

Structural studies on the ribosome have provided tremendous insights into how cognate codon–anticodon interactions in the decoding center lead to tRNA selection and, ultimately, to peptide chain elongation. Here, in the genetically and structurally tractable organism Thermus thermophilus, the authors isolate and identify streptomycin-dependent (SmD) ribosomal variants, as well as streptomycin-independent (SmI) revertants, with mutations affecting ribosomal protein S12 and the 16S rRNA, respectively. Single-molecule FRET and structural studies reveal here how these decoding-site proximal mutations can impact the ability of ribosomes to reach the GTPase-activated state required for progression in tRNA selection.
Keywords: ribosome, rRNA, decoding, single-molecule FRET, X-ray crystallography
Abstract
The ribosome decodes mRNA by monitoring the geometry of codon–anticodon base-pairing using a set of universally conserved 16S rRNA nucleotides within the conformationally dynamic decoding site. By applying single-molecule FRET and X-ray crystallography, we have determined that conditional-lethal, streptomycin-dependence mutations in ribosomal protein S12 interfere with tRNA selection by allowing conformational distortions of the decoding site that impair GTPase activation of EF-Tu during the tRNA selection process. Distortions in the decoding site are reversed by streptomycin or by a second-site suppressor mutation in 16S rRNA. These observations encourage a refinement of the current model for decoding, wherein ribosomal protein S12 and the decoding site collaborate to optimize codon recognition and substrate discrimination during the early stages of the tRNA selection process.
INTRODUCTION
The ribosome is a universal macromolecular machine that decodes the genetic information encoded in messenger RNA (mRNA) with remarkable precision. It does so by selecting aminoacyl-transfer RNAs (aa-tRNAs) from a pool of structurally similar substrates based on the geometry of base-pairing between the anticodon of the aa-tRNA and the codon of the mRNA within the aminoacyl (A) site of the small ribosomal subunit. The stereochemistry of this interaction is directly sensed by a set of universally conserved nucleotide bases located within a structurally dynamic “decoding site,” an asymmetric internal loop element within helix 44 (h44) of the 16S rRNA (Yoshizawa et al. 1999; Ogle et al. 2001; Hermann 2006; Lescoute and Westhof 2006; Sanbonmatsu 2006).
While differences in the dissociation rates of cognate and near-cognate aa-tRNAs contribute to decoding fidelity (Pape et al. 1999), the ribosome uses an induced-fit mechanism to discriminate between structurally similar tRNA substrates (Pape et al. 1999; Ogle et al. 2002; Geggier et al. 2010). This mechanism includes local conformational changes in the decoding site that preferentially occur in response to cognate codon recognition (Ogle et al. 2001). In a process related to codon–anticodon recognition, global structural transitions in the 30S ribosomal subunit from an open to a closed form allosterically trigger GTPase activation and GTP hydrolysis by elongation factor Tu (EF-Tu), to drive tRNA selection in the forward direction (Ogle et al. 2002; Voorhees et al. 2010). GTP hydrolysis divides decoding into two steps—initial selection and proofreading—providing the ribosome with two independent opportunities to reject incorrect substrates.
The two-state domain closure model (Ogle et al. 2002) has provided a highly effective structural framework for interpreting decades of experimental data, but recent findings indicate that the decoding mechanism remains to be fully elucidated (Gregory et al. 2009; Geggier et al. 2010; McClory et al. 2010; Zaher and Green 2010a,b; Schmeing et al. 2011; Demeshkina et al. 2012). This point is highlighted by observations of a kinetically unstable intermediate in the selection process that exhibits low FRET in single-molecule fluorescence resonance energy transfer (smFRET) investigations (Blanchard et al. 2004; Geggier et al. 2010). This transient intermediate represents a codon recognition complex early in the initial selection process that is distinct from those observed thus far by static structural methods such as cryo-electron microscopy or X-ray crystallography. Structural characterizations of this important intermediate in the selection process will be essential to further elucidating the induced-fit mechanism.
Crystal structures have revealed that the closed conformation of the 30S subunit is stabilized by contacts between conserved amino acid residues of ribosomal protein S12 and 16S rRNA h44 at the decoding site (Ogle et al. 2002). S12 is an essential protein (Sharma et al. 2007), and amino acid substitutions at its interface with the decoding site generally cause the ribosome to be hyperaccurate and resistant to the error-promoting antibiotic streptomycin (the SmR phenotype). Streptomycin-dependence (SmD) phenotypes are caused by conditional-lethal mutations that undermine the process of tRNA selection so severely that either streptomycin or a second-site, streptomycin-independence (SmI), mutation, is required for viability (for review, see Gale et al. 1981; Kurland et al. 1996). Despite the important role that SmD mutants have played in developing our understanding of decoding by the ribosome, the precise mechanism by which SmD mutations impose the dependence phenotype has yet to be established. In vitro studies have shown that SmD ribosomes have decreased A-site binding affinity (Ruusala et al. 1984; Bilgin et al. 1992; Karimi and Ehrenberg 1994), which, in the context of the induced-fit model, likely results from perturbations in the induced-fit mechanism underpinning initial selection such as the balance between the open and closed states (Ogle et al. 2002).
Here, we asked whether the SmD mutant phenotype could also result from distortion or destabilization of the decoding-site conformation required for proper tRNA selection in which residues A1492 and A1493 are extruded away from the helical axis of h44 toward the codon–anticodon binding site. The inherent plasticity of the decoding site is evident from structural investigations of decoding-site oligonucleotide analogs (Fourmy et al. 1996, 1998), ribosomes in cognate decoding complexes (Ogle et al. 2001; Schmeing et al. 2009), and ribosomes in complexes with antibiotics (Carter et al. 2000; Borovinskaya et al. 2008; Stanley et al. 2010; Demirci et al. 2013) or initiation factor IF1 (Carter et al. 2001). The effects of SmD mutations on global aspects of ribosome conformation have been revealed by chemical probing (Allen and Noller 1989) and crystallography (Vila-Sanjurjo et al. 2003), but the mechanistic underpinnings of the SmD phenotype remain unclear. To advance our understanding of this elusive aspect of the decoding mechanism, we examined SmD and SmI mutant ribosomes using a combination of smFRET and X-ray crystallography. Our observations provide a mechanistic explanation for the SmD phenotype and indicate a direct role for ribosomal protein S12 in structuring the decoding site during the early stages of the decoding process.
RESULTS
Isolation and genetic analysis of SmD and SmI mutants of T. thermophilus
To determine the structural basis for the streptomycin-dependence phenotype, we isolated SmD mutants of the thermophilic bacterium Thermus thermophilus. This was done using HG286 (genotype ΔrrsA::htk1) (Gregory and Dahlberg 2009), a derivative of the natural isolate HB8 (Oshima and Imahori 1974) in which the rrsA gene has been deleted and replaced with the heat-stable kanamycin adenyltransferase gene htk (Hashimoto et al. 2001), leaving a single 16S rRNA gene, rrsB. A spontaneous mutant of HG286, designated HG533 (ΔrrsA::htk1 rpsL-P90L), was identified in a selection for SmD mutants and was found to possess a single base mutation in rpsL that creates a P90L amino acid substitution in ribosomal protein S12. We had previously identified this same mutation in an SmD isolate of the Icelandic T. thermophilus strain IB-21 (Gregory et al. 2001). SmD mutants of Escherichia coli have been found with substitutions at several S12 positions, including P90, G91, P41, K43, P44, and N45 (Timms and Bridges 1993). These residues are highly conserved and are in close proximity to h44 and the decoding site (Fig. 1A,B; Schluenzen et al. 2000; Wimberly et al. 2000).
FIGURE 1.
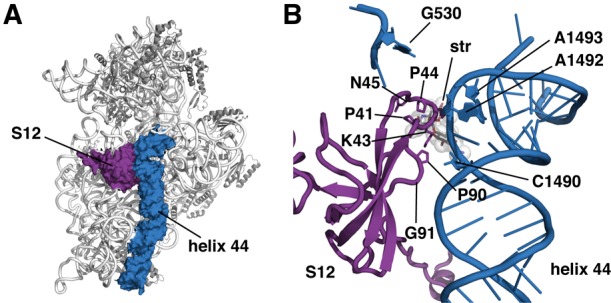
Location of SmD mutations affecting ribosomal protein S12. (A) Structure of the 30S ribosomal subunit from T. thermophilus (PDB entry 4DR1) (Demirci et al. 2013), indicating ribosomal protein S12 (purple) and 16S rRNA h44 (blue). (B) Close-up view of the decoding site, showing residues in ribosomal protein S12 (purple) mutated in SmD mutants identified in this and other studies. The upper portion of 16S rRNA h44 is colored blue and residues involved in monitoring codon recognition (G530, A1492, and A1493) and the site of the SmI mutation identified in this study are indicated.
In a previous study (Carr et al. 2005), we determined that bulkier side-chain substitutions at P90, including P90L, P90R, P90E, P90M, and P90W, produced SmD phenotypes, whereas smaller side chains at this position, such as P90G, P90A, and P90C, produced the less severe SmR phenotype. These findings suggested that steric clashes of these side chains of protein S12 with h44 underlie the SmD phenotype. To explore this possibility, we examined both the rpsL-P90L and rpsL-P90W mutations. Because replacement of a proline codon (CCN) with a tryptophan codon (UGG) requires multiple base mutations, the P90W substitution has never been identified in genetic selections for spontaneous mutants. The rpsL-P90W allele was obtained from JC452, constructed previously by site-directed mutagenesis and gene replacement (Carr et al. 2005). As this allele was originally constructed in a T. thermophilus IB-21 genetic background (Kristjansson et al. 1986), we transferred this mutant allele to T. thermophilus HB8 by transformation, selecting streptomycin dependence to produce HG944 (rpsL-P90W). Genomic DNA from HG944 was then used to transform HG286 to streptomycin dependence to produce HG962 (ΔrrsA::htk1 rpsL-P90W).
Using an enrichment scheme targeting rrsB, we derived, from HG 533, a spontaneous SmI double mutant bearing a C1490U base substitution in 16S rRNA h44 that reverses the SmD phenotype of the P90L mutation. The presence of a single 16S rRNA locus in the genome of mutants derived from HG286 allows the expression of potentially recessive rRNA mutations and also provides a pure population of mutant ribosomes for subsequent in vitro experiments. For this enrichment, a hygromycin B–resistant (HmR) mutant of HG286, HG470 (ΔrrsA::htk1 rrsB-U1406A) (Gregory and Dahlberg 2009), was transformed to SmD with genomic DNA from HG533 to produce HG574 (ΔrrsA::htk1 rpsL-P90L rrsB-U1406A). Using genomic DNA from a pool of several hundred spontaneous SmI derivatives of HG533, HG574 was transformed to SmI and screened for loss of the HmR phenotype. Given the tight genetic linkage between mutations in rrsB (Gregory and Dahlberg 2009), loss of the HmR phenotype indicated the presence of an SmI allele in rrsB. Among the transformants found to be hygromycin B sensitive was HG589 (ΔrrsA::htk1 rpsL-P90L rrsB-C1490U) in which the U1406A mutation had been replaced by a C1490U mutation in rrsB. For reasons that are presently unclear, despite several attempts, we were unable to identify spontaneous SmI derivatives of the P90W mutant HG962. 30S ribosomal subunits from HG533, HG589, and HG962 were used for kinetic and structural analyses.
Single-molecule imaging of the selection process on the T. thermophilus ribosome
We performed single-molecule FRET experiments using T. thermophilus 70S ribosomes programmed with mRNA and with native E. coli fMet-tRNAfMet in the P-site fluorescently tagged with a Cy3 fluorophore at the naturally occurring s4U8 modification via maleimide chemistry as previously described (Blanchard et al. 2004; Geggier et al. 2010). The process of aa-tRNA selection was imaged under pre-steady-state conditions using T. thermophilus EF-Tu in a ternary complex with GTP and E. coli Phe-tRNAPhe labeled as previously described (Blanchard et al. 2004; Geggier et al. 2010) with Cy5 at the naturally occurring acp3U47 residue. As anticipated from analogous experiments with E. coli ribosomes (Blanchard et al. 2004; Geggier et al. 2010), the accommodation of aa-tRNA into wild-type T. thermophilus ribosomes proceeded through low- and intermediate-FRET intermediate configurations on-path to a high-FRET state (Fig. 2A,B). Previous investigations of aa-tRNA selection using the E. coli components suggest that such configurations correspond to codon-recognition (CR), GTPase-activated (GA), and fully accommodated states (AC), respectively (Blanchard et al. 2004; Geggier et al. 2010).
FIGURE 2.

Single-molecule observations of tRNA selection on T. thermophilus 70S ribosomes. (A) Representative single-molecule fluorescence (Cy3 [green]; Cy5 [red]) and FRET trajectories [FRET = ICy5/(ICy3 + ICy5)] obtained during stopped-flow delivery of the ternary complex with Cy5-labeled Phe-tRNAPhe to ribosome complexes containing Cy3-labeled P-site fMet-tRNAfMet. The first 2 sec of the FRET trajectory (yellow) is expanded beneath, showing clear progression from zero-FRET to a fully accommodated (AC), high-FRET state via on-path intermediates in the tRNA selection process. (B) Post-synchronized FRET trajectories combined in two-dimensional contour plots from wild-type ribosomes decoding near-cognate UCU codon (left), cognate UUC codon with GTP replaced by the nonhydrolyzable GTP analog GDPNP (middle), and cognate UUC codon (right). (C) Decoding by P90L ribosomes (left), P90L ribosomes in the presence of 40 μM streptomycin (middle), and P90L/C1490U SmI mutant ribosomes (right). (D) tRNA incorporation by P90W ribosomes in the absence (left) or presence (right) of 40 μM streptomycin. (E) Representative traces state from P90L (left) and P90W (right) complexes showing molecules that successfully reached the fully accommodated (AC) state after fluctuating reversibly between FRET states assigned to codon recognition (CR) and GTPase-activated (GA) states. Fluorescence and FRET trajectories are shown as described in Figure 1A.
In line with these assignments, binding of the same ternary complex to ribosomes programmed with a near-cognate mRNA codon in the A-site was transient, where rejection principally occurred from short-lived, low- and intermediate-FRET configurations. Thus, under the conditions of these experiments, the aa-tRNA selection process on T. thermophilus ribosomes operates with high fidelity, where the ternary complex rapidly dissociates during initial selection prior to GTPase activation and GTP hydrolysis (Blanchard et al. 2004; Geggier et al. 2010). In contrast, the binding of the ternary complex to ribosomes programmed with a cognate mRNA codon in the A-site in the presence of the nonhydrolyzable GTP analog GDPNP resulted in the formation of a relatively stable complex exhibiting intermediate-FRET, as expected from previous descriptions of the structural and kinetic features of the GTPase-activated (A/T) complex (Moazed et al. 1988; Pape et al. 2000; Blanchard et al. 2004; Geggier et al. 2010). In this state, aa-tRNA adopts a bent configuration in the A-site that is stabilized by domain closure of the small subunit and recognition of the minor groove of the mRNA codon–tRNA anticodon pair by residues A1492 and A1493 (Valle et al. 2003; Schmeing et al. 2009).
Ribosomes from both SmD mutants exhibited marked defects in the selection process that significantly reduced the rate at which aa-tRNA achieved complete accommodation (Fig. 2C,D). Here, the cognate ternary complex appeared to fluctuate reversibly between low- and intermediate-FRET configurations for extended periods prior to successful accommodation (Fig. 2E) or dissociation. These results are in line with earlier experiments using E. coli SmD ribosomes that found a 20-fold decrease in GTP hydrolysis rates (Bilgin et al. 1992) and globally consistent with rapid kinetic measurements of restrictive SmR ribosomes from E. coli (Zaher and Green 2010b). Unexpectedly, both SmD mutants exhibited slow aa-tRNA dissociation rates (∼0.15 sec−1) from the ribosome in the presence of GTP, on par with that observed for GDPNP-stalled wild-type ribosomes (Fig. 2B–D, cf. the left panels). Visual inspection of individual smFRET trajectories from the SmD ribosome complexes exhibited reversible FRET fluctuations between low- and intermediate-FRET states (Fig. 2E). We conclude from these observations that the mechanistic defect during initial selection manifests as repeated, failed attempts of the ternary complex to achieve the GTPase-activated state at a step that is subsequent to codon recognition. As anticipated from earlier studies (Bilgin et al. 1992), streptomycin efficiently suppressed this functional defect in both the P90L and P90W mutant backgrounds (Fig. 2C, center panel; Fig. 2D, right panel). The C1490U substitution in 16S rRNA, selected for its ability to reverse the SmD phenotype, also restored function, albeit to a lesser extent. These observations argue that the SmD phenotype can be attributed to the stalling of the ternary complex on the ribosome during the initial selection process at a step between codon recognition and GTPase activation.
Crystallization of mutant ribosomes
We determined X-ray crystal structures of mutant 30S ribosomal subunits to uncover the structural basis for the tRNA selection defects observed through smFRET. Under the same conditions used to obtain crystals of wild-type 30S subunits (space group P41212, unit cell dimensions ∼403 Å × 403 Å × 176 Å, hereafter referred to as crystal form 1) (Clemons et al. 1999; Tocilj et al. 1999), SmD 30S subunits adopted a novel crystal form (space group P41212, unit cell dimensions ∼400 Å × 400 Å × 215 Å, hereafter referred to as crystal form 2) (Supplemental Fig. S1A). A contact involving helix 33a (the 30S subunit “beak”) and ribosomal protein S11 of a neighboring subunit (Supplemental Fig. S1B) is absent from crystal form 2 due to an ∼10 Å shift in the position of this helix in the 30S subunit head. This new crystal form contains a contact between ribosomal proteins S10 of neighboring subunits and a contact between ribosomal proteins S4 of adjacent subunits (Supplemental Fig. S1C,D) that are not found in crystal form 1. Overall, crystal form 2 results from a variation in the packing interactions seen in the original crystal form, with the helix 6 “spur” inserted into the P-site of a neighboring subunit (Supplemental Fig. S1E,F; Carter et al. 2000).
While SmD mutant subunits in the apo form adopted crystal form 2, SmI mutant subunits adopted crystal form 1. Notably, both SmD and SmI ribosomes adopted crystal form 1 when cocrystallized with streptomycin. Thus, the crystallization behavior of SmD and SmI ribosomes correlated with their in vivo phenotypes, suggesting that changes in structural dynamics occurring near the decoding site are propagated to distant sites within the 30S subunit, thereby affecting intersubunit packing. The P90L mutant 30S subunits adopted both crystal forms with approximately equal frequency, while the P90W mutant 30S subunits almost exclusively adopted crystal form 2. P90W ribosomes in crystal form 1 and wild-type ribosomes in crystal form 2 could also be found, implying that the two crystal forms are manifestations of an equilibrium between distinct 30S subunit conformations that is shifted by SmD mutations, SmI mutations, or streptomycin.
Structural impact of the SmD mutations
The universally conserved 16S rRNA bases A1492 and A1493 in h44, as well as G530 in helix 18, are inherently dynamic and change conformation to directly monitor codon–anticodon base-pairing geometry (Ogle et al. 2001). The present findings suggest that ribosomal protein S12 is involved in orchestrating this process. In crystal form 2, the structural impact of both SmD mutants was revealed as a distortion of the decoding site, immediately adjacent to ribosomal protein S12, and includes A1492 and A1493. These effects are readily visible upon comparison of composite omit maps of the wild-type and P90W mutants (Fig. 3A–D). In the P90W mutant, the backbone of A1492 was found to be compacted into a kinked conformation, where the phosphate atom shifted by 9.5 Å perpendicular to the helix axis (Fig. 4A; Table 1). This rotates the base further into the helix toward A1408, whose N1 is shifted 3.6 Å toward the minor groove. The C1409–G1491 base pair is also displaced away from its native position. The N1 of G1491 and the N3 of C1409 shift 7.0 Å and 6.1 Å, respectively, toward the minor groove (Fig. 4B). The A1493 base also rotates toward the helix axis, with the N1 shifted by 3.3 Å, and G1494 is disordered in this structure. The altered backbone conformation of A1492 positions both the ribose and the base such that the reorientation necessary for codon recognition is likely to be substantially compromised if not completely abolished. Similar effects were observed for both SmD mutants (Table 1).
FIGURE 3.

Distortion of the decoding site by SmD mutations in ribosomal protein S12, as observed in composite omit maps contoured at 1σ, of the wild-type (A,D,G), the P90L mutant (B,E,H), and the P90W mutant (C,F,I). (A–C) The decoding site viewed through ribosomal protein S12 (pink). The density for ribosomal protein S12 is excluded from this view for clarity. The position of A1492 is indicated. (D–F) View of the decoding site after an ∼100° rotation about the vertical axis of h44 of the view shown in panels A–C, and showing interaction between S12 and the decoding site. (G,H) Composite omit maps, contoured at 1σ, of ribosomal protein S12 residues 82–96. The densities for the proline and the leucine and tryptophan substitutions at position 90 are clearly visible.
FIGURE 4.
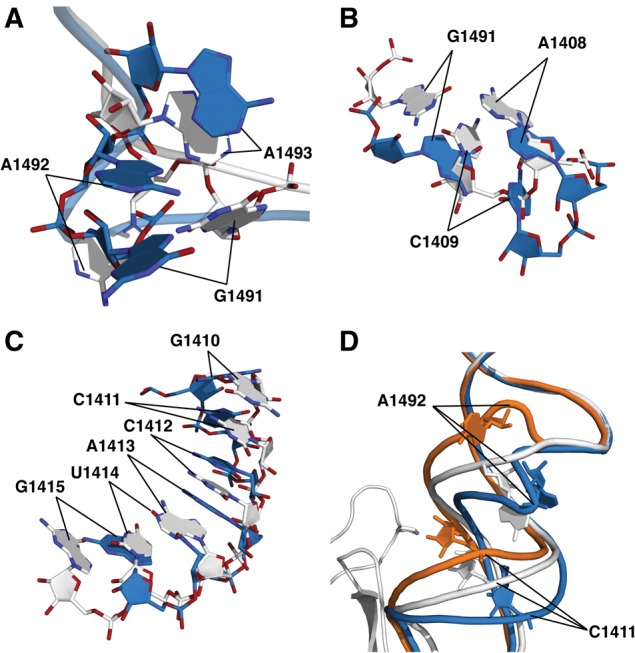
The conformational distortion caused by the SmD mutations. (A) Alignment of wild-type and the P90W mutant in crystal form 2, showing the shifts in position of A1491–A1493. (B) Alignment of wild-type and the P90W mutant in crystal form 2 indicating the shifts of A1408 and the C1409–G1491 base pair. (C) Alignment of wild-type and the P90W mutant in crystal form 2, showing a shift in helix register of bases in the vicinity of G1410–G1415. (D) Alignment of wild-type and P90W mutant in crystal form 2 with the published cocrystal structure of the 30S subunit bound to initiation factor IF1 (PDB entry 1HR0; orange) (Carter et al. 2001). The P90W mutation and IF1 induce shifts centered around A1492 and C1411 that occur in opposite directions. (Gray) The wild-type 30S; (blue) the P90W mutant; (orange) 30S–IF1 complex.
TABLE 1.
Displacement of 16S rRNA h44 backbone phosphate atoms in wild-type 30S and in the P90L and P90W mutants, all in crystal form 2, and comparison with displacements caused by IF1
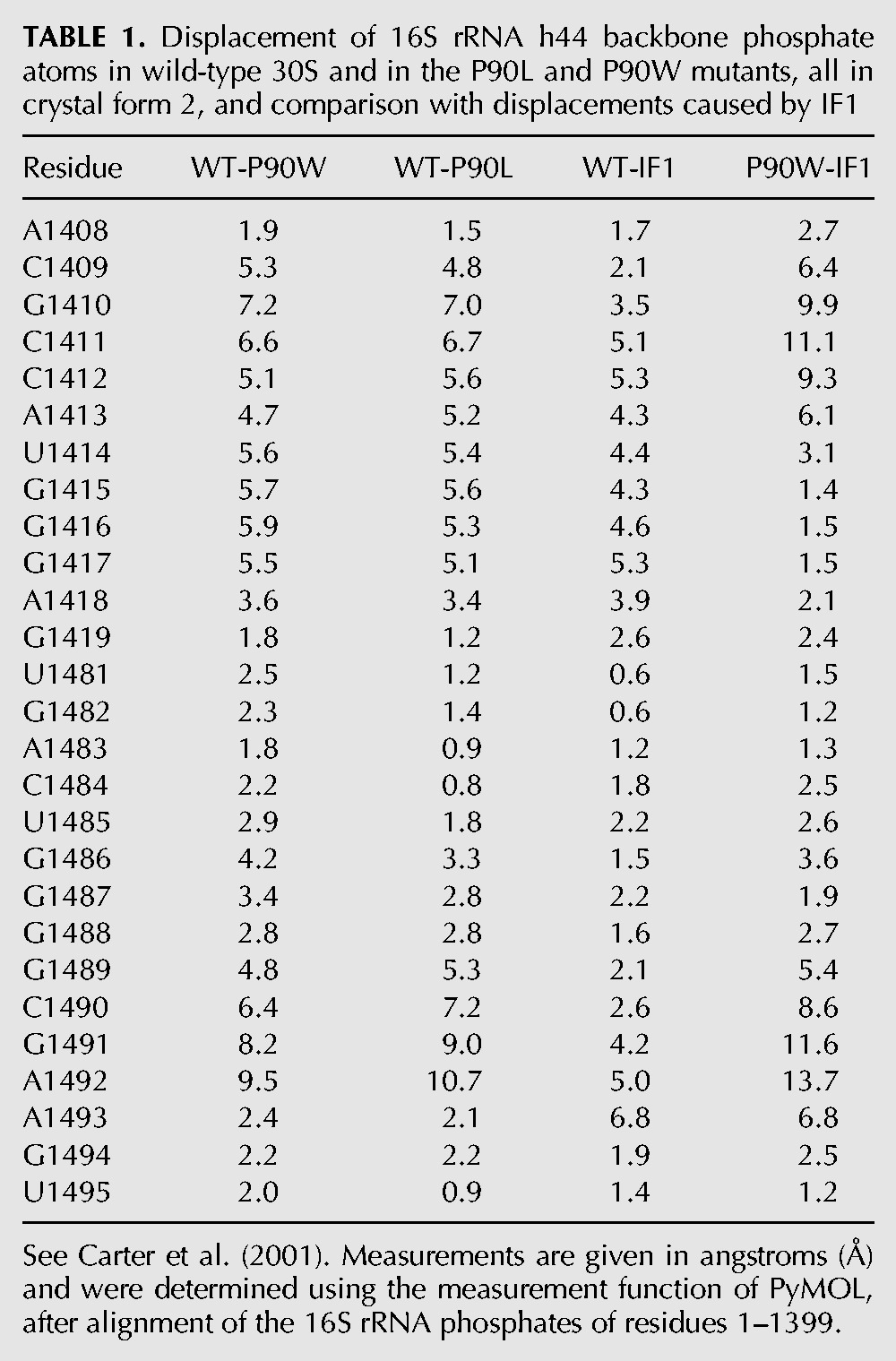
The conformational effects of the SmD mutations on h44 were also seen to extend beyond the decoding site. The lateral repositioning of the upper part of h44 causes the bases from G1410 to G1415 to shift in register (Fig. 4C). Distortion of the decoding site and the adjacent G1410 region of h44 was previously shown to be induced by translation initiation factor IF1 (Carter et al. 2001), which prevents binding of aa-tRNA to the A-site of the 30S initiation complex (Fig. 4D). However, SmD mutations and IF1 cause opposing shifts in h44 near its interface with protein S12. Specifically, G1410 is shifted 3.5 Å upward by IF1, while it is shifted 6.6 Å in the opposite direction by the P90W substitution (Table 1). The positions of G1491 and A1492 differ the most (11.6 Å and 13.7 Å, respectively) between the IF1 and P90W mutant structures, indicating the potential range of flexibility of the decoding site.
Wild-type ribosomes found in crystal form 2 did not show this same distortion (Fig. 5A,B; Supplemental Fig. S2), indicating that the conformation of h44 is independent of crystal packing forces. While only a small backbone shift near A1492 was visible in the subpopulation of P90W SmD ribosomes adopting crystal form 1 (Fig. 5C), a more pronounced displacement was clearly evident in the P90L mutant in this crystal form (Fig. 5D). These observations suggest that the native and distorted conformations represent two ground-state configurations of the 30S subunit existing in an equilibrium that is susceptible to structural perturbations of the decoding site.
FIGURE 5.
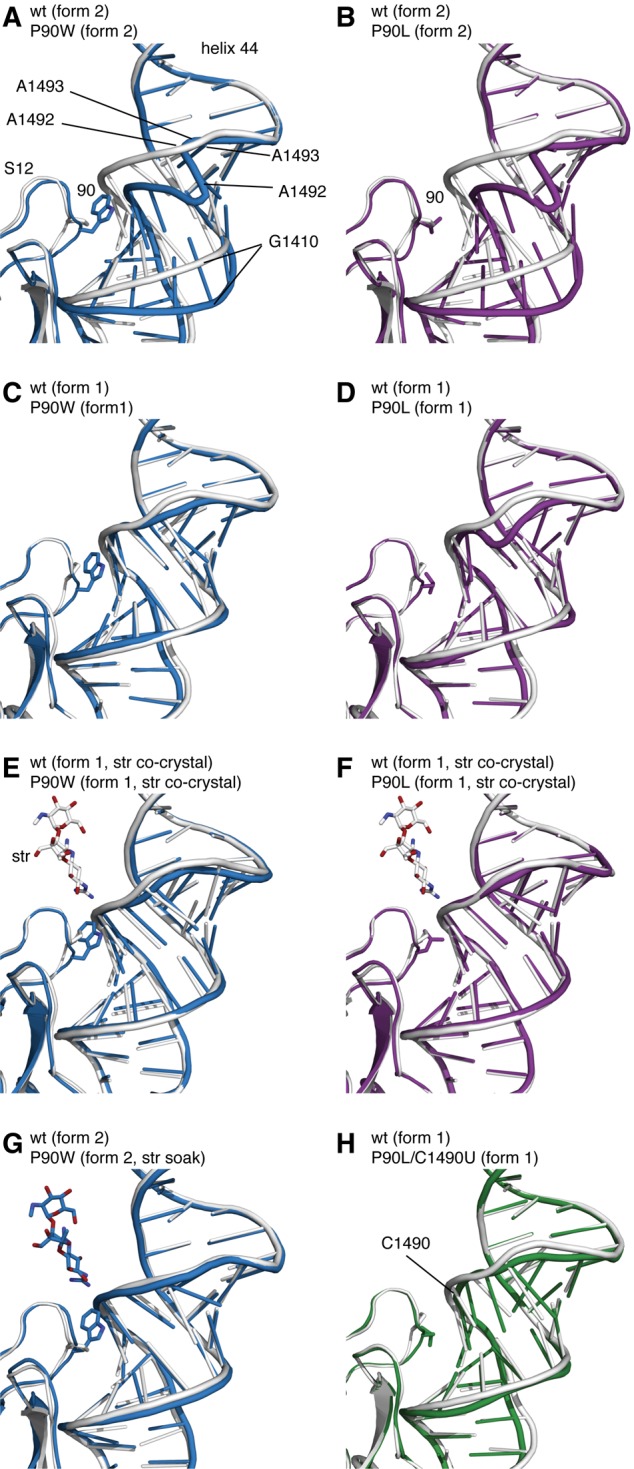
Structural distortion of the decoding site by the P90W SmD substitution and the effect of streptomycin binding. (Gray) The wild-type 30S subunit (PDB accession code 4DR1) (Demirci et al. 2013); (blue) the P90W mutant; (purple) the P90L mutant; (green) the P90L/C1490U mutant. Wild type aligned with the P90W mutant (A) or P90L mutant (B) in crystal form 2. Wild type aligned with the P90W mutant (C) or the P90L mutant in (D) in crystal form 1. Alignment of wild type with the P90W mutant (E) or the P90L mutant (F), each cocrystallized with streptomycin. (G) Alignment of wild type and the P90W mutant after addition of streptomycin in preformed crystals in form 2. (H) Alignment of wild type with the P90L/C1490U SmI mutant in crystal form 1.
Consistent with this model and the ability of streptomycin to suppress the tRNA selection defects of SmD mutants, cocrystallization of SmD ribosomes with streptomycin favored the formation of crystal form 1, in which the distortions in the decoding site were reversed (Fig. 5E,F). Importantly, preformed P90W crystals in form 2, when soaked with streptomycin, also showed a reversal of decoding-site distortions and a near-native conformation (Fig. 5G). This finding indicates that the structural changes resulting from SmD mutations are readily reversible. The ability of streptomycin to favor a near-native conformation of the decoding site of the SmD ribosome is in line with our own recent crystallographic data showing the drug to stabilize binding of near-cognate anticodon stem–loop analogs (ASL) by favoring a near-native conformation of the decoding site of wild-type ribosomes (Demirci et al. 2010). The binding energy of streptomycin, which interacts with several structural elements of the 30S subunit, is apparently sufficient to overcome the less favorable P90 side-chain conformation resulting from its steric clash with h44. These data also suggest that streptomycin lowers the activation energy for the transition between the distorted and native conformations.
The decoding site of the P90L/C1490U SmI double mutant did not exhibit the distorted conformation seen in the SmD mutants but instead resembled that of the wild-type ribosome (Fig. 5H). C1490 is located close to P90 such that substitution of one residue should influence the other. C1490 pairs with G1410 in the wild-type 30S structure, and the geometry of this base pair is affected by the C1490U substitution. This substitution could act either by compensating for the steric effect of the P90L substitution or by destabilizing the distorted conformation of h44 and freeing it to adopt the more favorable native conformation. Although the resolution of the SmI mutant structure is too low (3.7 Å) to make a definitive statement about the structural basis for suppression of the SmD phenotype, alignment of the wild-type and mutant structures shows that the 2′-OH at position 1490 appears to be shifted 2.7 Å away from the leucine at position 90. This is consistent with the change from a C-G Watson-Crick pair to a U-G wobble pair. While the backbone between positions 1490 and 1493 is still displaced due to the P90L substitution (2.7 Å between the G1491 phosphate atoms), the direction of this displacement differs from that seen in the P90L single mutant. Thus, while the SmI mutant is able to decode in the absence of streptomycin, the native conformational equilibrium has not been fully reestablished. These structural data mirror the smFRET findings, suggesting a structural basis for the observed defects in decoding and its rescue by either streptomycin or an SmI mutation.
DISCUSSION
When bound simultaneously to the ribosome and EF-Tu, aa-tRNA assumes a bent configuration of the anticodon helix in order to execute codon–anticodon pairing (Valle et al. 2003; Schmeing et al. 2009). The energetic penalty associated with tRNA bending is derived, in part, from the stabilizing contacts between the codon–anticodon complex and the decoding site, suggesting that both contribute to the mechanism of GTPase activation. Consistent with this notion, tRNA mutations that facilitate tRNA bending accelerate the rate constant for GTPase activation (Cochella and Green 2005; Schmeing et al. 2011). SmD mutations that distort the decoding-site conformation could therefore block GTPase activation by impairing the ribosome's capacity to achieve the native codon–anticodon interactions and tRNA bending that are prerequisites for productive early selection culminating in GTP hydrolysis. As both the efficiency and fidelity of GTPase activation during tRNA selection likely require a properly organized decoding site, we posit that the functional defects associated with SmD mutations are a direct consequence of the deformation of the decoding site. In this view, while aa-tRNA is able to interact with the mRNA codon in the A-site, structural transitions in the ribosome and tRNA during initial selection, which follow codon recognition, are either inhibited or altered in a manner that effectively prevents GTPase activation. Given the apparent stability of ternary complexes on the SmD ribosomes, we suggest that the step affected likely relates to domain closure events.
X-ray crystallographic studies of 30S subunits with ASL and mRNA bound to the A-site have been instrumental in elucidating the mechanism of codon recognition (Ogle et al. 2001; Murphy and Ramakrishnan 2004; Murphy et al. 2004; Dunham et al. 2007; Kurata et al. 2008). In the present study, binding of cognate ASL and mRNA to SmD mutant 30S crystals failed to produce the corresponding electron density, either in the absence or presence of streptomycin. We interpret this observation to indicate that codon–anticodon interaction alone is insufficient to counteract the distortion of the decoding site caused by the SmD mutations. Earlier studies showed that binding of near-cognate ASL and mRNA to 30S subunits produces only weak electron density, unless binding is stabilized by the error-inducing aminoglycoside antibiotic paromomycin (Ogle et al. 2002). We did not attempt to stabilize binding of ASL and mRNA with paromomycin or other aminoglycosides, as these drugs relieve the SmD phenotype (Sokolski et al. 1962; Gorini et al. 1967; Quesnel et al. 1971); indeed, paromomycin-dependence mutations and streptomycin-dependence mutations are one and the same (Quesnel et al. 1971; Carr et al. 2005). Presumably paromomycin and other aminoglycosides satisfy drug dependence by at least partly restoring a native decoding-site conformation and/or by stabilizing codon–anticodon interaction.
The decreased binding affinity of cognate ASL and mRNA for SmD ribosomes could be exaggerated by the inherently lower binding stability of ASLs compared with the ternary complex. This resembles observations with near-cognate codon–anticodon interaction; while near-cognate ASLs do not produce electron density in 30S subunit crystals in the absence of paromomycin (Ogle et al. 2002), near-cognate aa-tRNA in the fully accommodated (A/A) state can be observed bound to 70S ribosomes (Demeshkina et al. 2012). While we did not observe binding of ASL and mRNA to SmD ribosomes, we were able to observe binding of the ternary complex to SmD 70S ribosomes in single-molecule FRET experiments. Earlier biochemical studies showed that binding of the cognate ternary complex to 70S ribosomes is decreased 2.3-fold by an SmD mutation (Ruusala et al. 1984). The distortions of the decoding site we describe here were not detected in the earlier crystal structure of the 70S ribosome from an E. coli SmD mutant (Vila-Sanjurjo et al. 2003). This could be accounted for by stabilization of the native decoding-site conformation by the presence of aa-tRNA once it has achieved stable binding in the accommodated state. Future studies of 70S SmD ribosomes are a next logical step to address these issues, including 70S ribosomes with fully accommodated aa-tRNA bound to the A-site (Voorhees et al. 2009) and 70S ribosomes with the ternary complex in the A/T state.
Recently, the crystal structures of 70S ribosomes from two ram mutants having single-base substitutions in helices 12 and 14 of 16S rRNA were determined and shown to resemble the GTPase-activated state (Fagan et al. 2013). The large distance of the ram mutations from the decoding site suggests that they act via long-range signaling to the decoding site or by affecting conformational signals involved in GTPase activation. We did not observe any effect of the rpsL-P90L mutation on the conformation of 16S rRNA in the vicinity of the ram mutations; the presence of paromomycin in the decoding site of the former structures precludes any direct comparison of the decoding sites of the ram and SmD ribosomes.
While our data do not directly address how SmD mutations impact the domain closure process, impairment of this transition cannot be the sole basis for the SmD phenotype because it fails to explain the distorted conformation of the decoding site or how this distortion is relieved by streptomycin or second-site mutations. Furthermore, the observation that mutations at K42 of S12 can act as internal suppressors of SmD mutations at P90 (Björkman et al. 1999; Carr et al. 2005) and the existence of error-prone mutations in S12 (Agarwal et al. 2011) are also not explained by a simple steric model. Instead, our observations suggest that SmD mutations interfere with the ability of S12 to structure the decoding site. This notion is indeed consistent with the observation that SmR ribosomes stemming from S12 mutations exhibit defects in codon recognition (Zaher and Green 2010b). Thus, in addition to the current model in which the S12–h44 interactions serve only to stabilize the closed 30S subunit conformation, our findings show that ribosomal protein S12 also plays a direct role in preorganizing the decoding site in the open state so that it is properly poised for substrate binding and codon recognition.
A model in which S12 contributes to the structuring of the decoding site in preparation for aa-tRNA binding and to the subsequent domain closure process provides a new framework for explaining the phenotypes of the many mutations impacting decoding accuracy that have been identified in S12 or in the decoding site itself. Other amino acid substitutions in S12, such as those at the conserved R85, exhibit an intermediate, streptomycin pseudo-dependence (SmP) phenotype (Gregory et al. 2001), while those at R53 are extremely error-restrictive (Tubulekas et al. 1991). Base substitutions in h44 have been shown to act as SmI mutations (Allen and Noller 1991), nonsense suppressors or antisuppressors (Gregory and Dahlberg 1995; Murgola et al. 1995), or missense suppressors (McClory et al. 2010). It seems plausible that restrictive mutations could impact decoding by favoring various non-native decoding-site conformations, while nonsense or missense suppressor mutations could act by favoring a conformation normally achieved only upon cognate codon recognition. The inherent flexibility of the decoding site is essential for the induced-fit mechanism by playing a direct role in distinguishing between structurally similar cognate and near-cognate substrates (Ogle et al. 2002). This same fine balance among decoding-site conformations also makes the ribosome highly susceptible to perturbations by small molecules and mutations.
MATERIALS AND METHODS
Bacterial strains and culturing
All strains were derived from T. thermophilus HB8 (ATCC27634) (Oshima and Imahori 1974) and cultivated in TEM medium (ATCC medium 1598). Streptomycin sulfate was obtained from Sigma-Aldrich. Transformation with genomic or plasmid DNA was performed as previously described (Koyama et al. 1986). Transformants were purified by twice serially streaking for single colonies. All mutant alleles were confirmed by DNA sequencing.
Purification of T. thermophilus initiation factors (IF1, IF2, and IF3) and EF-Tu
Genomic DNA was extracted from T. thermophilus HB8 using the PureLink Genomic DNA Mini kit (Invitrogen). Genes encoding IF1, IF2, IF3, and EF-Tu were PCR-amplified from genomic DNA and cloned into the Champion pET SUMO Expression System vector (Invitrogen). E. coli strain BL21(DE3) carrying the expression plasmids was cultured at 37°C to an OD600 of 0.5 and induced with 0.5 mM IPTG. Cells were harvested after 4 h of expression and lysed in 50 mM potassium phosphate buffer (pH 7.5), 50 mM potassium chloride, and 5 mM β-mercaptoethanol. Proteins were purified using Ni-NTA resin (QIAGEN) following the manufacturer's instructions.
Formation of ribosomal complexes for single-molecule FRET experiments
Wild-type and mutant T. thermophilus ribosome complexes were assembled with biotinylated mRNA and fMet-tRNAfMet (labeled with Cy3 at s4U8) in the presence of IF1, IF2, and IF3 as described (Blanchard et al. 2004). The ternary complex formed from Phe-tRNAPhe (labeled with Cy5 at acp3U47) and EF-Tu in the presence of GTP was delivered by stopped-flow to surface-immobilized 70S ribosomes (Blanchard et al. 2004). Fluorescence signals were recorded at 15-msec time resolution under 200 mW, 532 nM laser illumination. FRET trajectories were determined according to the equation FRET = ICy5/(ICy3 + ICy5). Single-molecule FRET trajectories were post-synchronized to the first observation of FRET above the 0.13 FRET threshold and combined into a two-dimensional contour plot. Complexes contained either near-cognate mRNA (codon UCU) or cognate mRNA (codon UUC) in the A-site. Where indicated, complexes contained 40 μM streptomycin. The same ribosome preparations were used for both FRET experiments and crystallization (see below).
Preparation and crystallization of 30S ribosomal subunits
30S ribosomal subunits were purified and crystallized essentially as described (Clemons et al. 1999). Ribosome–streptomycin complexes were cocrystallized by mixing 30S subunits with 400 μg/mL streptomycin, filtering with a 0.22-μm filter, and incubating for 10 min at 55°C before cooling to 4°C. All crystals were sequentially transferred to a final buffer with 26% (v/v) 2-methyl-2,4-pentanediol (including streptomycin for cocrystals) for cryoprotection.
Diffraction data from apo and streptomycin-soaked P90W mutant crystals in form 2 were each collected from single crystals with a Pilatus 6M detector at beamline X06SA at the Swiss Light Source in Villigen. Diffraction data for wild-type subunits in crystal form 2 were collected from a single crystal with a Pilatus 6M detector at beamline X25 at the National Synchrotron Light Source at Brookhaven National Laboratory. All other data sets were collected from single crystals with an ADSC 315 detector at beamline ID-24-C at the Advanced Photon Source in Argonne.
P90L and P90W streptomycin cocrystal data sets were processed with HKL2000 (Otwinowski and Minor 1997), and all other data sets were processed with XDS (Kabsch 2010). Wild-type, P90W, and P90L apo structures in crystal form 2 were solved by molecular replacement, using wild-type 30S structure in crystal form 1 (PDB entry 4DR1) (Demirci et al. 2013) as a search model with Phaser (McCoy et al. 2007). The best solution was refined against wild-type, P90W, and P90L mutant data sets with the PHENIX software package (Adams et al. 2010). During initial refinement of P90W and P90L structures, the entire 16S rRNA h44 region was omitted, and the new h44 model was built into unbiased difference density using RCrane (Keating and Pyle 2012). For all other data sets, either the wild-type apo (PDB entry 4DR1) or wild-type streptomycin-bound 30S structure (PDB entry 4DR3) (Demirci et al. 2013) was used as the starting model for refinement with the PHENIX software package. After simulated-annealing refinement, individual coordinates, individual B factors, and TLS parameters were refined. Potential positions of magnesium or potassium ions were compared with those in a high-resolution (2.5 Å) 30S subunit structure (PDB entry 2VQE) (Kurata et al. 2008) in Coot (Emsley et al. 2010), and positions with strong difference density were retained. All magnesium atoms were replaced with magnesium hexahydrate, and water molecules located outside significant electron density were manually removed. Structure alignments were performed using the alignment algorithm of PyMOL (Schroedinger 2012) with the default 2σ rejection criterion and 50 iterative alignment cycles. Alignments used to detect repositioning of the 30S subunit head domain in crystal forms 1 and 2 were performed using 16S rRNA phosphate atoms from residues 1–926 (excluding the head domain and helices 44 and 45). Alignments used for analyzing structural changes in the decoding site included only 16S rRNA phosphate atoms from residues 1–1399 (excluding helices 44 and 45). All figures were produced with PyMOL (Schroedinger 2012).
DATA DEPOSITION
Coordinates of all structures have been deposited into the Protein Data Bank with the accession codes 4JI0 (P90L apo, crystal form 1), 4JI1 (P90L streptomycin cocrystal, form 1), 4JI2 (P90W apo, crystal form 1), 4JI3 (P90W streptomycin cocrystal form 1), 4JI4 (P90L/C1490U apo, crystal form 1), 4JI5 (wild-type apo, crystal form 2), 4JI6 (P90L apo, crystal form 2), 4JI7 (P90W apo, crystal form 2), and 4JI8 (P90W streptomycin soak, crystal form 2).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants GM019756 and GM019756-37S1 (to A.E.D.), GM094157 (to S.T.G. and G.J.), and GM079238 (to S.C.B.) from the US National Institutes of Health. Part of this work was conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by grants from the National Center for Research Resources (5P41RR015301-10) and the National Institute of General Medical Sciences (8 P41 GM103403-10) from the US National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. We thank D.S. Terry for writing the software used in the analysis of FRET data and J.E. Dahlberg for critical reading of the manuscript.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal D, Gregory ST, O'Connor M. 2011. Error-prone and error-restrictive mutations affecting ribosomal protein S12. J Mol Biol 410: 1–9. [DOI] [PubMed] [Google Scholar]
- Allen PN, Noller HF. 1989. Mutations in ribosomal proteins S4 and S12 influence the higher order structure of 16S ribosomal RNA. J Mol Biol 208: 457–468. [DOI] [PubMed] [Google Scholar]
- Allen PN, Noller HF. 1991. A single base substitution in 16S ribosomal RNA suppresses streptomycin dependence and increases the frequency of translational errors. Cell 66: 141–148. [DOI] [PubMed] [Google Scholar]
- Bilgin N, Claesens F, Pahverk H, Ehrenberg M. 1992. Kinetic properties of Escherichia coli ribosomes with altered forms of S12. J Mol Biol 224: 1011–1027. [DOI] [PubMed] [Google Scholar]
- Björkman J, Samuelsson P, Andersson DI, Hughes D. 1999. Novel ribosomal mutations affecting translational accuracy, antibiotic resistance and virulence of Salmonella typhimurium. Mol Microbiol 31: 53–58. [DOI] [PubMed] [Google Scholar]
- Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. 2004. tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol 11: 1008–1014. [DOI] [PubMed] [Google Scholar]
- Borovinskaya MA, Shoji S, Fredrick K, Cate JHD. 2008. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA 14: 1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr JF, Gregory ST, Dahlberg AE. 2005. Severity of the streptomycin resistance and streptomycin dependence phenotypes of ribosomal protein S12 of Thermus thermophilus depends on the identity of highly conserved amino acid residues. J Bacteriol 187: 3548–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AP, Clemons WM Jr, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407: 340–348. [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Hartsch T, Wimberly BT, Ramakrishnan V. 2001. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science 291: 498–501. [DOI] [PubMed] [Google Scholar]
- Clemons WM Jr, May JL, Wimberly BT, McCutcheon JP, Capel MS, Ramakrishnan V. 1999. Structure of a bacterial 30S ribosomal subunit at 5.5 Å resolution. Nature 400: 833–840. [DOI] [PubMed] [Google Scholar]
- Cochella L, Green R. 2005. An active role for tRNA in decoding beyond codon:anticodon pairing. Science 308: 1178–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G. 2012. A new understanding of the decoding principle on the ribosome. Nature 484: 256–259. [DOI] [PubMed] [Google Scholar]
- Demirci H, Murphy FV, Belardinelli R, Kelley AC, Ramakrishnan V, Gregory ST, Dahlberg AE, Jogl G. 2010. Modification of 16S ribosomal RNA by the KsgA methyltransferase restructures the 30S subunit to optimize ribosome function. RNA 16: 2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci H, Murphy FV, Murphy EL, Gregory ST, Dahlberg AE, Jogl G. 2013. A structural basis for streptomycin-induced misreading of the genetic code. Nat Commun 4: 1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham CM, Selmer M, Phelps SS, Kelley AC, Suzuki T, Joseph S, Ramakrishnan V. 2007. Structures of tRNAs with an expanded anticodon loop in the decoding center of the 30S ribosomal subunit. RNA 13: 817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan CE, Dunkle JA, Maehigashi T, Dang MN, Devaraj A, Miles SJ, Qin D, Fredrick K, Dunham CM. 2013. Reorganization of an intersubunit bridge induced by disparate 16S ribosomal ambiguity mutations mimics an EF-Tu-bound state. Proc Natl Acad Sci 110: 9716–9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourmy D, Recht MI, Blanchard SC, Puglisi JD. 1996. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 274: 1367–1371. [DOI] [PubMed] [Google Scholar]
- Fourmy D, Yoshizawa S, Puglisi JD. 1998. Paromomycin binding induces a local conformational change in the A-site of 16 S rRNA. J Mol Biol 277: 333–345. [DOI] [PubMed] [Google Scholar]
- Gale EF, Cundliffe E, Reynolds PE, Richmond MH, Waring MJ. 1981. The molecular basis of antibiotic action. Wiley, London. [Google Scholar]
- Geggier P, Dave R, Feldman MB, Terry DS, Altman RB, Munro JB, Blanchard SC. 2010. Conformational sampling of aminoacyl-tRNA during selection on the bacterial ribosome. J Mol Biol 399: 576–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorini L, Rosset R, Zimmermann RA. 1967. Phenotype masking and streptomycin dependence. Science 157: 1314–1317. [DOI] [PubMed] [Google Scholar]
- Gregory ST, Dahlberg AE. 1995. Nonsense suppressor and antisuppressor mutations at the 1409–1491 base pair in the decoding region of Escherichia coli 16S rRNA. Nucleic Acids Res 23: 4234–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory ST, Dahlberg AE. 2009. Genetic and structural analysis of base substitutions in the central pseudoknot of Thermus thermophilus 16S ribosomal RNA. RNA 15: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory ST, Cate JHD, Dahlberg AE. 2001. Streptomycin-resistant and streptomycin-dependent mutants of the extreme thermophile Thermus thermophilus. J Mol Biol 309: 333–338. [DOI] [PubMed] [Google Scholar]
- Gregory ST, Carr JF, Dahlberg AE. 2009. A signal relay between ribosomal protein S12 and elongation factor EF-Tu during decoding of mRNA. RNA 15: 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Yano T, Kuramitsu S, Kagamiyama H. 2001. Disruption of Thermus thermophilus genes by homologous recombination using a thermostable kanamycin-resistant marker. FEBS Lett 506: 231–234. [DOI] [PubMed] [Google Scholar]
- Hermann T. 2006. A-site model RNAs. Biochimie 88: 1021–1026. [DOI] [PubMed] [Google Scholar]
- Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi R, Ehrenberg M. 1994. Dissociation rate of cognate peptidyl-tRNA from the A-site of hyper-accurate and error-prone ribosomes. Eur J Biochem 226: 355–360. [DOI] [PubMed] [Google Scholar]
- Keating KS, Pyle AM. 2012. RCrane: Semi-automated RNA model building. Acta Crystallogr D Biol Crystallogr 68: 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, Hoshino T, Tomizuka N, Furukawa K. 1986. Genetic transformation of the extreme thermophile Thermus thermophilus and of other Thermus spp. J Bacteriol 166: 338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristjansson JK, Hreggvidsson GO, Alfredsson GA. 1986. Isolation of halotolerant Thermus spp. from submarine hot springs in Iceland. Appl Environ Microbiol 52: 1313–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata S, Weixlbaumer A, Ohtsuki T, Shimazaki T, Wada T, Kirino Y, Takai K, Watanabe K, Ramakrishnan V, Suzuki T. 2008. Modified uridines with C5-methylene substituents at the first position of the tRNA anticodon stabilize U•G wobble pairing during decoding. J Biol Chem 283: 18801–18811. [DOI] [PubMed] [Google Scholar]
- Kurland CG, Hughes D, Ehrenberg M. 1996. Limitations in translational accuracy. In Escherichia coli and Salmonella: Cellular and molecular biology (ed. Neidhardt FC, et al. ), pp. 979–1004. American Society for Microbiology, Washington, DC. [Google Scholar]
- Lescoute A, Westhof E. 2006. The A-minor motifs in the decoding recognition process. Biochimie 88: 993–999. [DOI] [PubMed] [Google Scholar]
- McClory SP, Leisring JM, Qin D, Fredrick K. 2010. Missense suppressor mutations in 16S rRNA reveal the importance of helices h8 and h14 in aminoacyl-tRNA selection. RNA 16: 1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Cryst 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D, Robertson JM, Noller HF. 1988. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature 334: 36362–36364. [DOI] [PubMed] [Google Scholar]
- Murgola EJ, Pagel FT, Hijazi KA, Arkov AL, Xu W, Zhao SQ. 1995. Variety of nonsense suppressor phenotypes associated with mutational changes at conserved sites in Escherichia coli ribosomal RNA. Biochem Cell Biol 73: 925–931. [DOI] [PubMed] [Google Scholar]
- Murphy FV, Ramakrishnan V. 2004. Structure of a purine–purine wobble base pair in the decoding center of the ribosome. Nat Struct Mol Biol 11: 1251–1252. [DOI] [PubMed] [Google Scholar]
- Murphy FV, Ramakrishnan V, Malkiewicz A, Agris PF. 2004. The role of modifications in codon discrimination by tRNALysUUU. Nat Struct Mol Biol 11: 1186–1191. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Brodersen DE, Clemons WM, Tarry MJ, Carter AP, Ramakrishnan V. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292: 897–902. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111: 721–732. [DOI] [PubMed] [Google Scholar]
- Oshima T, Imahori K. 1974. Description of Thermus thermophilus (Yoshida and Oshima) comb. nov., a nonsporulating thermophilic bacterium from a Japanese thermal spa. Int J Syst Bacteriol 24: 102–112. [Google Scholar]
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina M. 1999. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J 18: 3800–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina MV. 2000. Conformational switch in the decoding region of 16S rRNA during aminoacyl-tRNA selection on the ribosome. Nat Struct Biol 7: 104–107. [DOI] [PubMed] [Google Scholar]
- Quesnel LB, York P, Skinner VM. 1971. Drug dependence and phenotypic masking in E. coli. Nature 233: 121–122. [DOI] [PubMed] [Google Scholar]
- Ruusala T, Andersson D, Ehrenberg M, Kurland CG. 1984. Hyper-accurate ribosomes inhibit growth. EMBO J 3: 2575–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbonmatsu KY. 2006. Energy landscape of the ribosomal decoding center. Biochimie 88: 1053–1059. [DOI] [PubMed] [Google Scholar]
- Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. 2000. Structure of functionally activated small ribosomal subunit at 3.3 Å resolution. Cell 102: 615–623. [DOI] [PubMed] [Google Scholar]
- Schmeing TM, Voorhees RM, Kelley AC, Gao Y-G, Murphy FV, Weir JR, Ramakrishnan V. 2009. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science 326: 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeing TM, Voorhees RM, Kelley AC, Ramakrishnan V. 2011. How mutations in tRNA distant from the anticodon affect the fidelity of decoding. Nat Struct Mol Biol 18: 432–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroedinger L. 2012. The PyMOL Molecular Graphics System, Version 1.5. http://www.pymol.org/.
- Sharma D, Cukras AR, Rogers EJ, Southworth DR, Green R. 2007. Mutational analysis of S12 protein and implications for the accuracy of decoding by the ribosome. J Mol Biol 374: 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolski WT, Yeager RL, McCoy JK. 1962. Cross-resistance studies with neomycin antibiotics. Nature 195: 623–624. [DOI] [PubMed] [Google Scholar]
- Stanley RE, Blaha G, Grodzicki RL, Strickler MD, Steitz TA. 2010. The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat Struct Mol Biol 17: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms AR, Bridges BA. 1993. Double, independent mutational events in the rpsL gene of Escherichia coli: An example of hypermutability? Mol Microbiol 9: 335–342. [DOI] [PubMed] [Google Scholar]
- Tocilj A, Schlünzen F, Janell D, Glühmann M, Hansen HA, Harms J, Bashan A, Bartels H, Agmon I, Franceschi F, et al. 1999. The small ribosomal subunit from Thermus thermophilus at 4.5 Å resolution: Pattern fittings and the identification of a functional site. Proc Natl Acad Sci 96: 14252–14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubulekas I, Buckingham RH, Hughes D. 1991. Mutant ribosomes can generate dominant kirromycin resistance. J Bacteriol 173: 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J. 2003. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol 10: 899–906. [DOI] [PubMed] [Google Scholar]
- Vila-Sanjurjo A, Ridgeway WK, Seymaner V, Zhang W, Santoso S, Yu K, Cate JHD. 2003. X-ray crystal structures of the WT and a hyper-accurate ribosome from Escherichia coli. Proc Natl Acad Sci 100: 8682–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. 2009. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat Struct Mol Biol 16: 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. 2010. The mechanism for activation of GTP hydrolysis on the ribosome. Science 330: 835–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339. [DOI] [PubMed] [Google Scholar]
- Yoshizawa S, Fourmy D, Puglisi JD. 1999. Recognition of the codon–anticodon helix by ribosomal RNA. Science 285: 1722–1725. [DOI] [PubMed] [Google Scholar]
- Zaher HS, Green R. 2010a. Kinetic basis for global loss of fidelity arising from mismatches in the P-site codon:anticodon helix. RNA 16: 1980–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, Green R. 2010b. Hyperaccurate and error-prone ribosomes exploit distinct mechanisms during tRNA selection. Mol Cell 39: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.