

1-methyl-7-nitroisatoic anhydride (1m7) is a useful reagent for probing RNA structure and serves as the basis for SHAPE analysis. This paper reports a reliable safe and inexpensive method for converting the precursor (4NIA) to 1m7. This method may make SHAPE analysis available to most molecular biology laboratories.
Keywords: 1-methyl-7-nitroisatoic anhydride, 1m7, RNA structure, SHAPE
Abstract
Estimating the reactivity of 2′-hydroxyl groups along an RNA chain of interest aids in the modeling of the folded RNA structure; flexible loops tend to be reactive, whereas duplex regions are generally not. Among the most useful reagents for probing 2′-hydroxyl reactivity is 1-methyl-7-nitroisatoic anhydride (1m7), but the absence of a reliable, inexpensive source has prevented widespread adoption. An existing protocol for the conversion of an inexpensive precursor 4-nitroisatoic anhydride (4NIA) recommends the use of NaH in dimethylformamide (DMF), a reagent combination that most molecular biology labs are not equipped to handle, and that does not scale safely in any case. Here we describe a safer, one-pot method for bulk conversion of 4NIA to 1m7 that reduces costs and bypasses the use of NaH. We show that 1m7 produced by this method is free of side products and can be used to probe RNA structure in vitro.
INTRODUCTION
As appreciation for the many roles that RNA plays in the expression and control of genetic information has grown, so has interest in understanding the native folded structures of RNA. Among the many methods for detecting RNA folded structure, chemical probing is the most cost-effective and accessible. It requires dilute solutions of RNA, a few chemicals, simple electrophoresis equipment, and takes a few days to acquire and analyze the data. It is adaptable to a variety of experimental conditions and can be used to monitor dynamic changes in RNA folding as well.
The principle is fairly simple, and has been applied to many different RNA functional groups using various chemical probes (Cory et al. 1968; Peattie and Gilbert 1980; Inoue and Cech 1985; Wang and Padgett 1989; Kenri et al. 1992; Merino et al. 2005; Wilkinson et al. 2005; Regulski and Breaker 2008). After modification, the reacted sites are mapped along the RNA chain by either breaking the end-labeled RNA chain at the site of modification (chemically or enzymatically), or by extending a complementary, end-labeled primer with reverse transcriptase to the site of modification-induced cleavage. In cases where the modification itself blocks extension by reverse transcriptase, chain cleavage is unnecessary. The products of these reactions are separated by electrophoresis at nucleotide resolution on gels or capillaries, and the distance between the label (it can be radioactive or fluorescent) and the modified site identifies the modified nucleotide.
Since the fundamental structural contribution to RNA folding is provided by base-pairing, a major goal is to get the base pairs correct. Base-paired nucleotides will be mostly unreactive toward probes like dimethyl sulfate (DMS), which methylates N1 of A and N3 of C, groups that participate in Watson-Crick–type hydrogen bonding. For example, DMS-reactive bases are likely to have their W-C edges solvent exposed (Cory et al. 1968; Peattie and Gilbert 1980; Inoue and Cech 1985; Wilkinson et al. 2005). While the ultimate goal is to get the correct structure, it is often more practical to use the data to exclude structural models.
As a structural model develops beyond secondary structure, the interpretation of chemical probing data becomes more involved. Tertiary structures that pin together remote domains, such as loop–loop interactions or the packing of helices, use a rich diversity of molecular contacts, involving additional non-RNA constituents such as positioned water molecules and metal ions. Thus in addition to base-specific modification reagents, chemical probes that attack groups on the sugar provide a view of backbone features. Protection of the backbone from hydroxyl radical (OH•) cleavage (likely initiated at the ribose 4′-C) arises from helical packing or protein binding (Wang and Padgett 1989; Huttenhofer and Noller 1992). More recently, probes of the ribose 2′-OH group have helped provide information on the orientation and accessibility of the backbone. The “in-line probing” method assesses the slow spontaneous hydrolysis rate (likely “probe” is hydroxide ion, OH−) along the backbone, reflecting positions where the 2′-hydroxyl is more frequently oriented to attack the adjacent phosphate, resulting in strand cleavage (Regulski and Breaker 2008). Since the critical orientation for this reaction is infrequent for nucleotides participating in double-stranded RNA, in-line probing identifies loops and unconstrained nucleotides in the structure. More direct and experimentally versatile probes of 2′-hydroxyl position and dynamics are the acylation reagents developed primarily by the Weeks lab for their SHAPE methods (Merino et al. 2005).
SHAPE stands for selective 2′-hydroxyl acylation assessed by primer extension (Merino et al. 2005; Wilkinson et al. 2006; Mortimer and Weeks 2007, 2008); note also that exonuclease mapping can substitute for primer extension (Steen et al. 2010). The ability to probe the reactivity of the 2′-hydroxl along the chain provides a valuable view of the backbone not afforded by either hydroxyl radical probing or in-line probing. Duplex regions are relatively unreactive, as the acylation reagents are bulky and the nearby phosphate discourages proper orientation of the reagent to the 2′-hydroxyl of paired nucleotides. As detailed in work from the Weeks lab (Wilkinson et al. 2006, 2009; Gherghe et al. 2008a,b; Wang et al. 2008; Deigan et al. 2009; Low and Weeks 2010; Steen et al. 2012; Hajdin et al. 2013; Karabiber et al. 2013), the propensity of each 2′-hydroxyl to be acylated is correlated with certain RNA structural features, and these correlations can be applied to the development and evaluation of structure models within some range of confidence (Kladwang et al. 2011; Parisien and Major 2012; Washietl et al. 2012; Zarringhalam et al. 2012; Leonard et al. 2013; Ramachandran et al. 2013).
A key innovation in the 2′-hydroxyl acylation probing method is the use of different acylation reagents with different rates of reaction (Mortimer and Weeks 2007, 2008). The reagent-specific rate of reaction with water mirrors the rate of reaction with RNA 2′-hydroxyls; thus, the reaction is quenched in aqueous solution on different reagent-specific time scales. Selecting a reagent with an appropriate reaction rate offers the ability to capture long exposures or tight snapshots of RNA structure. This is useful in cases where the molecular population resides in a uniform structure at any one instant but dynamically explores alternatives, or for experiments designed to follow dynamic changes in RNA structure during assembly and function. Three main SHAPE reagents have been characterized that differ from each other in their reaction with both water and RNA 2′-hydroxyls. The original reagent N-methylisatoic anhydride (NMIA) is slow (t(1/2) = 4 min, 5 half lives = 20 min). For medium fast probing (t(1/2) = 14 sec, 5 half lives = 70 sec), 1-methyl-7-nitroisatoic anhydride (1m7) is recommended, and faster still (t(1/2) = 0.25 sec at 37°C, 5 half lives = 1–2 sec), benzoyl cyanide (BzCN) is gaining wide use, despite its toxicity (Mortimer and Weeks 2007, 2008). For general use the reagent of choice is 1m7; however, obtaining it is problematic.
We requested a sample of 1m7 from the Weeks lab, but instead they shared their protocol for converting 4-nitroisatoic anhydride (4NIA) to 1m7. The protocol uses sodium hydride (NaH), an air-reactive chemical supplied under oil and requiring special handling. In addition, the reaction is done in dimethyl formamide (DMF) as a solvent. While use of NaH in DMF has a long history, it has more recently been recognized as particularly dangerous (http://www.crhf.org.uk/incident101.html, as noted by the UK Chemical Reaction Hazards Forum), especially when reactions are scaled to pilot plant level, as would be necessary for commercial production (Buckley et al. 1982; DeWall 1982). Here we describe an alternative protocol for 1m7 synthesis that incorporates an approach that relies on N, N′-diisopropylethylamine (DIEA) as a base in place of NaH (Beutner et al. 2007). The protocol can be performed safely at gram scale with greatly improved (97%) yield in a lab with a standard fume hood, with potential for safer scale-up to commercial production. The 1m7 produced by this method is free of contaminants that would interfere with SHAPE as demonstrated by probing of E. coli initiator tRNA.
RESULTS AND DISCUSSION
The reaction scheme to convert 4NIA to 1m7 is shown in Figure 1. The first step entails reacting 4-nitroisotoic anhydride with DIEA in order to generate a nucleophilic nitrogen anion in the ring of 4NIA. In the second step, this anion attacks methyl iodide and becomes methylated. In the presence of water, the basic anion will become protonated and the subsequent methylation step will not occur. Therefore, to achieve high efficiency in the conversion, it is important to exclude water from all reagents and glassware used in the reaction (see Materials and Methods for additional cautionary tips). To develop a useful modification of the reaction from Beutner et al. (2007) to allow production of 1m7 with appropriate yield and purity, we performed a small-scale reaction. The starting material, 4NIA (0.50 g, 2.4 mmol), was transferred to a flame-dried, cooled 100-mL round bottom flask and dissolved with anhydrous DMF (∼15 mL). A teflon-coated magnetic stir bar was added to the flask and the solution was stirred using a magnetic stirring plate. Due to the volatile nature of the liquid reagents used in this reaction, the flask was sealed with a rubber septum. Dry (anhydrous) diisopropylethylamine (DIEA; 1.2 mL, 7.2 mmol) was added to the flask and the mixture was stirred for 10 min at room temperature. After 10 min, anhydrous methyl iodide (0.75 mL; 12 mmol) was added dropwise and stirring was continued at room temperature for 2 h. After 2 h, 30 mL of 0.5 N HCl was poured into the flask and the resulting mixture was stirred for 5 min. The reaction mixture was then transferred to a separatory funnel and the product, 1m7, was isolated by extracting the reaction mixture three times with ethyl acetate (∼25–30 mL each). The (lower) aqueous layer was disposed of as hazardous waste, and the ethyl acetate layers were combined in a clean separatory funnel. The ethyl acetate layer was back-extracted with a second 30-mL aliquot of 0.5 N HCl in order to remove any trace of DMF that might have partitioned into the ethyl acetate layer. Finally, the (upper) ethyl acetate layer was transferred to a 250-mL round bottom flask and the solvent was evaporated using a rotary evaporator. The product was obtained as a yellow-orange solid.
FIGURE 1.
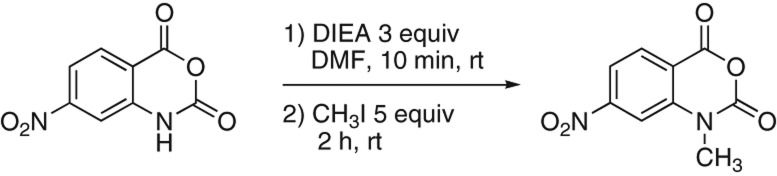
Reaction scheme for 1m7 synthesis. (Left) 4-nitroisatoic anhydride (4NIA); (right) 1-methyl-7-nitroisatoic anhydride (1m7). (DIEA) N, N′-diisopropyl ethylamine; (DMF) dimethyl formamide; (CH3I) methyl iodide; (rt) room temperature; (equiv) mole equivalents.
Both proton and carbon NMR spectra were obtained in order to confirm the identity of the product and assess its purity (Fig. 2). Spectra were obtained in Fourier Transform mode on a Varian 500 MHz nuclear magnetic resonance spectrometer. Chemical shifts are reported in parts per million (ppm) with the solvent resonance as the internal standard (Fig. 2). The number of resonances observed (six protons and nine carbons) and their chemical shifts are consistent with the structure of 1m7 (Fig. 2). Specifically, the proton spectrum shows the N-methyl group as a singlet at 3.70 ppm that integrates for three protons. Moreover, both the proton and carbon spectra are free of contaminating peaks, indicating that the 4NIA starting material was converted completely into 1m7 and that the purification protocol was successful in separating the 1m7 product from the reaction by-products and DMF solvent. We conclude the product is 1-methyl-7-nitroisatoic anhydride (1m7). The protocol efficiently converts 4NIA to 1m7 with a minimum of side product generation: Molar yield was calculated to be ∼97% (0.5 g of 4NIA MW 208.13 produced 0.520 g of 1m7 MW 222.15). The 2.3 mmol of 1m7 produced by this method when dissolved in anhydrous DMSO at a convenient stock concentration of 100 mM would yield enough reagent to perform 20,000 10-μL scale probing reactions. Although no toxicity studies have been published, it is prudent to consider 1m7 toxic. When dissolved in dimethyl sulfoxide (DMSO), measures must be taken to prevent it from coming into contact with the skin. DMSO can facilitate rapid penetration of the skin by any chemical dissolved therein. Gloves specifically designated for use with DMSO should be worn when handling DMSO stock solutions of 1m7.
FIGURE 2.

Nuclear magnetic resonance analysis of 1m7 product. (A) Proton NMR in deuterated chloroform (CDCl3). 1H spectrum of 1m7, 500 MHz: s, 3.703, 3H; d, 8.062, J = 2.0 Hz; dd, 8.145, J = 2.0, 8.5 Hz, 1H; d, 8.396, J = 8.5 Hz, 1H. Internal solvent standard is at 7.27 ppm. (Inset) Structure shows the positions of the six H atoms in 1m7. The resonance at 1.62 is DHO, formed by deuterium exchange between the NMR solvent and a small amount of H2O that enters the NMR solvent from the atmosphere, and thus does not arise from the product. (B) Carbon NMR in deuterated dimethyl sulfoxide (DMSO-d6) 13C spectrum of 1m7, 125 MHz: 32.05, 109.81, 116.56, 117.48, 131.08, 143.00, 147.31, 152.35, 157.85 ppm. Internal solvent standard is at 38.95–39.88 ppm. (Inset) Structure shows the positions of the nine C atoms in 1m7. (s) Singlet; (d) doublet; (dd) doublet of doublets; (MHz) megahertz; (Hz) hertz; (J) coupling constant.
To demonstrate the performance of 1m7 made by this method, we probed the reactivity of 2′-hydroxyls in initiator tRNA-fMet purified from E. coli and monitored the reaction by primer extension (SHAPE) (Fig. 3). Denatured and snap-cooled initiator tRNA (100 nM) was exposed to an initial concentration of 10 mM 1m7 product for 70 sec at 37°C, recovered, and subjected to primer extension using a primer complementary to the 3′ end of the tRNA (see Materials and Methods). As expected, the majority of nucleotides participating in standard A-form RNA duplex structure are unreactive toward the reagent (Fig. 3A, bars at right indicate duplexed regions that bound the indicated loops). High reactivity is evident for G19 in the D-loop and Ψ55 and C56 in the T-loop. In addition, the base before the anticodon (U33) and the first two bases of the anticodon (C34 and A35) are reactive. These are the major loop regions of the tRNA. Using the crystal structure of initiator tRNA as a guide, we located the reactive regions (Fig. 3B). The residues discussed above are colored pink in the cartoon in the top left panel. The bottom panel shows the positions of the reactive 2′-hydroxyls of G19, Ψ55, and C56. The 2′-hydroxyl of Ψ55 extends away from the top of the T-loop. G19 and C56 pair with each other to help pin the D- and T-loops together, and their 2′-hydroxyls extend out from the corner of the L-shaped tRNA formed by their interaction. The reactive 2′-hydroxyls in the anticodon loop also project into accessible positions (Fig. 3B, top right panel). This experiment shows that 1m7 produced by this alternative reaction scheme generates 2′-hydroxyl acylation data as generally expected without otherwise damaging the RNA, confirming its utility for RNA structure probing.
FIGURE 3.
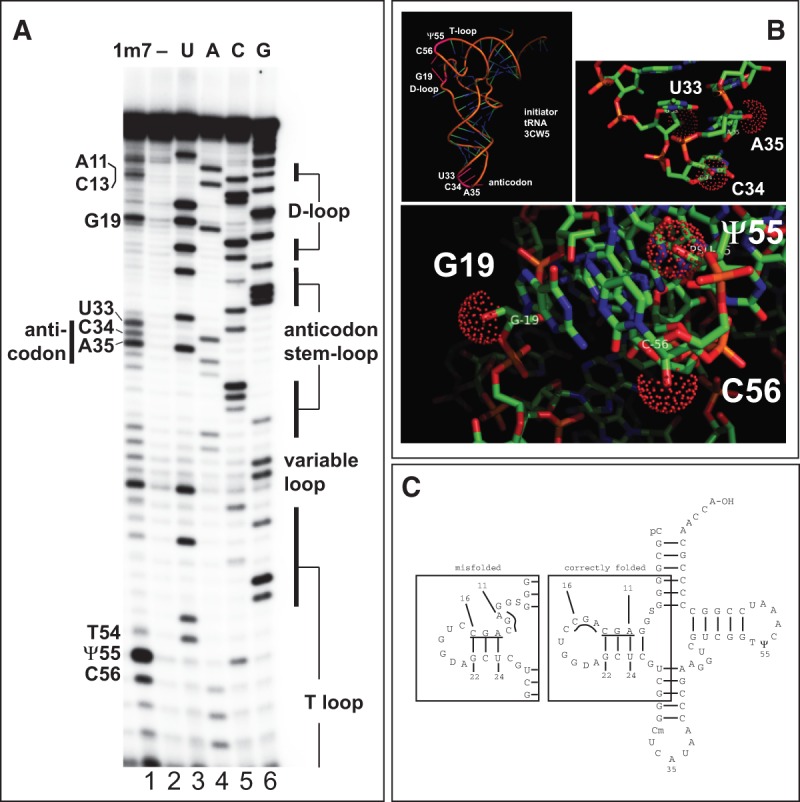
1m7 probing of E. coli initiator tRNA-fMet. (A) Primer extension products from 1m7-treated tRNA-fMet. Adduct formation leads to primer extension stops (cf. lane 1 [10 mM 1m7 for 70 sec] and lane 2 [no 1m7]). Lanes 3–6 are primer extensions that include one of four dideoxynucleoside triphosphates (ddNTPs) to generate a sequencing ladder; (lane 3) ddATP; (lane 4) ddTTP; (lane 5) ddGTP; (lane 6) ddCTP. The primer extension stop created by a ddNTP incorporation is 1 nt longer than the primer extension stop created by the adduct at the same base. The labels on the left indicate the identity of key modified nucleotides. On the right, duplex regions of the tRNA are indicated. (B) Sites of 1m7 reactive positions mapped to the crystal structure of tRNA-fMet (3CW5). (Top left panel) Cartoon of initiator tRNA-fMet. Two regions of efficient 2′-hydroxyl acylation, where the D-loop and T-loop interact, and in the anticodon loop, are labeled in pink (see text). (Top right panel) Detail of the anticodon region. Reactive 2′-hydroxyls on residues in (C34 and A35), and near (U33) the anticodon loop are indicated by red spheres. (Bottom panel) Detail of the region where the T-loop and the D-loop interact. Reactive 2′-hydroxyls on G19 (D-loop), C56 and pseudouridine-55 (Ψ55) in the T-loop are indicated by red spheres. (C) Evidence for misfolding of the D-stem in snap-cooled tRNA-fMet. Secondary structure model derived from the crystal structure (3CW5) is shown at right. Based on SHAPE reactivity in A, residues 11–13 are reactive, whereas adjacent residues 14–16 are less so, suggesting the folding shown in the box at left (“misfolded”). See text for details.
Higher than expected reactivity occurs in residues on the 5′ strand (but not the 3′ strand) of the D-stem (Fig. 3A). In the crystal structure, the sequence 11AGC13 pairs with 22GCU24 and should be protected (Fig. 3C). However, the three residues following 11–13 are also AGC and thus a slipped-pairing misfolded form of the RNA could exist in this snap-cooled sample with residues 14–16 paired to 22–24, exposing residues 11–13 in an unconstrained loop (Fig. 3C), explaining the reactivity of their 2′-hydroxyls. This points out the challenge of interpreting probing results when mixed populations of structures might be present (for example, see Wilkinson et al. 2005).
For conversion of smaller amounts of 1m7 in labs where organic chemistry is not generally practiced, the following protocol yields enough probe for ∼1000 reactions. As with the larger-scale reactions, the reagents must be made anhydrous using calcium hydride and stored over molecular sieves for 24 h prior to the start of the reaction (see Materials and Methods). The volatile nature of the liquid reagents used in this reaction requires the procedure to be carried out in a fume hood. The reaction is set up in a flame-dried and cooled 20-mL scintillation vial that should remain tightly capped except when adding reactants (this is not an exothermic or gas-generating reaction). Transfer 20 mg of 4-nitroisotoic anhydride (96 μmol) to the scintillation vial and dissolve with dry DMF (∼0.5–1 mL). Add a small magnetic stir bar to the vial and stir the solution using a magnetic stirring plate. Add diisopropylethylamine (DIEA; 3 equiv; 290 μmol; 48 μL) to the vial and continue stirring for an additional 10 min at room temperature. After 10 min, add the methyl iodide (5 equiv; 480 μmol; 30 μL) in one aliquot and continue stirring at room temperature for 2 h. Terminate the reaction by adding 3 mL of 0.5 N HCl to the reaction vial and stirring the resulting mixture vigorously for 5 min (it no longer needs to be closed). The yellow-orange 1m7 is extracted from the aqueous acid/DMF mixture into ethyl acetate. Add a 5-mL aliquot of ethyl acetate to the vial and emulsify the two immiscible layers by vigorously stirring on the stir plate for ∼30 sec. Allow a few moments for the organic and aqueous layers to separate, and then use a Pasteur pipet to carefully transfer the top layer (ethyl acetate) to a clean and tared scintillation vial. Repeat the extraction two more times, and combine the ethyl acetate layers. Evaporate the ethyl acetate solvent in the hood using a rotary evaporator or by blowing a gentle stream of dry nitrogen over the top of the vial. Weigh the scintillation vial with the product and calculate the yield. Yields should approach 95%.
Larger-scale conversion starting with >5 g of 4NIA is also feasible. This requires handling of larger (although not necessarily proportionately scaled) volumes of organic liquids and reactants, and would more reasonably be done in a working organic chemistry lab. In summary we present a simple, safe method for converting an inexpensive precursor 4NIA to 1m7 for selective 2′-hydroxyl acylation studies to probe RNA structure. The reaction (Fig. 1) can be scaled down and performed in a molecular biology lab without NaH and produces 1m7 that is sufficiently contaminant-free (Fig. 2) to allow identification of RNA solution structural features (Fig. 3).
MATERIALS AND METHODS
Handling organic solvents/reagents
The solvent DMF and the reactant DIEA both have strong odors and methyl iodide is especially volatile. Therefore, all solvents and liquid reagents should be handled and mixed in a fume hood. In particular, methyl iodide (boiling point ∼42.5°C) is a carcinogen used as a fumigant pesticide and should not be inhaled. DMSO is not particularly toxic, but efficiently carries dissolved compounds into the skin. The solvents used here may react with latex gloves commonly found in molecular biology or biochemistry laboratories, and thus gloves with appropriate protection (nitrile or butyl) must be used. 4NIA and 1m7 are irritants and react with water to produce CO2 and organic decomposition products of uncertain toxicity, thus measures should be taken to avoid contact with solutions or breathing dust from the solid. Safety glasses and lab coats should be worn while handling these materials.
Removing water from glassware and organic solvents
The glassware, 3–4 Å molecular sieves (crystalline aluminosilicates that can be dehydrated and used to capture water), solvent, and liquid reagents used in this process must be dried prior to use, since water reverses the abstraction of the proton from 4NIA that is critical to initiate the reaction. Glassware should be clean and well rinsed with ethanol, and then “flame dried” while empty. Never flame dry glassware that retains any amount of solvent of any kind. It is only necessary to briefly heat the glassware (2–3 min, depending on vessel size) to drive off bound moisture. A source of dry nitrogen gas is useful for generating a cooling stream without reintroducing moisture. Nitrogen gas from a tank is usually dry enough to use without an in-line drying cartridge. Always ensure the glassware has cooled to room temperature before adding any solvent. Glassware and molecular sieves can be dried in advance (pre-weigh the molecular sieves into the flask and flame dry them at the same time), cooled, stored filled with dry nitrogen (or other anhydrous unreactive gas such as argon), and capped with rubber septa.
“Drying” a liquid solvent in this context means rendering it anhydrous by removing dissolved H2O. This is done using calcium hydride (CaH2), a mild desiccant that reacts with water to form H2 gas and Ca(OH)2 (calcium hydroxide). Although it is considerably safer than sodium hydride, it is best to minimize exposure to air by storing it in a bench-top or a cabinet dessicator, in a parafilm-sealed bottle containing inert gas. Calcium hydride should be added to organic solvents in small portions, in a fume hood. If the solvent contains any water, small bubbles of H2 gas will be observed rising from the solid organic insoluble calcium hydride. When the bubbles stop, the water is gone. After decanting the bulk of the dried solvent to another vessel, the remaining calcium hydride can be quenched by adding tert-butanol very slowly until no additional bubbles are observed rising from the solid material. At this point, complete the quench by adding several milliliters of water in a dropwise manner. Calcium hydride and calcium hydroxide are visually indistinguishable, so other than the evolution of gas, there is no visual cue indicating the completion of this process. It is advisable to leave the flask containing the quenched calcium hydride in the fume hood for up to 24 h to ensure that all gas has been sufficiently vented. The remaining sludge should be disposed of as hazardous waste.
To dry the DMF solvent, two flame-dried, cooled 100-mL flasks are prepared, one containing 4–5 g of dried molecular sieves. The DMF (25 mL) is added to the flask without molecular sieves, a stir bar is added, and then a small amount (∼0.25 g) of the mild desiccant calcium hydride (see above) is introduced with stirring. After 30 min, ∼20 mL of dry DMF is decanted into the flask containing molecular sieves, the air in the flask is replaced with dry nitrogen as a rubber septum is fitted to create a seal. The solvent is stored over the molecular sieves for 24 h before use.
Methyl iodide was dried using a procedure similar to that described above for DMF. Instead of flasks, two 20-mL scintillation vials were used for this procedure. Molecular sieves (∼1 g) were added to one of the vials and then both vials were flame dried using the process described above. After cooling the vials, ∼1.5 mL of methyl iodide was added to the empty vial and a small portion of calcium hydride was added (∼0.1 g). The methyl iodide/CaH2 mixture was stirred gently at room temperature for 30 min using a magnetic stir bar and a magnetic stirring plate. After 30 min, a pasture pipet was used to carefully transfer the methyl iodide to the scintillation vial containing the molecular sieves. Dry nitrogen gas was added to the headspace above the liquid by blowing a gentle stream of nitrogen gas into the vial while the cap was being secured tightly. The anhydrous methyl iodide was stored over molecular sieves for 24 h prior to use in the reaction. Methyl iodide should only be handled in a fume hood while wearing safety glasses, a lab coat, and chemically resistant gloves. Store methyl iodide in an airtight, light-resistant container by applying parafilm around the cap and wrapping the vial in aluminum foil. Keep the container in a cool, well-ventilated area.
To dry the DIEA, ∼1–2 g of 3–4 Å molecular sieves were added to a 20-mL scintillation vial, and both were flamed dried as described above. Once the vial and sieves were cool, ∼3 mL of DIEA was transferred to the vial, the airspace above the liquid was replaced by dry nitrogen, and the vial was capped for 24 h before use in the reaction. When adding DIEA to flame-dried molecular sieves, it is critical that the vial and sieves are at room temperature to prevent decomposition or evaporation. DIEA is an odorous and toxic liquid and it should only be handled in a fume hood using appropriate gloves.
Source of reagents
We purchased 4-nitroisatoic anhydride (4NIA) from AstaTech (by quote, 10 g for $267). N,N′-dimethyl formamide (DMF) was purchased from Acros Organics (Cat # 615211000). Diisopropylethylamine (DIEA) was purchased from Advanced ChemTech (Cat # RB8433). Methyl iodide (Iodomethane) was purchased from Alfa Aesar (Cat # 43681).
Ethyl acetate was purchased from VWR. DMSO (99.5%, over molecular sieves) was from Fluka.
Nuclear magnetic resonance
Proton and carbon NMR spectra were obtained in Fourier Transform mode on a Varian 500 MHz nuclear magnetic resonance spectrometer. Chemical shifts are reported in parts per million (ppm) with the solvent resonance as the internal standard: CDCl3 at 7.27 ppm (proton spectrum); DMSO-d6 at 38.95–39.88 ppm (carbon spectrum).
Chemical probing and primer extension of initiator tRNA (SHAPE)
A stock of 1-μM initiator tRNA-fMet (Sigma, Cat # R8019-50UN) was prepared. For each reaction, 1 μL of tRNA stock was added to 5 μL of 0.5XTE buffer in separate tubes. The samples were heated to 95°C for 1 min, then immediately placed on ice for 5 min. Then 3 μL of folding mix were added to each (333 mM HEPES at pH 8.0, 20 mM MgCl2, 333 mM NaCl). To access the reactivity of 2′-hydroxyls, 1 μL of DMSO (no reagent control) or 1 μL of a 100 mM stock of 1m7 in DMSO was added (initial concentration in the reaction was 10 mM) and the samples were incubated at 37°C for 70 sec. The reaction was stopped by the addition of 200 μL of H2O, 20 μL of 3 M sodium acetate (pH 5.3), and 500 μL of ethanol. Ethanol precipitates were collected by centrifugation and the pellets were rinsed with 75% ethanol, briefly speed vacuumed, and resuspended in 6 μL of RNase-free H2O.
Sites of modification in the tRNA were identified by reverse transcriptase primer extension blocks and mapped by comparison with dideoxynucleotide incorporation sites. A primer complementary to the 3′ end of initiator tRNA-fMet (5′-GGTTGCGGGGGCCGG-3′) was end labeled using polynucleotide kinase and [γ-32P]rATP. The 10-μL annealing reaction was done by mixing 3 μL of 5X RT buffer with no MgCl2 (5X RT no Mg = 375 mM KCl, 250 mM Tris-Cl at pH 8.3) and 1 μL of 1 μM [32P]-labeled DNA primer with 6 μL of modified RNA. The reaction was heated to 95°C for 2 min and cooled down to 48°C for 5 min, and then 4 μL of an RT enzyme mix was added (7 mM DTT, 2.5 mM dNTP, 5.8 mM MgCl2, 0.5 μL of Superscript II reverse transcriptase [Invitrogen]) for a total reaction volume of 14 μL. The reaction was incubated at 42°C for 30 min and stopped by ethanol precipitation. Reaction products were resuspended in formamide and dyes, heated to 95°C for 2 min, cooled, and loaded onto an 8-M urea-15% polyacrylamide gel. The sequencing ladder was made by setting up four equivalent reactions using untreated tRNA and including 1 μL of a stock of one of the four dideoxynucleoside triphosphates (stock was 5 mM for ddATP and ddTTP, 3 mM for ddGTP and ddCTP) in the annealing reaction in place of water. These amounts of dideoxynucleoside triphosphates were empirically determined to generate a useful distribution of base specific stops in each reaction. A band in the modification lane due to 2′-hydroxyl acylation of a given nucleotide will be shorter by one residue than the band produced by ddNTP-incorporation induced termination, allowing the modified nucleotide to be identified. E. coli expresses two forms of tRNA-fMet (Cory et al. 1968). In mapping the reactivity data to the structure of tRNA-fMet, we made use of the crystal structure for tRNA2-fMet (Barraud et al. 2008), although most preparations from E. coli (including the one we used here from Sigma) are composed of the predominant form tRNA1-fMet (Cory et al. 1968). These tRNAs differ only at position 46 in the variable loop, where tRNA2-fMet has A and tRNA1-fMet has 7meG (Cory et al. 1968). There is no crystal structure for tRNA1-fMet; however, this substitution likely has no more than a subtle effect on tRNA structure or function, since E. coli expressing either form alone are viable (Kenri et al. 1992).
ACKNOWLEDGMENTS
This work was funded by NIH grant GM040478 to M.A.
REFERENCES
- Barraud P, Schmitt E, Mechulam Y, Dardel F, Tisne C. 2008. A unique conformation of the anticodon stem-loop is associated with the capacity of tRNAfMet to initiate protein synthesis. Nucleic Acids Res 36: 4894–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner GL, Kuethe JT, Yasuda N. 2007. A practical method for preparation of 4-hydroxyquinolinone esters. J Org Chem 72: 7058–7061. [DOI] [PubMed] [Google Scholar]
- Buckley J, Webb RL, Laird T, Ward J. 1982. Report on thermal reaction. Chem Eng News 60(28): 5. [Google Scholar]
- Cory S, Dube SK, Clark BF, Marcker KA. 1968. Separation of two initiator transfer RNAs from E. coli. FEBS Lett 1: 259–261. [DOI] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, Weeks KM. 2009. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci 106: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWall G. 1982. Sodium hydride and DMF. Chem Eng News 60(37): 5. [Google Scholar]
- Gherghe CM, Mortimer SA, Krahn JM, Thompson NL, Weeks KM. 2008a. Slow conformational dynamics at C2′-endo nucleotides in RNA. J Am Chem Soc 130: 8884–8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherghe CM, Shajani Z, Wilkinson KA, Varani G, Weeks KM. 2008b. Strong correlation between SHAPE chemistry and the generalized NMR order parameter (S2) in RNA. J Am Chem Soc 130: 12244–12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM. 2013. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci 110: 5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhofer A, Noller HF. 1992. Hydroxyl radical cleavage of tRNA in the ribosomal P site. Proc Natl Acad Sci 89: 7851–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Cech TR. 1985. Secondary structure of the circular form of the Tetrahymena rRNA intervening sequence: A technique for RNA structure analysis using chemical probes and reverse transcriptase. Proc Natl Acad Sci 82: 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabiber F, McGinnis JL, Favorov OV, Weeks KM. 2013. QuShape: Rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA 19: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenri T, Imamoto F, Kano Y. 1992. Construction and characterization of an Escherichia coli mutant deficient in the metY gene encoding tRNA(f2Met): Either tRNA(f1Met) or tRNA(f2Met) is required for cell growth. Gene 114: 109–114. [DOI] [PubMed] [Google Scholar]
- Kladwang W, VanLang CC, Cordero P, Das R. 2011. Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry 50: 8049–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CW, Hajdin CE, Karabiber F, Mathews DH, Favorov OV, Dokholyan NV, Weeks KM. 2013. Principles for understanding the accuracy of SHAPE-directed RNA structure modeling. Biochemistry 52: 588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JT, Weeks KM. 2010. SHAPE-directed RNA secondary structure prediction. Methods 52: 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. 2005. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 127: 4223–4231. [DOI] [PubMed] [Google Scholar]
- Mortimer SA, Weeks KM. 2007. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J Am Chem Soc 129: 4144–4145. [DOI] [PubMed] [Google Scholar]
- Mortimer SA, Weeks KM. 2008. Time-resolved RNA SHAPE chemistry. J Am Chem Soc 130: 16178–16180. [DOI] [PubMed] [Google Scholar]
- Parisien M, Major F. 2012. Determining RNA three-dimensional structures using low-resolution data. J Struct Biol 179: 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peattie DA, Gilbert W. 1980. Chemical probes for higher-order structure in RNA. Proc Natl Acad Sci 77: 4679–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Ding F, Weeks KM, Dokholyan NV. 2013. Statistical analysis of SHAPE-directed RNA secondary structure modeling. Biochemistry 52: 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulski EE, Breaker RR. 2008. In-line probing analysis of riboswitches. Methods Mol Biol 419: 53–67. [DOI] [PubMed] [Google Scholar]
- Steen KA, Malhotra A, Weeks KM. 2010. Selective 2′-hydroxyl acylation analyzed by protection from exoribonuclease. J Am Chem Soc 132: 9940–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen KA, Rice GM, Weeks KM. 2012. Fingerprinting noncanonical and tertiary RNA structures by differential SHAPE reactivity. J Am Chem Soc 134: 13160–13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Padgett RA. 1989. Hydroxyl radical “footprinting” of RNA: Application to pre-mRNA splicing complexes. Proc Natl Acad Sci 86: 7795–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wilkinson KA, Weeks KM. 2008. Complex ligand-induced conformational changes in tRNA(Asp) revealed by single-nucleotide resolution SHAPE chemistry. Biochemistry 47: 3454–3461. [DOI] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Stadler PF, Kellis M. 2012. RNA folding with soft constraints: Reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res 40: 4261–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. 2005. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts. J Am Chem Soc 127: 4659–4667. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. 2006. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc 1: 1610–1616. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Vasa SM, Deigan KE, Mortimer SA, Giddings MC, Weeks KM. 2009. Influence of nucleotide identity on ribose 2′-hydroxyl reactivity in RNA. RNA 15: 1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarringhalam K, Meyer MM, Dotu I, Chuang JH, Clote P. 2012. Integrating chemical footprinting data into RNA secondary structure prediction. PLoS One 7: e45160. [DOI] [PMC free article] [PubMed] [Google Scholar]