

The debate continues concerning translation in the nucleus. In this work from the Brogna lab, the authors use fluorescent complementation between the large and small subunits of the ribosome to show that ribosome joining can occur in the nucleus, and accumulate in the nucleolus in particular, in fly tissues and cultured cells. Puromycin incorporation occurred at these sites indicating the possibility that the association is functional.
Keywords: nuclear ribosomal subunits, ribosomal subunits joining, nascent transcripts, translation
Abstract
In eukaryotes the 40S and 60S ribosomal subunits are assembled in the nucleolus, but there appear to be mechanisms preventing mRNA binding, 80S formation, and initiation of translation in the nucleus. To visualize association between ribosomal subunits, we tagged pairs of Drosophila ribosomal proteins (RPs) located in different subunits with mutually complementing halves of fluorescent proteins. Pairs of tagged RPs expected to interact, or be adjacent in the 80S structure, showed strong fluorescence, while pairs that were not in close proximity did not. Moreover, the complementation signal is found in ribosomal fractions and it was enhanced by translation elongation inhibitors and reduced by initiation inhibitors. Our technique achieved 80S visualization both in cultured cells and in fly tissues in vivo. Notably, while the main 80S signal was in the cytoplasm, clear signals were also seen in the nucleolus and at other nuclear sites. Furthermore, we detected rapid puromycin incorporation in the nucleolus and at transcription sites, providing an independent indication of functional 80S in the nucleolus and 80S association with nascent transcripts.
INTRODUCTION
Joining of the 40S and 60S ribosomal subunits to form the 80S ribosome is the hallmark of translation initiation. The general consensus is that 80S, which correspond to ribosomes at the initiation, elongation, termination, or post-termination stages of translation, are present only in the cytoplasm in eukaryotes (Jackson et al. 2010). The 40S and 60S ribosomal subunits are synthesized in the nucleolus by a complex mechanism, starting with cotranscriptional processing of the tricistronic precursor of the 18S, 5.8S, and 28S rRNAs (Venema and Tollervey 1999; Osheim et al. 2004; Kos and Tollervey 2010). The nucleus is therefore replete with 40S and 60S subunits at various stages of maturation. The current view is that intra-nuclear mechanisms prevent these subunits from associating with mRNA and so prevent 80S assembly before they are exported to the cytoplasm.
Many studies in Saccharomyces cerevisiae and to a lesser extent in other organisms indicate that nonribosomal assembly factors (AFs) bind to nuclear pre-40S and pre-60S subunits, keeping them inactive as well as preventing them from assembling into 80S (Panse and Johnson 2010; Strunk et al. 2011). Other proteins are required for translocation of the subunits through the nuclear pore complexes (NPCs) and may also contribute to keeping the nuclear subunits inactive (Tschochner and Hurt 2003; Zemp and Kutay 2007; Henras et al. 2008).
The rRNA of the pre-40S subunits is not fully processed in the nucleus and this may be another factor that prevents nuclear assembly of 80S (Venema and Tollervey 1999). In S. cerevisiae the exported pre-40S subunit has a 20S pre-rRNA that is trimmed to 18S in the cytoplasm (Udem and Warner 1973). In mammalian cells, 18S rRNA was long believed to be fully processed in the nucleus (Penman et al. 1966), but it now seems that their pre-40S subunits are also exported to the cytoplasm with an extended 18S pre-rRNA (Rouquette et al. 2005). It was initially concluded that extended 18S is excluded from polysome-associated 40S subunits in S. cerevisiae (Udem and Warner 1973) and in mammalian cells (Rouquette et al. 2005). However, a recent study reached the opposite conclusion: Immature 40S subunits containing 20S pre-rRNA can bind translation factors and engage in translation initiation in S. cerevisiae, although the resulting 80S is less efficient in translation and rapidly destroyed by a specialized mRNA decay mechanism (Soudet et al. 2010). In a similar vein, two very recent studies have concluded that processing of S. cerevisiae 20S pre-rRNA in fact requires the pre-40S particle to associate in the cytoplasm with both the translation initiation factor eIF5b and 60S subunit; it was suggested that this might represent the final proofreading step before 40S engages in translation (Lebaron et al. 2012; Strunk et al. 2012). The subunits appear to form an 80S-like structure that is similar in composition to genuine 80S, including translation elongation factors eEF1A and eEF1B. Since this structure lacks initiator tRNA, it was proposed that it is not engaged in translation and that its disassembly would be required before 40S mRNA binding, reassembly, and productive translation initiation (Strunk et al. 2012). Earlier studies in Dictyostelium discoideum had reached a similar conclusion but had more radically proposed that 18S maturation occurs as in Escherichia coli primarily after 80S formation, possibly at the first translation initiation stage (Mangiarotti et al. 1997; Shajani et al. 2011). Therefore, although for many years it has been assumed that incomplete 18S processing prevents nuclear subunits from associating, there is now abundant evidence that the 40S subunit containing 20S pre-rRNA can interact with 60S. The accepted view, however, is that this interaction occurs only in the cytoplasm.
Despite the consensus that ribosomal subunits are inactive in the nucleus, it has been previously reported that many ribosomal proteins (RPs), rRNA, and some translation factors (including eIF5b) seem to associate with nascent transcripts at polytene chromosomal transcription sites in Drosophila (Brogna et al. 2002; Coleno-Costes et al. 2012; Rugjee et al. 2013). These observations suggest the presence of ribosomal subunits at these sites. This may even be a general feature of eukaryotes since several RPs associate with nascent transcripts also in budding and fission yeast (Schroder and Moore 2005; De and Brogna 2010; De et al. 2011). The Drosophila study also reported rapid incorporation of radioactive amino acids at the chromosomes and nucleolus in polytenic nuclei (Brogna et al. 2002). However, the issue of whether, or to what extent, nuclear ribosomal subunits can join into functional 80S remains an important open question, alongside the related, but in part separate, issue of whether some proteins can be synthesized in the nucleus in different cell types and organisms (Dahlberg and Lund 2012; Reid and Nicchitta 2012).
RESULTS
Visualization of interaction between ribosomal subunits in Drosophila S2 cells
Joining of the 40S and 60S subunits into a functional 80S ribosome is the hallmark of translation initiation, thus we were interested in developing a method to visualize this interaction in cells. We identified pairs of RPs that form intersubunit protein–protein bridges in cryo-EM reconstructions of the yeast and mammalian 80S ribosome and also in the crystal structures of Thermus thermophilus 70S and yeast 80S (Spahn et al. 2001; Yusupov et al. 2001; Chandramouli et al. 2008). We then tagged these with complementary constructs that would signal their proximity in the assembled 80S. Two appropriate protein–protein contacts involve the 60S ribosomal protein L11 (RpL11 termed RpL5 in bacteria). RpL11 is located on the central protuberance (CP) of the 60S subunit and is adjacent to 40S head proteins RpS15 and RpS18 (in bacteria, RpS19 and RpS13, respectively) (Fig. 1A).
FIGURE 1.

BiFC visualization of interaction between RPs in Drosophila S2 cells. (A) Models of 40S (left) and 60S subunits, oriented as predicted by the EM structure of the yeast 80S (Spahn et al. 2001). The structures were generated with PyMol, by modifying a PyMol Session downloaded from http://www.mol.biol.ethz.ch/groups/ban_group/Ribosome, based on the PDB files 2XZM (40S) and 4A17, 4A19 (60S). S15, S18, and L11 are indicated with different colors. Structures of the complementary YN and YC halves of YFP are shown above in yellow. (B) Diagram of the BiFC constructs with sequences of the spacers (above). Diagrams show the general structure of the C-terminal fusion constructs; N-terminal versions (data not shown) carry the same linkers. Western blot of whole-cell extracts from cells transfected with the indicated constructs and detection with GFP polyclonal antibody (middle panel). The molecular weights of the fusion proteins are S18–YN, 35.8 kDa; RpL11–YC/YC–RpL11 31.5 kDa; YN–RpS15 35.0 kDa. (Bottom) Western blot with an antibody against endogenous S18 (17.6 kDa), which also detects the S18–GFP fusion (39.1 kDa). The ratio of S18–GFP to endogenous S18 is 0.8, after correcting for transfection efficiency (23%). Lane 4 shows separate Western blotting of an extract of cells transfected with S18–YN- and L11–YC-expressing plasmids. (C) YFP signal visualized in cells cotransfected with pUAST–RpS18–YN, pUAST–RpL11–YC, and p-Act–GAL4. The top row shows cells with typical cytoplasmic YFP pattern (Type 1), and the bottom row cells with nucleolar signal (Type 2; arrow indicates the nucleolus). YFP signals are shown on the left, DAPI staining in the middle, and the merged images on the right. All micrographs are confocal images taken with a 60× oil immersion objective. (D) Images of cells transfected with other BiFC pairs, tagged at the N- or C-terminal as indicated. (E) Indirect immunostaining of cells transfected with the constructs indicated using a polyclonal GFP antibody and counterstained with DAPI.
Initially we set out to test whether these two bridges can be detected in vivo in Drosophila cells. We tagged RpS15 (S15), RpS18 (S18), and RpL11 (L11) with the two halves of the yellow fluorescent protein (YFP): These only combine to generate functional YFP when they are brought into close proximity by interacting partners (Hu et al. 2002). This technique, termed bimolecular fluorescence complementation (BiFC), was developed to study protein–protein interactions in living cells in a manner more sensitive and straightforward than fluorescence energy transfer (FRET) methods (Hu et al. 2002; Kerppola 2008). To maximize the chance of detecting 40S/60S interactions, the BiFC fragments of YFP (YN, N-terminal-half tagged onto 40S RPs; and YC, C-terminal-half tagged onto 60S RPs) were fused to either end of the RPs. To increase the mobility of the YN and YC fragments, short peptide linkers were included (examples are in Fig. 1B). The resulting constructs, using RpL11 as an example, are abbreviated as L11–YC (tagged at the C terminus) and YC–L11 (N-terminally tagged). Initially, BiFC was assessed in Drosophila S2 cells transiently transfected with constructs driven by the UAS promoter and a Gal4-expressing plasmid (Materials and Methods). The constructs led to a good expression of the expected polypeptides (Fig. 1B, middle panel), but notably at levels that are below that of endogenous RPs. S18–GFP, for example, which could be detected with both anti-GFP and anti-S18 antibodies, is expressed at ∼80% of the endogenous protein level after normalization by transfection efficiency (Fig. 1B, bottom panel). The level of S18–YN is also lower than endogenous S18 (Fig. 1B, bottom panel, lane 4).
BiFC fluorescence was readily detected in transfected cells (Fig. 1C). The S18–YN/L11–YC pair produced the strongest YFP fluorescence, apparent in both fixed (Fig. 1C) and live cells (shown below). The BiFC signal was much weaker than that observed when similarly expressing standard GFP fusions of the same RPs (Supplemental Fig. S1A; Rugjee et al. 2013), but it was clearly visible by epifluorescence microscopy (data not shown) and was unambiguous by confocal imaging (Fig. 1C). In most cells, the signal was predominantly cytoplasmic (these were termed Type 1 cells), but some fluorescence was detectable in the nucleus and, in a minority of cells, was particularly apparent in the nucleolus (Type 2 cells) (Fig. 1C; Movie S1). Frequency quantification of this observation is given further down. The signal in the nucleolus was often more intense at its periphery. In some experiments, especially when using more efficient transfection reagents or stronger promoters (Materials and Methods), there were a few cells in which the signal was mainly nuclear: Typically these had shrunken nuclei, suggesting they were damaged or dying.
Of the other BiFC pairs tested, S18–YN + YC–L11 and YN–S15 + L11–YC produced slightly weaker signals than S18–YN/L11–YC, but with a similar distribution (Fig. 1D). The other YFP-tagged RP combinations yielded weaker fluorescence even though the proteins were expressed at similar levels (Fig. 1B).
To drive BiFC, the RP-linked YFP segments must be brought together when 80S assembles
While we estimated that the tagged proteins are expressed well below that of endogenous RPs, they are still expected to be at relatively high concentrations. A concern when using the BiFC technique is that high concentrations of the BiFC fragments alone might functionally interact even when they are not tethered to interacting proteins (Cabantous et al. 2005; Kerppola 2008). The pattern of signals depicted in Figure 1 argues against this type of fluorescence being an artifact. Like the corresponding GFP-tagged versions we have tested, all of the BiFC–RPs are at higher concentrations in the nucleus than in the cytoplasm (Fig. 1E), yet the YFP fluorescence signal arising from their interactions is most intense in the cytoplasm. Thus BiFC–RP concentrations cannot explain the generation of functional YFP. To investigate more directly the degree to which the BiFC signal depended on proximity of the partner proteins, we assayed additional RP pairs using information from the most recent eukaryotic 80S structures (Klinge et al. 2012). These included pairs that are apart on the 80S, and thus would not be expected to generate a signal (Fig. 2A): L5 is next to L11 and therefore close to S15 and S18, S6 is at the opposite side of the ribosome at the “feet” of the 40S and is in close proximity to L24 and, to a lesser extent, L22, but the other pairs (S11/L32, S13/L11, S13/L5, and S9/L11) are widely separated. When BiFC-tagged versions of these RPs were expressed in S2 cells the expected polypeptides were produced (Fig. 2B): They accumulated throughout the cell, with highest concentrations in the nucleus (Fig. 2D). The YN–S15/L5–YC, S6–YN/L22–YC, and S6–YN/L24–YC pair produced strong BiFC fluorescence, similar to the earlier RP pairs illustrated in Figure 1C,D, whereas the widely separated RP pairs (S11–YC/L32–YN, S13–YN/L11–YC, and S13–YN/ L5–YC) generated no signal or very dim fluorescence (Fig. 2C).
FIGURE 2.

Only adjacent RPs drive BiFC. (A) Models of 40S and 60S with tagged RPs highlighted in different colors. (B) Western blot of whole-cell extracts of cells transfected with the constructs indicated. Bottom panel shows blot with YN and YC fragments not fused to other peptides. The fusion protein molecular weights are YN–S15, 35 kDa; L5–YC, 52.2 kDa; S13–YN, 35.2 kDa; L11–YC and YC–L11, 31.5 kDa; S9–YN, 40.8 kDa; L32–YN, 34.2 kDa; S11–YC, 28.5 kDa; S6–YN, 39 kDa; L22–YC and S6–YN, both 39 kDa; L24–YC, 24 kDa; YN, 18.2 kDa; YC, 10.6 kDa. (C) Confocal images showing YFP signal in cells transfected with the constructs indicated. Arrows show nucleoli. (D) Indirect immunostaining with polyclonal GFP antibody with DAPI staining.
The positive pairs produced an apparent signal in the nucleolus in some cells like the S18/L11 pair (cells with clear nucleolar signal are shown in Supplemental Fig. S2). Importantly, the distant RP pairs also did not produce any signal in the DAPI-stained region of the nucleus or, except for one pair, in the nucleolus, even though the proteins are abundant throughout the nucleus (Fig. 2C). The exception was S9–YN/L11–YC, which produced only a very faint cytoplasmic signal although a clear signal was visible in the nucleolus (Fig. 2C).
We also expressed the YN and YC fragments alone (Fig. 2B). These produced much weaker fluorescence, with a pattern very different from that displayed by adjacent RP pairs. Most of their fluorescence was in the DAPI-stained region of the nucleus (Fig. 2C), which based on the immunostaining is where the BiFC-tagged peptides are more concentrated (Figs. 1E, 2D).
These results suggested that our BiFC assay might be an effective technique to investigate the association between 40S and 60S subunits to form 80S ribosomes. They established that strong BiFC fluorescence was only generated when the pairs of RPs involved lie close to one another at the intersubunit boundary in assembled 80S. The concentration of the expressed RP constructs is highest in the nucleus, where ribosomal subunits are made, but the BiFC signal was mainly in the cytoplasm, although also apparent in and around the nucleolus in some cells. These results are consistent with the BiFC being the result of ribosomal subunit joining and suggest that this interaction may also occur to some degree in the nucleus, in the nucleolus in particular.
The BiFC interaction between RpS18 and RpL11 occurs in the 80S and is prevented by inhibitors of translation
We used S18–YN/L11–YC, the RP pair that produced the strongest BiFC signal, to investigate whether the BiFC signal is actually produced by functionally joined ribosomal subunits. First, we assessed whether the tagged proteins are assembled into ribosomal subunits and into 80S. We analyzed polysomal fractions prepared from cytosolic extracts of cells transfected with either S18–YN or L11–YC individually or with both constructs together. When expressed individually, S18–YN and L11–YC are predominately found in polysomal fractions (Fig. 3A,B). When expressed together, a significant amount of the polypeptides is still found in polysomal fractions, yet both proteins seem to be more abundant in lighter fractions corresponding to monosomes or individual subunits (Fig. 3C). It is possible that, when both subunits are tagged, 80S are less efficient in translation elongation than when carrying just one tag, because the S18–L11 intersubunit linkage produced by BiFC probably hinders 80S translocation (see below). It is thus conceivable that most of the BiFC polypeptides found in the polysomal fractions derive from 80S with only one of the subunits tagged. EDTA treatment of the cell extract caused a visible shift of the BiFC polypeptides toward lighter fractions (Supplemental Fig. S3), further suggesting that the tagged proteins are incorporated into polysomes. The effect is most apparent with S18–YN and L11–YC expressed individually (Supplemental Fig. S3A,B) but is also apparent in extracts of cells expressing the two proteins together (Supplemental Fig. S3C). Pretreatment of the cells with puromycin, followed by further incubation of the extract with the drug in high-salt conditions, also resulted in a visible shift of the BiFC polypeptides toward lighter fractions (Supplemental Fig. S3D). However, peaks of S18–YN and L11–YC persist in fractions corresponding to 80S (Supplemental Fig. S2D, fractions 5 and 6); this observation further suggests that the proteins in these fractions are in 80S, which cannot be easily dissociated, possibly because they are bridged by the BiFC interaction. In summary, these observations suggest that the tagged RPs can be incorporated into ribosomal subunits that can join into 80S and, at least when only one of the subunits is tagged, can join actively translating polysomes. That these tagged proteins are individually functional was later confirmed by genetic tests in flies (described below).
FIGURE 3.

The S18 and L11 proteins interact only on the 80S. (A–C) Sucrose gradient fractionation profiles of extracts from cell expressing S18–YN (A), L11–YC (B), or both (C). Western blotting of the different fractions with a polyclonal GFP antibody recognizing both YN and YC (see Materials and Methods) are shown under each profile. Positions of polysomes, monosomes (80S), and 60S and 40S subunits are indicated. (D) Polysome profile of the extract used for the fluorimetry shown below, from cells expressing both S18–YN and L11–YC. Vertical arrows indicate fractions containing 80S. (E) YFP emission values, 500–550 nm, of the fractions collected from the gradient shown in D; fractions, F3 (heaviest) to F13 (lightest), are in different colors. Emission values were normalized by subtraction of the background reading of a similarly prepared control extract from untransfected cells. YFP emission (525-nm peak) is apparent in fractions F8 and F9.
To assess more directly whether the interaction between S18–YN and L11–YC happens on the 80S, cell extracts were fractionated as above (Fig. 3D) and emission spectra were recorded (Fig. 3E). Characteristic YFP fluorescence (excitation 488 nm, emission peak 525 nm) was detected in the two fractions corresponding to the 80S peak (Fig. 3E), but there was no obvious signal above background in lighter fractions corresponding to free 60S or 40S subunits. BiFC seems therefore to be primarily a result of association between ribosomal subunits to form 80S, feasibly as a consequence of translation initiation.
The observation that the BiFC signal is most apparent in 80S fractions indicates that the assay is correctly reporting the joining of the subunits that is established at translation initiation. To assess more directly the dependence of the BiFC signal on translation, we first compared the effects of emetine and puromycin. These translation inhibitor drugs have different mechanisms of action (Tscherne and Pestka 1975): Emetine blocks ribosomal polypeptide elongation, freezes 80S, and stabilizes polysomes (Grollman 1968), whereas puromycin is an aminoacyl–tRNA-like molecule that causes premature translation termination by serving as acceptor for the peptidyl–tRNA and leads to rapid breakdown of polysomes (Nathans and Lipmann 1961). As exemplified in Supplemental Figure S3D, puromycin causes dissociation of the two subunits at least in vitro (Blobel and Sabatini 1971).
Cells transfected with S18–YN and L11–YC were treated with emetine or puromycin before and after cell lysis, and fluorescence of cell extracts was quantified. There was a clear increase in the BiFC signal in emetine-treated cells and, to a lesser extent, a decrease with puromycin (Fig. 4A). Puromycin possibly did not produce a stronger reduction of the signal because the BiFC linkage, as indicated by the in vitro data mentioned above, prevents complete 80S disassociation. The enhancing effect of emetine was also very apparent by microscopical inspection of the cells: A clear increase in fluorescence was visually apparent in all experiments in which the cells were pre-incubated with emetine (Fig. 4B shows a distribution of the mean intensity of the signal in 100 cells from the same transfection, with or without drug treatment). Notably, enhancement of the signal was visible after only 5-min emetine treatment (Supplemental Fig. S5). Additionally, a more readily quantifiable effect of the emetine treatment was a threefold increase in the proportion of Type 2 cells—those with obvious BiFC fluorescence from in and around the nucleolus (Fig. 4C,D).
FIGURE 4.

Translation affects S18–L11 interaction. (A) Fluorescence emission spectra, 500–550 nm, in extracts of transfected cells pre-incubated prior to lysis with or without the indicated drug. The three experiments were performed with different batches of transfected cells, which were split into aliquots. Incubation with emetine (5 µg/mL or 20 µg/mL) was for 30 min or 2 h with puromycin (100 µg/mL) for 30 min, or 1 or 2 h; and with 2 µM pactamycin or 1 µM harringtonine for 30 min. The fluorescence of each sample was normalized as in Figure 3B. (B) Boxplots showing whole-cell mean fluorescence intensities of 100 transfected cells after 50 µg/mL emetine treatment, for 30 min and 1 h, and a control without the drug. Cells were manually defined as regions of interest (ROI) and mean fluorescence values were obtained using the Automated Measurement Results function of the Nikon NIS-BR Software. Values were corrected by subtracting the intensities in identical ROIs defined in adjacent regions without cells. (C) Confocal images of cells pretreated prior to fixation with 50 µg/mL emetine for the time indicated. Arrow indicates nucleolus. (D) Relative frequencies of Type 1 (blue) and Type 2 (red) cells with and without the indicated treatment with 50 µg/mL emetine. One hundred cells were counted from two separate transfections; standard deviations are indicated.
Pactamycin and harringtonine, the other drugs that were tested, are both protein synthesis inhibitors that lead to depletion of polysomes in eukaryotic cells (Kappen et al. 1973; Tscherne and Pestka 1975; Fresno et al. 1977). As with puromycin, we found that both reduced the BiFC signal in extracts (Fig. 4A, third panel). It has generally been assumed that this disappearance of polysomes is due to inhibition of translation initiation, but the mechanism is not yet clear. Recent structural and biochemical studies have indicated that in bacteria pactamycin may inhibit the first translocation step rather than initiation (Dinos et al. 2004). Our observations, however, are more consistent with both drugs reducing translation initiation in eukaryotes, as the earlier studies cited above had concluded.
In summary, the effects of these drugs are consistent with the view that the BiFC signal is a consequence of translation-dependent assembly of 80S, and they suggest that this might occur both in the cytoplasm and in the nucleus. In particular, the apparent enhancement of the signal upon emetine treatment indicates that the BiFC interaction occurs during translation elongation.
The RpS18–RpL11 BiFC interaction might not prevent ribosome translocation
Following the observation that no significant BiFC fluorescence could be detected in polysomal fractions (Fig. 3), we initially reasoned that this might be due the BiFC linkage restricting the rotational movement of the subunits, which occurs during translocation following peptide elongation (Dunkle and Cate 2010). However, an additional reason might simply be that the intersubunit rotation breaks the YFP BiFC interaction during elongation, drastically decreasing signal detection in polysomes. To investigate this possibility further, we tested a more sensitive BiFC reporter. We have fused S18 and L11 with BiFC-compatible fragments of Venus fluorescent protein, which were reported to yield a brighter and more specific BiFC interaction in Drosophila (Hudry et al. 2011). Noticeably, S18–VN and L11–VC produced a much brighter fluorescence signal in S2 cells; the subcellular pattern of the signal is similar to that of the previous YFP-based system but apparent nucleolar signal is visible in much larger fraction of cells (∼40%) (Supplemental Fig. S4A,B). Notably, with this new system ∼20% of the BiFC signal was detected in polysomal fractions (Fig. 5A; Supplemental Fig. S4C). This latter observation suggests that the BiFC linkage, which may be interfering with translocation, leaves the 80S at least in part functional. Unlike the YFP-based system, S18–VN/L11–VC produced signal also in lighter nonribosomal fractions; these perhaps correspond to more long-lived degradation intermediates of BiFC-joined 80S (see Discussion). Additionally, puromycin treatment caused a clear shift of the BiFC signal toward lighter fractions (Fig. 5B; Supplemental Fig. S4D), consistent with the signal reporting translating 80S.
FIGURE 5.

Tagging of S18 and L11 with Venus BiFC fragments allows 80S detection in polysomes. (A) Polysomes profile of the extract from transfected S2 cells expressing both S18–VN and L11–VC. Transfected cells were treated with 20 μg/mL emetine for 30 min prior to lysis. Bar chart shows normalized Venus emission (528 nm) values for the different fractions. Emission values calculated from the emission spectra shown in Supplemental Figure S4C which were normalized by subtracting the background reading in parallel fractions from a control lysate of untransfected S2 cells. Positions of polysomes, monosomes (80S), and 60S and 40S subunits are indicated. (B) Polysome profiling as above of cells pretreated with 100 μg/mL puromycin for 30 min. The lysate was treated with the same concentration of puromycin and 375 mM KCl for 30 min at room temperature prior to loading of the gradient (see Materials and Methods). Normalized emission values were calculated as above from Supplemental Figure S4D.
Development of transgenic Drosophila which allows visualization of interaction between RpS18 and RpL11 in flies
To apply the same BiFC technique in flies, we generated transgenic lines with the S18–YN and L11–YC pair that worked best in cell culture: These and most other RPs are encoded by essential single-copy genes in Drosophila (Marygold et al. 2007). To assess the functionality of these modified RPs, transgenic lines were crossed with strains carrying homozygous lethal mutations in the corresponding endogenous genes, RpS18c02853 and RpL11k16914 (Materials and Methods). In each case we could rescue homozygous mutant adult flies by complementation with the corresponding BiFC transgene and an Actin-Gal4 driver (5% for S18–YN and 6% for L11–YC, out of 14% expected). The rescued flies did not show any obvious external morphological phenotype, but we have not been able to breed them (perhaps due to sterility). This level of complementation reinforces the earlier conclusion that, at least individually, the tagged proteins must be functionally incorporated into ribosomes.
Having established their functional competence in vivo, we coexpressed the two transgenes in various tissues with appropriate Gal4 drivers (see Materials and Methods). The results of such experiments are illustrated here by reference to results obtained with salivary glands coexpressing fkh-Gal4; the level of S18–YN was ∼5% endogenous S18 (Supplemental Fig. S6A). S18–YN was less abundant despite both transgenes being regulated by the identical UAS promoter sequences (Supplemental Fig. S6B). Salivary glands are easy to culture and the large polytenic nuclei allow optimal imaging of the intra-nuclear BiFC signal. We found that the BiFC signal is most apparent in the cytoplasm in both fixed (Fig. 6A) and live salivary glands (Fig. 6B), and that there is no YFP signal in cells that express only one of the BiFC peptides (Supplemental Fig. S6C). As expected, the signal in the cytoplasm appears to be excluded from the tightly packed secretory vesicles (Fig. 6A,B); the number of vesicles varies with the developmental age of the larva (Fig. 6A, cf. top and bottom rows). Similarly expressed transgenic S18–GFP and L11–RFP are more abundant on the surface of the polytenic chromosomes than in the cytoplasm (Supplemental Fig. S1B), and the BiFC-tagged RPs should be similarly distributed, so as in S2 cells, there is no direct correlation between the concentrations of the BiFC peptides and the fluorescence signal of their complex.
FIGURE 6.

Visualization of 80S in salivary glands. (A) Main panel shows a confocal image of YFP fluorescence in a fixed 3rd instar larva salivary gland, expressing S18–YN and L11–YC (driven by fork-head Gal4). These gland cells are replete with secretory vesicles. Right inset shows a high-magnification cell (from a different gland) in which fibrillarin (red signal) was detected by indirect immunostaining with a Cy5-conjugated antibody. Bottom row shows a younger gland cell with only a few vesicles. DAPI is shown in blue. Arrow indicates the nucleolus. (B) Confocal YFP image of live salivary glands. Bottom left shows a close-up view of the cell in the square; and bottom right is a confocal reflection image of the same cells showing a pseudo brightfield view of the chromosomes (the line indicates a segment of a chromosome arm with visible banding) and nucleolus (arrow). (C) Confocal images of glands treated with a high concentration of actinomycin D (10 μg/mL) for 1 or 4 h, compared with an untreated control gland. (D) Confocal images of a gland treated with a low concentration of actinomycin D (0.2 μg/mL) for 4 h. Right insets show higher magnification images of the boxed cells from the left-hand images. Arrow indicates the surface of the nucleolus and the pin its dimmer core.
Within the nucleus a strong BiFC signal is most apparent in the nucleolus, as indicated by its colocation with the nucleolar protein fibrillarin (Fig. 6A) or by parallel brightfield imaging which clearly shows the nucleolus and chromosomes (Fig. 6B, bottom panels). Weak fluorescence is also detectable in the chromosomes region, particularly in live cells (Fig. 6B, bottom image). In human cells it has long been reported that there are connections between the nuclear envelope and the nucleolus (Bourgeois et al. 1979); here, however, we found no evidence of such contacts. This observation is more apparent in cells in which the lamina was marked by immunostaining and the nucleolus with S9–GFP: There is no evidence by confocal microscopy of either a contact with the nucleolus or nuclear envelope invaginations (see Supplemental Fig. S7).
The nucleolar 80S signal is transcription-dependent
The BiFC signal appears to be transcription-dependent. Following a 1-h treatment with a high concentration of actinomycin D, which blocks transcription by all three RNA polymerases (evidence that this is also the case in salivary glands is given in Supplemental Fig. S8), there was a small increase in the signal around the nuclear envelope and nucleolus (Fig. 6C, left panels). However, the nucleolar signal disappeared completely after 4 h with the same concentration of actinomycin D (Fig. 6C, right panels). With a lower concentration, expected to inhibit Pol I but not Pol II (Supplemental Fig. S8), fluorescence persisted except for the inner core of the nucleolus (Fig. 6D). These data suggest that maintenance of the BiFC signal requires Pol II transcription but that, perhaps except in the core of the nucleolus, it does not require ongoing rRNA transcription. The nucleolus signal was also sensitive to actinomycin D and DRB in S2 cells (Supplemental Fig. S4B).
Leptomycin B treatment increases the nucleolar 80S signal
The data presented above point to the existence of 80S ribosomes within the nucleus. The BiFC signal therein is most apparent in or around the nucleolus, but there appeared to be some occasional fluorescence on the chromosomes or in the interchromosomal space. We tested whether blocking the CRM1-dependent export of ribosomal subunits from the nucleus with leptomycin B (LMB) (for review, see Henras et al. 2008) might increase the nuclear signal. Brief LMB treatment brought about a clear increase in the nucleolar signal and a small increase around the nuclear periphery (Fig. 7A, middle panel): In some glands we also detected an increase in fluorescence around the chromosomal region (Fig. 7A, bottom panel; Fig. 7B). Longer LMB treatments of S2 cells also increased the proportion of Type 2 cells showing nucleolar or perinucleolar signal (Fig. 7C, two middle panels). In a small fraction of the treated cells fluorescence was apparent throughout the nucleus but weak in the cytoplasm (Fig. 7C, right panel), perhaps as a result of a toxic effect of LMB.
FIGURE 7.

Leptomycin B treatment increases nuclear 80S signal. (A) Large panels are confocal images showing YFP fluorescence in salivary glands incubated for 30 min either without (top panel) or with (bottom panels) 50 nM LMB. Magnified insets show single cells and DAPI signal (blue). (B) Images of salivary glands incubated for 5 h in LMB. (C) Confocal images of S2 cells transfected with the indicated constructs and incubated as indicated with LMB for either 4 h (second panel from left) or 5 h. Arrows indicate nucleoli. (D) Confocal imaging of glands expressing S9–GFP incubated in vitro for 30 min with or without LMB as indicated.
The most characteristic effect of a short LMB treatment of salivary glands or S2 cells was an increase in nucleolar BiFC fluorescence. Notably, similar treatment of salivary glands from a transgenic line expressing S9–GFP also increased nucleolar fluorescence (Fig. 7D). Overall, the results of the LMB treatment are consistent with the view that ribosomal subunits can interact in the nucleus, and that blocking their export makes this a more frequent event. Moreover, the experiments indicate that the clearest effect of LMB is to cause accumulation of ribosomal subunits in the nucleolus, rather than in the nucleoplasm as studies in yeast and in other systems have suggested (for review, see Henras et al. 2008).
Peptidyl transferase activity associates with nascent transcripts
Our BiFC assay only detected a weak 80S signal in the nuclear region occupied by the chromosomes. As we discuss later, this could be due, at least in part, to a slow kinetic of formation of the fluorescent complex, which has been reported to vary greatly between experimental systems (Kerppola 2009); the observation that emetine could enhance the signal within 5 min (Supplemental Fig. S5) suggests that it may take only a few minutes to produce fluorescence from the time of subunit joining. We therefore used the recently described technique of ribopuromycylation (David et al. 2012) to assess independently whether 80S are present in the nucleus and at chromosomal sites in particular, where they would presumably be loaded onto nascent pre-mRNA transcripts as suggested by previous studies (see Introduction). This method uses a fluorescence-labeled puromycin-specific antibody to visualize puromycylated nascent peptides that are immobilized on ribosomes by emetine treatment.
To assess the feasibility of the ribopuromycylatin technique in Drosophila, S2 cells and salivary glands were incubated with puromycin, alone or in combination with other translation inhibitors, as suggested by the protocol used for mammalian cells (David et al. 2012). Western blotting of cell extracts from either treated S2 cells or salivary glands readily detected puromycylated polypeptides, in both the presence and absence of emetine, but no signal was apparent when puromycin was omitted, confirming that the antibody is specific also in Drosophila (Supplemental Fig. S9A,B). Furthermore, puromycylation was drastically reduced when the samples were pretreated with harringtonine or anisomycin, as suggested by David et al. (2012). However, to the contrary of what was reported in mammalian cells, emetine visibly reduced the extent of puromycylation, suggesting that the drug can reduce puromycylation in Drosophila cells (Supplemental Fig. S9A,B, cf. lanes 1 and 2). Puromycylated peptides are released from the ribosome in the absence of emetine, yet the kinetic prediction is that these are probably at the highest concentration at translation sites even in the absence of emetine. Notably, we found the pattern of puromycylation in intact glands very similar to that of the BiFC 80S signal: a strong signal in the cytoplasm and in the nucleolus (Supplemental Fig. S10A). This cytoplasm/nucleolus pattern is particularly apparent in the proximal portion of the gland lobe, which is made of smaller cells. On the other hand, in the larger cells, which form the remainder of the lobe, puromycylated peptides also accumulate around the chromosomes (Supplemental Fig. S10B) (as in these larger cells the nucleolus staining is restricted to its outer shell, we suspect some poor penetration of the antibody in the larger cells). Specifically, the relative intensity of the nuclear and nucleolar signals seems higher in the presence of emetine, consistent with these being translation sites and emetine delaying nascent peptide release (Supplemental Fig. S10). To assess ribopuromycylation at the chromosomes in more details polytene chromosome was analyzed; we briefly incubated salivary glands with emetine and puromycin as above prior to fixation and chromosome spreading (Materials and Methods). Anti-puromycin immunostaining showed an apparent banding pattern of puromycin incorporation along the entire chromosome arms (Fig. 8A, top panels). The signal was mostly weak, yet still visible both at bands (densely DAPI-stained chromatin) and at interbands (less densely packed and transcriptionally active chromatin) (Fig. 8A). However, at a few interbands the signal was intense (indicated by the arrow in Fig. 8A, magnified in upper insets). The immunostaining was virtually absent if puromycin was omitted (Supplemental Fig. S9C).
FIGURE 8.
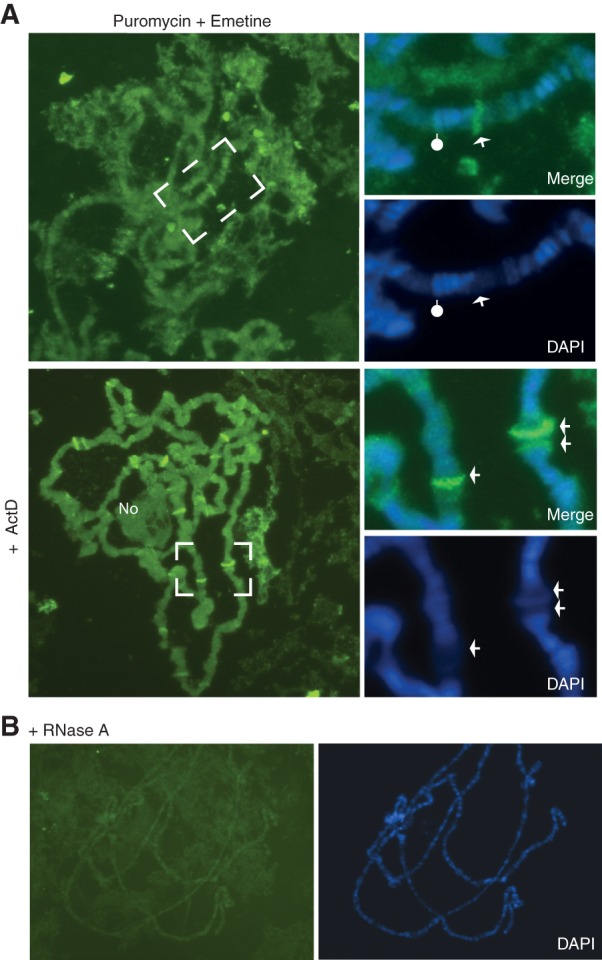
Puromycin incorporation at transcription sites. (A) Direct immunostaining of polytene chromosomes with Alexa488-conjugated anti-puromycin antibody. Top panels show salivary glands pre-incubated with puromycin and emetine, and the bottom panels show glands incubated with puromycin, emetine, and actinomycin D. Details are in Materials and Methods. Lower insets show higher magnification DAPI images of the regions indicated, and the upper insets are merged fluorescence and DAPI images. Arrows indicate interbands/puffs with strong anti-puromycin fluorescence; the pointer indicates a fluorescent band associated with a condensed chromosomal region. (B) Immunostaining of chromosomes as in A, of glands treated with RNase A (100 µg/mL for 2 min at room temperature to the first squashing step, before fixation).
The intensity of the immunostaining at interbands increased when an RNase inhibitor was added to the solution in which glands are pre-incubated before fixation and chromosome spreading (comparisons not shown, see Materials and Methods), so we reasoned that the signal was likely to be transcription-dependent. Having observed that actinomycin D treatment could transiently increase the BiFC 80S signal (Fig. 6C), we added this to the labeling incubation. Remarkably, immunostaining then became visibly more intense at many interbands, and particularly at transcription puffs (Fig. 8A, arrows in the lower panels). Puromycin incorporation was also detected at the nucleolus, which is often retained near the centromere in chromosome spreads (Fig. 8A, bottom panel). As predicted by the Western blot results shown above, anti-puromycin immunostaining did not occur in the absence of puromycin and was dramatically reduced following pre-incubation of glands with harringtonine or by treatment with RNase A (Fig. 8B; Supplemental Fig. S9C). These results are directly consistent with the earlier study which reported that RPs, rRNA, and translation factors are found at polytene chromosomes transcription sites (Brogna et al. 2002).
DISCUSSION
Here we have described the development of an assay in Drosophila cells that directly reports the interaction between BiFC-tagged RPs which are located in different subunits but become adjacent to the 80S. In particular, we have extensively characterized the interaction between two of these proteins, S18–YN and L11–YC. Flies in which S18–YN replaces the endogenous RpS18, or in which L11–YC replaces RpL11, are viable, thus ribosomes that include a subunit containing one of the BiFC-tagged partners must function at a near normal level. Our characterization indicates that the BiFC signal corresponds to 80S that have formed as a consequence of translation initiation. The BiFC signal is found in ribosomal fractions and is translation-dependent: Translation initiation inhibitors reduced the signal while the elongation inhibitor emetine increased it. Other observations also suggest that it is unlikely that a considerable fraction of the signal originated from nonspecific subunit interactions unrelated to translation. For example, it is a well-established fact that, when translation initiation inhibitors or puromycin are added to cells, the ribosomal subunits run off the polysomes and accumulate as nontranslating 80S couples, which can only dissociate into separate subunits in high salt conditions (Supplemental Fig. S3D; data not shown; Blobel and Sabatini 1971; Ramirez et al. 1991; Jackson 2007). Here, however, these treatments resulted in a reduction of the BiFC fluorescence rather than an increase (Fig. 4A; data not shown). The data, therefore, argue that the BiFC signal does not originate from an unspecific interaction in nontranslating 80S couples.
Although the observation that the S18–YN/L11–YC signal is predominantly in 80S fractions (Fig. 3D) might be interpreted as the BiFC linkage halting elongation, clear polysomal signal was detected using the same RPs tagged with Venus BiFC fragments (Fig. 5). We suspect that the explanation for these observations is that the intersubunit rotation that occurs during translocation breaks the YFP BiFC linkage but less so that formed by the Venus complex (Dunkle and Cate 2010). The BiFC complex is stable in vitro, but there are reports that the YC/YN complex can undergo dissociation in vivo (for review, see Kerppola 2009). The bulk of the S18–YN/L11–YC signal probably corresponds to elongation-engaged ribosomes temporarily delayed in the proximity of the initiation site, precluding loading of subsequent ribosomes and allowing time for the preceding ribosomes (which mostly will only have one tagged subunit) to terminate translation and run off the mRNA. The rapid and apparent enhancement of the signal produced by exposure to the elongation inhibitor emetine, also strongly suggests that the BiFC interaction occurs on translating 80S, which have both subunits tagged; emetine probably enhances the interaction because it locks the intersubunit rotation and stabilizes the 80S (Schneider-Poetsch et al. 2010).
Moreover, besides the issue of to what extent the BiFC-tagged 80S remains functional, collectively these observations indicate that the assay is correctly reporting that a translation-dependent interaction between subunits has occurred and that 80S have been formed. Furthermore, that the BiFC linkage might hinder subunit movements rather than being a limitation of technique is what probably fixes the ribosome long enough for the BiFC fluorescence to be produced, and is key for the sensitivity of our BiFC assay. Our data suggest that the level of BiFC-joined 80S is maintained at a steady state by a balance between subunits joining and the breaking down of BiFC-joined ribosomes. Perhaps there is also a pool of jammed BiFC-joined 80S that failed elongation and are degraded by mechanisms targeting aberrant ribosomes in conjunction with other mRNP surveillance processes (LaRiviere et al. 2006; Fujii et al. 2009). The kinetic of this clearance processed might depend on the strength of the BiFC linkage, which might be less effective with Venus-based BiFC.
Notably, while the 80S signal is, as expected, typically most intense in the cytoplasm, it can also be detected in the nucleus. Within the nucleus the 80S signal is most apparent in the nucleolus. One possibility is that a pre-ribosomal particle similar to the 90S complex found at the initial stages of ribosome biogenesis in yeast might generate the nucleolar BiFC fluorescence (Grandi et al. 2002). But our observations argue against this interpretation. Firstly, RP pairs that are not adjacent to the 80S structure do not generally produce nucleolar BiFC fluorescence even though the peptides concentrate in the nucleolus at levels similar to those BiFC pairs that produce the signal (the S9/L11 exception suggests that there may be some complexes in the nucleolus in which the two proteins are closer than they are in the cytoplasmic 80S). Secondly, emetine exposure also enhances the nucleolar signal, pointing to translation-dependent subunit joining and suggesting that there is a steady-state accumulation of mRNA in the nucleolus, similar to that reported in Arabidopsis (Kim et al. 2009). The fact that the strong nucleolar signal is sensitive to Pol II inhibition also suggests it corresponds to mRNA-associated 80S.
In summary, when one considers both the evidence for functional nuclear 80S provided by the BiFC assay and our finding that puromycin is readily incorporated in the nucleolus and at chromosomal transcription sites, it seems even more likely that these nuclear 80S are translating. The data we have presented here are in full agreement with the previous polytene chromosomes study, which in addition to showing evidence of ribosomal subunits at transcription sites, also reported rapid amino acid incorporation at the chromosomes and nucleolus (Brogna et al. 2002). This conclusion is also similar to that recently reached by David et al. (2012) and by earlier studies (Iborra et al. 2001; for historical review, see Reid and Nicchitta 2012). It has been argued that translation only occurs in the cytoplasm because key translation factors such as eEF1 are actively exported from the nucleus in mammalian cells (Bohnsack et al. 2002; Calado et al. 2002), but our data would be hard to explain if the cells we studied had no residual nuclear eEF1. Whether any proteins can be fully synthesized in the nucleus, particularly in the 80S-rich nucleolus, remains to be investigated. The observation that LMB enhances nuclear and nucleolar signals argues against these corresponding 80S re-imported from the cytoplasm (Fig. 7). It is possible that the nuclear 80S we have characterized are participating in a quality control check on the function of newly made ribosomal subunits similar to that which has been proposed to work in yeast cytoplasm (Lebaron et al. 2012; Strunk et al. 2012). This proposed mechanism is said to involve 80S-like structures not containing mRNA; however, the 80S that we have studied appear to associate with mRNA and to engage, at least to some degree, in translation.
MATERIALS AND METHODS
Plasmid constructions and fly strains
To generate plasmids expressing the BiFC-tagged RPs, YN and YC segments plus linker sequences were PCR-amplified from previously described plasmids, pBiFC Jun–YN and pBiFC Fos–YC (Hu et al. 2002). Venus VN (1–173) and VC (155–238) fragments were amplified from pAVW (Drosophila Genomic Resource Center, DGRC). RPs coding regions were PCR-amplified from various available cDNA libraries using primers tagged with appropriate restriction enzyme sequences (libraries available from DGRC). RP and BiFC segments were sequentially cloned in the pUAST vector (Brand and Perrimon 1993) or pAc5.1/V5–His A (Invitrogen). All constructs have been sequence-verified. Transgenic flies were produced by P element-mediated transformation of a standard yw strain (Bestgene). The fkh-Gal4 was typically used to drive expression in salivary glands (Henderson and Andrew 2000). The RpS18 mutant (RpS18c02853/CyO) was obtained from Exelixis, and that for RpL11 (RpL11k16914/CyO) from the Bloomington Drosophila Stock Center.
Cell culture and microscopy
Drosophila melanogaster Schneider line-2 cells (S2 cells) were typically grown inside six-well plates or 10-cm dishes in insect-XPRESS (Lonza) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin/glutamine mix (Invitrogen) at 27°C in a normal-atmosphere incubator. Transfection was typically performed using dimethyldioctadecylammonium bromide (DDAB) as previously described (Ramanathan et al. 2008)—more efficient commercial transfection reagents can be used but their use increase the frequency of cells with signs of stress (rounding up and shrinking of the nucleus). Post-transfection, cells were incubated for one or two nights at 27°C before usage. For assaying transcription dependency, transfected cells were treated with transcription inhibitors for 4 h, 2 d after transfection, at the indicated concentrations. For microscopy imaging, cells were either grown directly or attached post-transfection onto coverslips, then fixed with 4% formaldehyde in PBS for 20 min at room temperature, washed two times in PBS for 10 min, and permeabilized with an additional wash in cold PBS + 0.1% Tween for 10 min on ice. Post-permeabilization, coverslips were washed three more times in PBS for 10 min at room temperature or in cooled PBS; DAPI (4–6-diamidino-2-phenyl indole, Sigma-Aldrich) was added (0.1 µg/mL) to the second wash. Briefly, drained coverslips were mounted with a drop of fluorescence mounting medium (PromoFluor, Promokine). Microscopy imaging was carried out with either a Nikon Eclipse Ti epifluorescence microscope, equipped with a ORCA-R2 camera (Hamamatsu Photonics) or a Leica SP2-AOBS confocal microscope.
Immunostaining and Western blotting
Transfected cells attached to coverslips were fixed with 4% formaldehyde and 0.1% Triton X-100 in PBS for 30 min at room temperature and washed three times in PBS/0.1% Triton for 5 min. Blocking was in 4% BSA for 30 min at room temperature. Primary incubation was with a rabbit anti-GFP (1:200 in 4% BSA, Molecular Probes/Invitrogen) for 3 h at room temperature in a humid chamber. The cells were washed three times with PBS/0.1% Triton and then incubated with a secondary antibody, typically 1:250 dilution of Cy5- or Cy5-conjugate anti-rabbit (Jackson Laboratories or Invitrogen in 4% BSA). The cells were washed three times with PBS /Triton twice for 10 min each, DAPI was added in the second wash as above. Briefly, drained coverslips were mounted as described above. Transfected cells attached to the bottom of the well were drained of the media, resuspended in SDS loading buffer, and analyzed by standard SDS-PAGE followed by Western blotting with a GFP polyclonal antibody (goat anti-GFP, AbD Serotec).
Polysomes analysis
Cells were typically pretreated (15 min) with 100 μg/mL cycloheximide to stabilize polysomes, then pelleted by centrifugation at 4°C and washed in ice-cold PBS, pelleted again, and lysed in: 20 mM HEPES pH 7.4, 2 mM magnesium acetate, 2 mM MgCl2, 100 mM potassium acetate, 1 mM dithiothreitol (DTT), 250 μg/mL heparin, 20 units/mL of RiboLock RNase inhibitor (Fermentas), 0.6% Triton X-100, 100 μg/mL cycloheximide, EDTA free Complete Protease Inhibitor Cocktail (Roche), and 1 mM phenylmethanesulfonyl fluoride (PMSF). Cell lysates were cleared of nuclei, mitochondria, and other insoluble material by centrifugation at maximum speed for 20 min in a microcentrifuge at 4°C. Typically 10 OD260 units of extracts were loaded on 11 mL of a linear 10%–50% or 10%–30% sucrose gradient (in 20 mM HEPES pH 7.4, 2 mM magnesium acetate, 2 mM MgCl2, 100 mM potassium acetate) and centrifuged at 38,000 rpm for 2 h and 40 min in a SW40Ti rotor (Beckman). Puromycin treatment consisted of 1-h pre-incubation of the cells with 100 µg/mL puromycin, lysis in buffer containing the same drug concentration plus 375 mM KCl without cycloheximide, and 30-min incubation at room temperature prior to loading and centrifugation. EDTA treatment consisted of treatment of the cleared cell lysate (from cells not treated with cycloheximide) with 30 mM EDTA and 30-min incubation on ice prior to loading. Puromycin and EDTA-treated samples were fractionated in Mg2+-free gradients. After centrifugation fractions (typically 0.9 mL) were recovered from the bottom of the tube using a capillary attached to a peristaltic pump and absorbance monitored with a plotter-connected UV-1 monitor with a 254-nm filter (both from Pharmacia). Fractions were precipitated by adding 0.1 volumes of 100% trichloroacetic acid (TCA), vortexing, incubating overnight at 4°C, and centrifuging at maximum speed for 30 min in a microfuge. The supernatant was then carefully removed and the precipitate washed with ice-cold acetone and centrifuged for 2 min at 4°C; the pellet was analyzed by standard SDS-PAGE and Western blotting with GFP polyclonal antibody as above.
Fluorimetry
To measure BiFC fluorescence in cell extracts, transfected cells were split into aliquots (0.5–1 × 107 cells) and were pre-incubated at room temperature with or without translation inhibitors, washed once in ice-cold PBS, centrifuged, and the pellet resuspended in 200 μL of polysome lysis buffer (as above) supplemented with the corresponding translation inhibitor at the same concentration as in vivo. Samples were cleared by centrifugation at 4°C at maximum speed for 20 min, and fluorescence measured in a 100 μL micro quartz cuvette (Starna Scientific Ltd) using a PTI QuantaMaster 40 fluorimeter (Photon Technology International Inc), then analyzed with the provided FeliX32 software. To measure the YFP/ BiFC excitation spectrum the sample was excited at the fixed wavelength of 488 nm and excitation measured from 500 to 550 nm with the emission monochromator set at band pass of 3 nm. The fluorescence of every sample was automatically counted three times. Mean values were normalized by subtracting background readings of a parallel control extract of untransfected cells (adjusted to have same OD260 as the other samples).
Salivary glands 5-FUrd labeling and immunostaining
Larvae were grown at 18°C to slow development so as to increase the size of the salivary glands. Glands were routinely dissected in PBS and immediately fixed in cold PBS with 4% formaldehyde. For drug treatment experiments or metabolic labeling glands were dissected and incubated in Shield and Sang M3 insect medium (Sigma). For 5-fluorouridine (FUrd) labeling, dissected glands were incubated for 10 min with 2 mM FUrd (SIGMA, F5130), fixed with 4% formaldehyde diluted in PBS-Triton (0.3% Triton X-100) for 60 min on ice, and then washed four times in PBS-Triton for 5 min. Blocking is in 10% fetal bovine serum (FBS) diluted in PBS for 60 min at room temperature. FUrd was detected with an anti-BrdU (1:200 dilution in 0.3% Triton X-100 in PBS with 10% FBS, SIGMA B2531) incubated for 16 h at 4°C. The secondary antibody (1:250) was an Alexa Fluor 647 Goat Anti-Mouse (Invitrogen) incubated overnight at 4°C. The fibrillarin (Nop1p) antibody was diluted 1:500 and incubated overnight at 4°C. The lamin Dm0 antibody was ADL84.12 (DS Hybridoma Bank), used as above. Secondary antibodies (as above) were diluted (1:250) in blocking buffer and also incubated overnight at 4°C. DAPI staining was typically for 15 min.
Ribo-puromycylation and polytene chromosomes preparation
Salivary glands were dissected in M3 media from 3rd instar larvae grown at 18°C. The optimal labeling procedure consisted of incubation in media containing puromycin (50 µg/mL), emetine (100 µg/mL), and actinomycin D (ActD) 10 µg/mL for 15 min at room temperature. This was followed by three washes in ice-cold PBS, containing emetine and ActD, 5 min each, prior to chromosome squashing. Chromosome squashing involved a 2-min incubation of a pair of glands in a drop of 15 mM HEPES pH 7.4, 60 mM KCl, 15 mM NaCl, 1.5 mM Spermine, 1.5 mM Spermidine, 1% Triton X-100, 100 µM aurintricarboxylic acid (ATA), and EDTA-free Complete Protease Inhibitor Cocktail (Roche), followed by fixation and chromosome spreading as previously described (Rugjee et al. 2013). Immunostaining was with an Alexa 488 conjugated 2A4 monoclonal antibody (1:200) (from Dr. John Yewdell, NIH, USA). For whole salivary gland puromycylation, glands were dissected and incubated as above (but without actinomycin D) and then fixed in PBS containing 4% formaldehyde on ice for 30 min, and processed for immunostaining as described above, using Alexa 488 conjugated 2A4 monoclonal antibody overnight at 4°C.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tom Kerppola (University of Michigan) for sending plasmids; Alex David (CNRS, France) and John Yewdell (NIH) for swiftly providing the 2A4 mAb; Debbie Andrew (Johns Hopkins University) for the fkh-Gal4 line; Alicia Hidalgo for fly genetics advice; and Bob Michell and Tina McLeod for critically reading the manuscript. We also acknowledge several master's and undergraduate students who over the years have made various contributions to this project: Stephanie Cartwright, Carlo Casale, Chris Humphreys, Akinwusi Akinlolu Oloyede, and Shichao Rong. This work was supported by a Royal Society fellowship and a Wellcome Trust grant to S.B., as well as Saudi Embassy and Islamic Development Bank PhD scholarships to K.A-J. and A.A., respectively.
Footnotes
Freely available online through the RNA Open Access option.
REFERENCES
- Blobel G, Sabatini D. 1971. Dissociation of mammalian polyribosomes into subunits by puromycin. Proc Natl Acad Sci 68: 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack MT, Regener K, Schwappach B, Saffrich R, Paraskeva E, Hartmann E, Gorlich D. 2002. Exp5 exports eEF1A via tRNA from nuclei and synergizes with other transport pathways to confine translation to the cytoplasm. EMBO J 21: 6205–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois CA, Hemon D, Bouteille M. 1979. Structural relationship between the nucleolus and the nuclear envelope. J Ultrastruct Res 68: 328–340. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [DOI] [PubMed] [Google Scholar]
- Brogna S, Sato TA, Rosbash M. 2002. Ribosome components are associated with sites of transcription. Mol Cell 10: 93–104. [PubMed] [Google Scholar]
- Cabantous S, Terwilliger TC, Waldo GS. 2005. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol 23: 102–107. [DOI] [PubMed] [Google Scholar]
- Calado A, Treichel N, Muller EC, Otto A, Kutay U. 2002. Exportin-5-mediated nuclear export of eukaryotic elongation factor 1A and tRNA. EMBO J 21: 6216–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouli P, Topf M, Menetret JF, Eswar N, Cannone JJ, Gutell RR, Sali A, Akey CW. 2008. Structure of the mammalian 80S ribosome at 8.7 Å resolution. Structure 16: 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleno-Costes A, Jang SM, de Vanssay A, Rougeot J, Bouceba T, Randsholt NB, Gibert JM, Le Crom S, Mouchel-Vielh E, Bloyer S, et al. 2012. New partners in regulation of gene expression: The enhancer of trithorax and polycomb corto interacts with methylated ribosomal protein l12 via its chromodomain. PLoS Genet 8: e1003006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg J, Lund E. 2012. Nuclear translation or nuclear peptidyl transferase? Nucleus 3: 320–321. [DOI] [PubMed] [Google Scholar]
- David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW. 2012. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol 197: 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Brogna S. 2010. Are ribosomal proteins present at transcription sites on or off ribosomal subunits? Biochem Soc Trans 38: 1543–1547. [DOI] [PubMed] [Google Scholar]
- De S, Varsally W, Falciani F, Brogna S. 2011. Ribosomal proteins’ association with transcription sites peaks at tRNA genes in Schizosaccharomyces pombe. RNA 17: 1713–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinos G, Wilson DN, Teraoka Y, Szaflarski W, Fucini P, Kalpaxis D, Nierhaus KH. 2004. Dissecting the ribosomal inhibition mechanisms of edeine and pactamycin: The universally conserved residues G693 and C795 regulate P-site RNA binding. Mol Cell 13: 113–124. [DOI] [PubMed] [Google Scholar]
- Dunkle JA, Cate JH. 2010. Ribosome structure and dynamics during translocation and termination. Annu Rev Biophys 39: 227–244. [DOI] [PubMed] [Google Scholar]
- Fresno M, Jimenez A, Vazquez D. 1977. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem 72: 323–330. [DOI] [PubMed] [Google Scholar]
- Fujii K, Kitabatake M, Sakata T, Miyata A, Ohno M. 2009. A role for ubiquitin in the clearance of nonfunctional rRNAs. Genes Dev 23: 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schafer T, Kuster B, Tschochner H, Tollervey D, et al. 2002. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell 10: 105–115. [DOI] [PubMed] [Google Scholar]
- Grollman AP. 1968. Inhibitors of protein biosynthesis. V. Effects of emetine on protein and nucleic acid biosynthesis in HeLa cells. J Biol Chem 243: 4089–4094. [PubMed] [Google Scholar]
- Henderson KD, Andrew DJ. 2000. Regulation and function of Scr, exd, and hth in the Drosophila salivary gland. Dev Biol 217: 362–374. [DOI] [PubMed] [Google Scholar]
- Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65: 2334–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, Kerppola TK. 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell 9: 789–798. [DOI] [PubMed] [Google Scholar]
- Hudry B, Viala S, Graba Y, Merabet S. 2011. Visualization of protein interactions in living Drosophila embryos by the bimolecular fluorescence complementation assay. BMC Biol 9: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra FJ, Jackson DA, Cook PR. 2001. Coupled transcription and translation within nuclei of mammalian cells. Science 293: 1139–1142. [DOI] [PubMed] [Google Scholar]
- Jackson RJ. 2007. The missing link in the eukaryotic ribosome cycle. Mol Cell 28: 356–358. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappen LS, Suzuki H, Goldberg IH. 1973. Inhibition of reticulocyte peptide-chain initiation by pactamycin: Accumulation of inactive ribosomal initiation complexes. Proc Natl Acad Sci 70: 22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK. 2008. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys 37: 465–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK. 2009. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: Characteristics of protein fragment complementation. Chem Soc Rev 38: 2876–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Koroleva OA, Lewandowska D, Pendle AF, Clark GP, Simpson CG, Shaw PJ, Brown JW. 2009. Aberrant mRNA transcripts and the nonsense-mediated decay proteins UPF2 and UPF3 are enriched in the Arabidopsis nucleolus. Plant Cell 21: 2045–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge S, Voigts-Hoffmann F, Leibundgut M, Ban N. 2012. Atomic structures of the eukaryotic ribosome. Trends Biochem Sci 37: 189–198. [DOI] [PubMed] [Google Scholar]
- Kos M, Tollervey D. 2010. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37: 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRiviere FJ, Cole SE, Ferullo DJ, Moore MJ. 2006. A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell 24: 619–626. [DOI] [PubMed] [Google Scholar]
- Lebaron S, Schneider C, van Nues RW, Swiatkowska A, Walsh D, Bottcher B, Granneman S, Watkins NJ, Tollervey D. 2012. Proofreading of pre-40S ribosome maturation by a translation initiation factor and 60S subunits. Nat Struct Mol Biol 19: 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarotti G, Chiaberge S, Bulfone S. 1997. rRNA maturation as a “quality” control step in ribosomal subunit assembly in Dictyostelium discoideum. J Biol Chem 272: 27818–27822. [DOI] [PubMed] [Google Scholar]
- Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, Millburn GH, Harrison PM, Yu Z, Kenmochi N, Kaufman TC, et al. 2007. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol 8: R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathans D, Lipmann F. 1961. Amino acid transfer from aminoacyl-ribonucleic acids to protein on ribosomes of Escherichia coli. Proc Natl Acad Sci 47: 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheim YN, French SL, Keck KM, Champion EA, Spasov K, Dragon F, Baserga SJ, Beyer AL. 2004. Pre-18S ribosomal RNA is structurally compacted into the SSU processome prior to being cleaved from nascent transcripts in Saccharomyces cerevisiae. Mol Cell 16: 943–954. [DOI] [PubMed] [Google Scholar]
- Panse VG, Johnson AW. 2010. Maturation of eukaryotic ribosomes: Acquisition of functionality. Trends Biochem Sci 35: 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penman S, Smith I, Holtzman E. 1966. Ribosomal RNA synthesis and processing in a particulate site in the HeLa cell nucleus. Science 154: 786–789. [DOI] [PubMed] [Google Scholar]
- Ramanathan P, Guo J, Whitehead RN, Brogna S. 2008. The intergenic spacer of the Drosophila Adh-Adhr dicistronic mRNA stimulates internal translation initiation. RNA Biol 5: 149–156. [DOI] [PubMed] [Google Scholar]
- Ramirez M, Wek RC, Hinnebusch AG. 1991. Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol Cell Biol 11: 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid DW, Nicchitta CV. 2012. The enduring enigma of nuclear translation. J Cell Biol 197: 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouquette J, Choesmel V, Gleizes PE. 2005. Nuclear export and cytoplasmic processing of precursors to the 40S ribosomal subunits in mammalian cells. EMBO J 24: 2862–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugjee KN, Roy Chaudhury S, Al-Jubran K, Ramanathan P, Matina T, Wen J, Brogna S. 2013. Fluorescent protein tagging confirms the presence of ribosomal proteins at Drosophila polytene chromosomes. PeerJ 1: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder PA, Moore MJ. 2005. Association of ribosomal proteins with nascent transcripts in S. cerevisiae. RNA 11: 1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, Williamson JR. 2011. Assembly of bacterial ribosomes. Annu Rev Biochem 80: 501–526. [DOI] [PubMed] [Google Scholar]
- Soudet J, Gelugne JP, Belhabich-Baumas K, Caizergues-Ferrer M, Mougin A. 2010. Immature small ribosomal subunits can engage in translation initiation in Saccharomyces cerevisiae. EMBO J 29: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CM, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J. 2001. Structure of the 80S ribosome from Saccharomyces cerevisiae—tRNA-ribosome and subunit-subunit interactions. Cell 107: 373–386. [DOI] [PubMed] [Google Scholar]
- Strunk BS, Loucks CR, Su M, Vashisth H, Cheng S, Schilling J, Brooks CL III, Karbstein K, Skiniotis G. 2011. Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science 333: 1449–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunk BS, Novak MN, Young CL, Karbstein K. 2012. A translation-like cycle is a quality control checkpoint for maturing 40S ribosome subunits. Cell 150: 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne JS, Pestka S. 1975. Inhibition of protein synthesis in intact HeLa cells. Antimicrob Agents Chemother 8: 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschochner H, Hurt E. 2003. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol 13: 255–263. [DOI] [PubMed] [Google Scholar]
- Udem SA, Warner JR. 1973. The cytoplasmic maturation of a ribosomal precursor ribonucleic acid in yeast. J Biol Chem 248: 1412–1416. [PubMed] [Google Scholar]
- Venema J, Tollervey D. 1999. Ribosome synthesis in Saccharomyces cerevisiae. Annu Rev Genet 33: 261–311. [DOI] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. 2001. Crystal structure of the ribosome at 5.5 Å resolution. Science 292: 883–896. [DOI] [PubMed] [Google Scholar]
- Zemp I, Kutay U. 2007. Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett 581: 2783–2793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.