 1440–1444. [DOI] [PubMed] [Google Scholar]
- Starck J, Kallenius G, Marklund BI, Andersson DI, Akerlund T. 2004. Comparative proteome analysis of Mycobacterium tuberculosis grown under aerobic and anaerobic conditions. Microbiology 150: 3821–3829. [DOI] [PubMed] [Google Scholar]
- Vogel J, Argaman L, Wagner EG, Altuvia S. 2004. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr Biol 14: 2271–2276. [DOI] [PubMed] [Google Scholar]
- Voskuil MI, Visconti KC, Schoolnik GK. 2004. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis (Edinb) 84: 218–227. [DOI] [PubMed] [Google Scholar]
- Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136: 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J, Diederichs S. 2011. Argonaute proteins regulate microRNA stability: Increased microRNA abundance by Argonaute proteins is due to microRNA stabilization. RNA Biol 8: 1149–1157. [DOI] [PubMed] [Google Scholar]