

Abstract
Hemorrhagic shock is associated with metabolic defects, including hyperglycemia and insulin resistance but the mechanisms are unknown. We recently demonstrated that reduction of the extracellular domain of the insulin receptor by degrading proteases may lead to a reduced ability to maintain normal plasma glucose values. In shock, transfer of digestive enzymes from the lumen of the intestine into the systemic circulation after breakdown of the intestinal mucosal barrier causes inflammation and organ dysfunction. Suppression of the digestive enzymes in the lumen of the intestine with protease inhibitors is effective in reducing the level of the inflammatory reactions. To determine the degree to which blockade of digestive enzymes affects insulin resistance in shock, rats were exposed to acute hemorrhagic shock (mean arterial pressure of 30 mmHg for 2 hours) at which time all shed blood volume was returned. Digestive proteases in the intestine were blocked with a serine protease inhibitor (tranexamic acid in polyethylene glycol and physiological electrolyte solution) and the density of the insulin receptor was measured with immunohistochemistry in the mesentery microcirculation. The untreated rat without enzyme blockade had significantly attenuated levels of insulin receptor density as compared to control and treated rats. Blockade of the digestive proteases after 60 min of hypotension in the lumen of the small intestine lead to a lesser decrease in insulin receptor density compared to controls without protease blockade. Glucose tolerance test indicates a significant increase in plasma glucose levels two hours after hemorrhagic shock, which are reduced to control values in the presence of protease inhibition in the lumen of the intestine. The transient reduction of the plasma glucose levels after an insulin bolus is significantly attenuated after shock, but is restored in when digestive enzymes in the lumen of the intestine are blocked. These results suggest that in hemorrhagic shock elevated microvascular extracellular digestive enzyme activity causes insulin receptor dysfunction, hyperglycemia and reduced ability to regulate blood glucose values.
Keywords: Glucose tolerance, pancreatic enzymes, proteases, auto-digestion, mesentery microvessels, leukocytes, Wistar rat
INTRODUCTION
Shock and multi-organ failure is associated with an altered metabolic milieu and malnutrition (23) as well as acute insulin resistance (6, 27). The insulin resistance is present even in non-lethal shock (22) and can persist for many days (28). The acute insulin resistance is detected in multiple organs (30, 32) and may be accompanied by hyperglycemia, cytokine actions (8), loss of signaling pathways (19) and growth hormone resistance (17). A number of mechanisms have been proposed, including involvement of macrophages (18), oxygen free radicals and nitric oxide (34), cytokines (33) and ATP depletion (6). But no consensus exists for the underlying molecular mechanisms as the basis for a targeted approach to treat altered glucose metabolism following injury and/or critical illness (23). The inability to respond to insulin tends to be present during the inflammatory state and precedes extensive organ dysfunction and failure. In this study we will focus on the insulin receptor in the microcirculation during hemorrhagic shock.
In several forms of shock the powerful pancreatic digestive enzymes escape from the lumen of the intestine across the mucosal barrier into the wall of the intestine where they initiate an autodigestion process (5). The presence of pancreatic enzymes generates inflammatory mediators and causes organ dysfunction (9, 21). Blockade of the enzymes reduces the systemic inflammation and organ dysfunction (9). But none of these pathophysiological mechanisms have been directly linked to a process that may be responsible for insulin resistance.
We obtained evidence in a chronic model of insulin resistance that unchecked extracellular protease activity in plasma and on cell surfaces causes proteolytic cleavage of the extracellular domain of multiple membrane receptors (29), including the insulin receptor (11). Proteolytic cleavage of the insulin receptor interferes with signaling to GLUT 4 for transmembrane glucose transport after binding of insulin and protease blockade restores the glucose transport. So-called “soluble receptor” fragments are detected in plasma of patients with insulin resistance (16). We hypothesize that blockade of digestive enzymes by an inhibitor of pancreatic digestive serine proteases in the lumen of the intestine serves to prevent insulin resistance and corresponding metabolic damage after severe hemorrhagic shock.
Thus, the current experiments were designed to determine to what degree digestive enzymes cause acute reduction of extracellular immunobinding sites for the insulin receptor and glucose transport dysfunction in hemorrhagic shock. We tested the effectiveness of a treatment with protease inhibitors in the lumen of the intestine to retain insulin receptor density in the mesentery microcirculation hand in hand with retention of glucose transport and the response to intravascular glucose or insulin bolus administration.
METHODS
Animals
The protocol was reviewed and approved by the University of California San Diego Animal Subjects Committee. University practices for the procurement, housing, care and use of animals were in accordance with Federal, State and local laws, regulations and guidelines. Studies were carried out on mature male Wistar rats (Harlan Sprague Dawley, Indianapolis, IN; specific pathogen free) of age 15–18 weeks and weight 400–500 grams (462 ± 48 gm) under general anesthesia (pentobarbital sodium, 50 mg/kg, Abbott Laboratories, North Chicago, IL, i.m. after local anesthesia with 2% Lidocaine HCl, Hospira, Inc., Lake Forrest, IL). Animals were maintained on standard laboratory chow (8604 Teklad Rodent Diet, Harlan Laboratories, Indianapolis, IN) without restriction and water ad libitum and maintained without pathogen free conditions. Surgical procedures were carried out under sterile conditions and catheters were inserted into left femoral artery and vein (PE 50 tubing) for measurement of blood pressure and for blood withdrawal or drug delivery, respectively. The animal’s core temperatures were kept near 39°C by continuous placement on a water-heated pad throughout the experiment and placement of a surgical drape (Tiburon Square-Folded Drape Sheet, cat. no. 9461, Cardinal Health, 7000 Cardinal Place, Dublin, OH) over the animals with rectal temperature measurement. At the termination of the post-shock observation period the animals were euthanized (Beuthanasia IV, 120 mg/kg; Schering-Plough Animal Health Corp., Union, NJ).
Shock Protocol
Hemorrhagic shock was initiated by reduction of blood volume (~40% of whole blood volume based on 6% body weight) to achieve a blood pressure of 30 mmHg for a period of 2 hours, at which time the shed volume was returned. Blood was withdrawn via the femoral venous catheter over a period of about 5 minutes, stored at 4°C in heparin (0.5 U/ml), and rewarmed to 37°C just minutes before reinjection. The femoral blood pressure was monitored for two hours past fluid restoration at which time the animals were euthanized.
Enteral Protease Blockade
To block digestive enzymes in the lumen of the intestine, before start of blood pressure reduction a mid-line incision (~2.5 cm) was made to expose the small intestine. Exposure of the intestine was kept to a minimum due to fluid evaporation and cooling since under anesthesia core temperature could not be returned to initial control values if it had dropped. A serine protease inhibitor (tranexamic acid, TXA, cyclokapron, 0.025 g/ml, Pfizer, New York, NY) was used for blockade of digestive enzymes and administered in polyethylene glycol electrolytes solution (Go-LYTELY, Braintree Laboratories Inc, Braintree, MA) referred to in the following collectively as TXA. The TXA concentration was determined in pilot studies to be at a level to significantly reduce the trypsin activity in the lumen of the intestine (5). The solution was injected into the lumen of the intestine over its entire length from the duodenum to, and including, the cecum. A total fluid volume of about 15 ml was injected into the lumen of the intestine spaced out over the length of the small intestine in equal intervals of about 8 injections and 2 ml was injected into the cecum. Control animals received an identical solution but without the protease inhibitor. After enzyme blockade, the intestine was returned gently into the peritoneal cavity and the incision was closed with use of chromic gut suture (4-0, Ethicon Inc., Bridgewater Township, NJ) and Vicryl suture (4-0, Ethicon) to close abdominal wall and skin, respectively. The intestinal injection to block the digestive enzymes required less than 10 minutes. The treatment choices were interspersed in an alternating sequence between protease blockade and control and one animal was studied on a single day. Pilot studies with identical enteral TXA administration but without hypotension show no reduction in blood pressure or other symptoms over the same observation period as used in this study (results not shown).
Protease Activity Measurements
Plasma and intestinal samples (after rinsing of the luminal contents with saline) were tested for overall protease activity with a colorimetric detection kit (Product Code PC0100, Sigma-Aldrich, St. Louis, MO) using a casein substrate. Several proteases cleave this substrate (metallo-, serine, acid and sulfhydryl proteases) and protease activity is colorimetrically determined (in light absorption units) (UV/VIS Spectrometer Model Lambda 20, PerkinElmer, Waltham, MS) and expressed as µMole of tyrosine equivalents released (over a reaction period of 10 minutes). Protease measurements in the small intestine were obtained from the duodenum to the ileum and measured in about 38 individual intestinal length segments of 2.5 cm and averaged over the length of the intestine.
Blood Glucose Levels
Blood samples were tested for sugar levels using the Contour blood glucose meter (Bayer Diabetes Care, Tarrytown, NY).
Receptor Density Labeling
The cellular distribution of the insulin receptor on fixed (10% Formalin, neutral buffered) polymorphonuclear leukocytes on blood smears (derived from venous blood) and in whole mount mesentery specimens was delineated with immunohistochemistry by use of a primary antibody (Ra (N-20) sc-710, Santa Cruz Biotechnology, Santa Cruz, CA). The antibody maps towards the N-terminus at the insulin binding-site on the α-domain. Images were recorded after secondary antibody binding and delineated on isolated blood cells by a biotin/avidin immunolabeling technique (Vectastain Elite ABC Kit, Vector Laboratories, Inc., Burlingame, VA) and in whole mount tissues with a fluorescent antibody kit (Fl-1200, Vector Laboratories). The density of the insulin receptor on individual leukocytes was recorded on a brightfield microscope (60x objective) and in microvessels (arterioles, capillaries, venules) of mesentery tissue on a fluorescent microscope (20x objective) and digitally measured in form of substrate light absorption (after placement of a digital window on the image of the cell surface) or by fluorescent intensity, respectively, as described (11). Special care was taken that all cell and tissue specimen in the different groups were treated in an identical fashion to permit comparisons between groups. Light absorption was expressed as
where I is the light intensity over the tissue and Io the light intensity over the background (10). The leukocytes images were recorded by systematic scanning of the blood smears and inclusion of all polymorphonuclear leukocytes (identified by their segmented nucleus) during the scanning process without preselection (30 cells/ blood smear with two to three blood smears per animal).
Fluorescent Glucose Uptake
To measure glucose uptake, fluorescence-tagged analog of glucose (non-hydrolyzable glucose analog 6-NBD-deoxyglucose, Molecular Probes, Invitrogen, Eugene, OR; 1 µM in buffered saline) was used on fresh polymorphonuclear leukocytes (isolated by Histopaque-1077, 10771, Sigma-Aldrich, St. Louis, MO) from naïve control rats exposed for 30 min to plasma one hour after and as control before hemorrhagic shock. Thereafter the plasma in each group was removed, the cells were re-suspended in autologous Wistar plasma and the fluorescent glucose uptake was carried out for 10 min at room temperature. At that time the extracellular glucose was removed by centrifugation and the cells were suspended in buffered saline. The intracellular fluorescent intensity was determined immediately on individual leukocytes (minimum of 300 cells per sample) with fluorescent microscopy (60x objective). The images were digitized and fluorescent intensity was determined after subtraction of background fluorescent intensity (11).
Intravenous Glucose Tolerance Test
To determine the degree of glucose transport upon intravenous administration, a bolus of glucose (40% solution, 1 gm/kg in 0.5 gm/ml saline) was administered before induction of hemorrhagic shock and repeated in each animal at 2 hours after hemorrhagic shock and restoration of blood volume. Blood samples were taken from a femoral artery catheter at 5, 10, 15, 30, 45, 60, and 120 min after the bolus infusion. Blood glucose values were measured (Ascensia Contour Blood Glucose Monitoring System; Bayer HealthCare LLC, Mishawaka, IN), and averaged over four repeat measurements per sample from each animal at each time point.
Insulin Tolerance Test
The ability of insulin to stimulate a reduction in intra-arterial glucose levels was tested by a bolus injection of insulin (0.35 units/kg; Humulin R, Lilly USA, LLC, Indianapolis, IN; i.v.) in 0.5 ml of saline. Insulin bolus was administered before shock (as control), 2 hours after hemorrhagic shock in two groups with and without blockade of digestive enzymes, as described above. The plasma glucose values from a femoral artery sample were measured before and after insulin infusion over a period of two hours.
Statistics
Values are presented as mean ± SD and the number of animals used was between 3 and 8 in each group (CONTROL, SHOCK, SHOCK+TXA). Group differences in blood glucose levels before and after hypotension with and without protease blockade were tested by ANOVA with Bonferoni’s post-hoc T-test. Unpaired comparisons of mean values between groups were carried out by two-tail Student’s t-test unless stated otherwise. A probability of p < 0.05 was considered significant.
RESULTS
The protease activity in the wall of the intestine and in the plasma was significantly elevated at two hours after restoration of blood volume (Fig.1) in line with previous observations (9). Blockade of pancreatic digestive enzymes in the lumen of the intestine with TXA reduces the proteolytic activity in the wall of the intestine and in the plasma (Fig. 1) to the level of controls without shock.
Figure 1.

Protease activity levels (determined by serine substrates) (A) in the wall of the intestine and (B) in plasma of controls and animals subjected to hemorrhagic shock without and with protease inhibition in the lumen of the intestine with tranexamic acid in polyethylene glycol and electrolyte solution (TXA). Measurements were made before hypotension (CONTROL) and at 2 hours after hypotension (SHOCK), without and with enteral TXA blockade (CONTROL+TXA). Protease activity values of intestinal wall tissue in shock animals without enzyme inhibitor were significantly higher than both control animals and shock animals with treatment. Protease values in the plasma of shock animals without inhibitor were significantly higher than both control animals and shock animals treated with enzyme inhibitor. For tissue measurement there were 6 rats in CONTROL group, 5 in SHOCK group, 5 in SHOCK+TXA group, and for plasma measurement 14 rats in control group, 7 in SHOCK group, 6 in SHOCK+TXA group. Groups are significantly different as determined by ANOVA. *p < 0.05 SHOCK vs SHOCK+TXA, †p < 0.05 SHOCK vs Control.
At the same time point, the blood glucose levels are significantly elevated after hemorrhagic shock and the protease blockade serves to reduce these levels to those of non-shock controls (Table 1).
Table 1.
Blood Glucose Values (mg/dl)
| Before Shock | Post Shock* | |
|---|---|---|
| Control sham shock | 107±15 | 97±22 |
| Shock without protease treatment | 95±15 | 344±13 **,† |
| Shock + TXA treatment | 111±5 | 116±15 |
Two hours after ischemia
p < 0.002 versus Control by paired t-test
p < 0.02 versus Shock with TXA treatment by paired t-test
n = 3 rats per group; group averages are significantly different (p<0.001) by ANOVA.
The extracellular α-domain density of the insulin receptor on freshly collected leukocytes at two hours after hypotension was significantly reduced compared to the density before hypotension (Fig. 2, Shock vs Control), compatible with a potential receptor cleavage in the circulation after hemorrhagic shock. This reduction of the extracellular receptor immunobinding sites is blocked if digestive proteases are blocked in the lumen of the intestine. The circulating leukocytes after activation in hemorrhagic shock may be trapped in the microcirculation (1), so that an analysis of freshly collected leukocytes may be biased towards less activated cells still able to circulate. Therefore we also tested the hypothesis for receptor dysfunction by incubating naïve donor leukocytes from an animal without hypotension in the plasma collected from animals after two hours of hypotension. Already after 30 min incubation in shock plasma there was a significant reduction of the extracellular density of the insulin receptor α (Fig. 3A), a phenomenon that was accompanied by reduced glucose transport into the cell cytoplasm (Fig. 3B).
Figure 2.
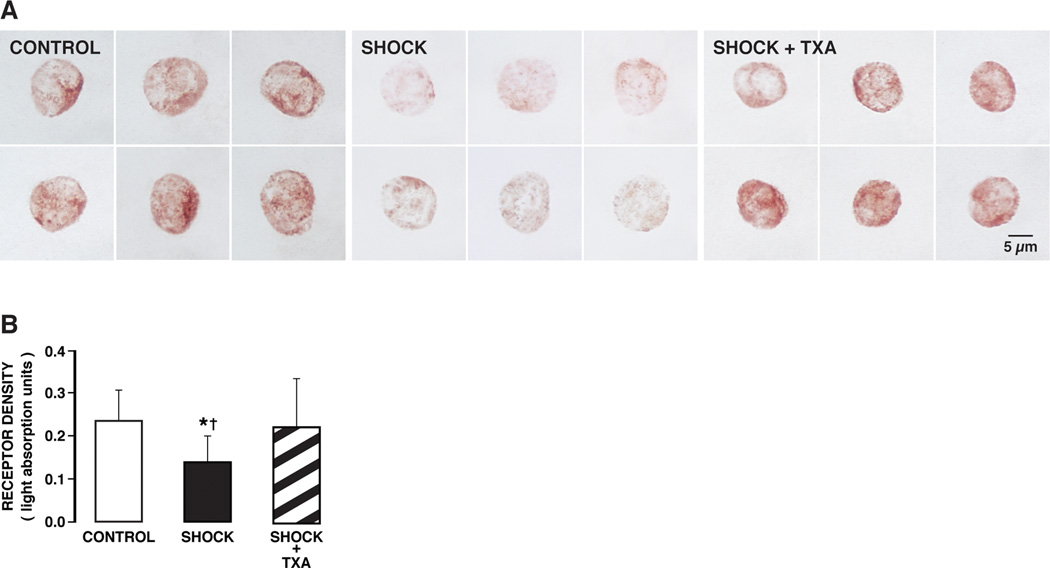
(A) Brightfield micrographs of formalin-fixed leukocytes immunolabeled with antibody against the extracellular α-domain of the insulin receptor with Vector NovaRED substrate in control animals (CONTROL) and 2 hours after hemorrhagic shock without (SHOCK) and with (SHOCK + TXA) pancreatic digestive enzyme inhibition. The density of the insulin receptor in control leukocytes shows uniform staining of leukocytes as indicated by Vector NovaRED substrate (left panel). The label density was reduced after hemorrhagic shock (middle panel). Hemorrhagic shock animals treated with pancreatic digestive enzyme inhibitor (SHOCK + TXA) (right panel) had insulin receptor densities in leukocytes comparable to control animals (left panel). (B) Light absorption values of the insulin receptor alpha in control animals and after hemorrhagic shock without (Go-Lytely) and with (SHOCK + TXA) pancreatic enzyme inhibition. The average light absorption values of the insulin receptor over randomly selected leukocytes were significantly higher in control and treated animals than without treatment. Pancreatic digestive enzyme blockade with TXA improves extracellular insulin receptor density after hemorrhagic shock. (n = 90 cells/group with 3 rats per group) *p < 0.05 SHOCK vs SHOCK+ TXA, †p < 0.05 SHOCK vs Control.
Figure 3.

(A). (Left) Micrographs of naïve circulating leukocytes with immuno-histochemical label against the extracellular binding α-domain of insulin receptor before (CONTROL) and after (SHOCK) incubation in plasma of rats collected two hours after hemorrhagic shock. Note the strong reduction of the extracellular domain binding sites of the receptor in shock plasma. (Right) Receptor density measured by quantitative light absorption under standard light conditions. (90 cells/rat with 3 rats). *p<0.01. B). (Left) Micrographs of cytoplasmic fluorescent intensity by non-hydrolyzable glucose analog after uptake into naïve circulating leukocytes before (Control) and after (Shock) incubation in plasma of rats collected two hours after hemorrhagic shock. The incubation in fluorescent glucose was for 30 min in all groups. (Right) Cytoplasmic fluorescent intensity in digital units measured under standard light conditions showing significantly reduced cytoplasmic glucose levels after shock (300 cells/rat with 3 rats). *p<0.03.
The density of the insulin receptor α domain on microvessels in the mesentery of control animals shows uniform staining of individual arterioles, capillaries and venules (Fig. 4A, CONTROL). After hemorrhagic shock there is a significantly attenuated fluorescent label density, indicating loss of insulin receptor binding sites in hemorrhagic shock (Fig. 4A, B, CONTROL vs SHOCK group). Treatment with pancreatic digestive enzyme inhibitor in the lumen of the intestine prevents insulin receptor cleavage in the mesentery microvessels to a significant degree, as indicated by fluorescent intensities that remained at the level of control animals (Fig. 4, CONTROL vs SHOCK + TXA group).
Figure 4.
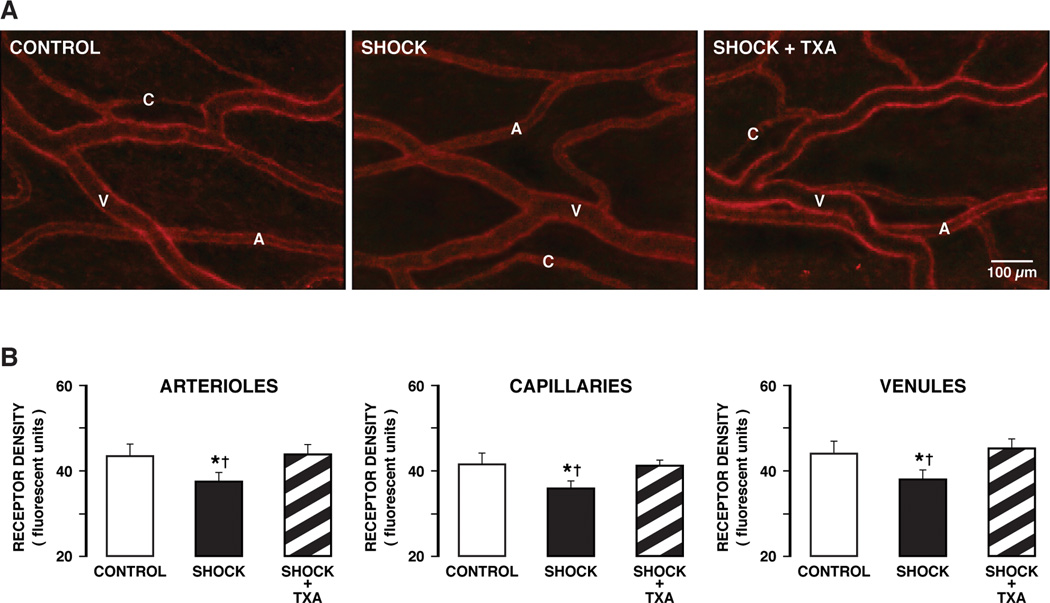
(A) Fluorescent micrographs of whole mount mesentery specimens showing the cellular distribution of the insulin receptor in control animals (CONTROLS) and 2 hours after hemorrhagic shock without (SHOCK) and with (SHOCK + TXA) pancreatic digestive enzyme inhibition. The density of the insulin receptor in control tissue shows staining uniformly along individual microvessels (A, arterioles; C, capillaries; V, venules). (B) Fluorescent intensity values of the insulin receptor label density over randomly selected arterioles, capillaries, and venules of the mesentery. The microvessel sample size was 10–15 for each vessel type (arterioles, capillaries, venules) in each group. (3 animals per group) †p< 0.05 SHOCK vs CONTROL, *p< 0.05 SHOCK vs SHOCK + TXA.
The glucose tolerance test after bolus infusion shows a dramatic increase in blood glucose vales after hemorrhagic shock as compared to values during a glucose tolerance test before hemorrhagic shock (Fig. 5, SHOCK vs CONTROL). After enteral blockade of pancreatic digestive enzymes the blood glucose values during the tolerance test are significantly reduced (Fig. 5, SHOCK + TXA + SHOCK) and not significantly different from those before shock (CONTROL vs TXA + SHOCK).
Figure 5.

Femoral artery blood glucose concentrations before shock (arrows) and following an intravenous glucose bolus infusion two hours before shock (CONTROL group) and two hours after hemorrhagic shock without (SHOCK group) and with TXA (in polyethylene glycol and electrolyte solution) treatment into the lumen of the intestine (SHOCK + TXA) to block serine protease. The SHOCK group reached blood glucose values which were above the maximum level of detection (600 mg/ml). Location of the arrow indicates control glucose values before shock. Time 0 refers to the instant after glucose bolus infusion. N = 3 rats in each group. Groups are significantly different as determined by ANOVA. *p< 0.05 SHOCK vs SHOCK + TXA or CONTROL. SHOCK + TXA group is not significantly different from the CONTROL group.
The insulin tolerance test showed that the transient reduction of the blood glucose value following an intravenous bolus injection of insulin was significantly reduced after hemorrhagic shock. But it was restored to control levels when pancreatic digestive enzymes were blocked inside the lumen of the small intestine (Fig. 6).
Figure 6.

Femoral artery blood glucose concentrations before shock (arrows) and following an intravenous insulin bolus infusion two hours before shock (CONTROL group) and two hours after hemorrhagic shock without (SHOCK group) and with TXA in polyethylene glycol and electrolyte solution treatment into the lumen of the intestine (SHOCK + TXA) to block serine protease. Time 0 refers to the instant after insulin bolus infusion. N = 3 rats in each group. Groups are significantly different as determined by ANOVA. *p< 0.05 SHOCK vs SHOCK + TXA or CONTROL. SHOCK + TXA group is not significantly different from the CONTROL group.
DISCUSSION
The current results point towards a mechanism that may underlie acute insulin resistance encountered in hemorrhagic shock. After shock digestive proteases appear in the wall of the intestine and in plasma, start to reduce the extracellular domain density of the insulin receptor and impair glucose transport, and thereby lead to a hyperglycemic and insulin-resistant state. The uptake of glucose after shock in response to a bolus of insulin in-vivo is impaired as is the spontaneous uptake of glucose by naive isolated leukocytes exposed to plasma of shock animals. Blockade of the digestive enzymes with tranexamic acid in polyethylene glycol electrolyte solution in the lumen of the small intestine attenuates breakdown of the mucosal barrier (4), the appearance of protease activity in the wall of the intestine and in plasma (5), subsequent proteolytic loss of insulin receptor binding sites and the post-shock insulin resistance.
Enteral blockade of digestive enzymes serves to attenuate the production of systemic inflammatory mediators and generation of a microvascular inflammatory cascade in the intestine and in peripheral organs (9, 13, 25). Therefore the current evidence is in line with the hypothesis that escape of the digestive enzymes during intestinal ischemia, when the mucosal permeability is increased (15), initiates an inflammatory reaction, with leukocyte adhesion to microvascular endothelium, cell apoptosis, cytokine release (13), and proteolytic breakdown of membrane components, such as epithelial adhesion molecules, mucin, TLR4 (4, 5) as well as the insulin receptor. In the presence of unchecked degrading enzymes in the plasma, and possibly also in endothelial cells and circulating leukocytes, other membrane receptors and plasma proteins may be subject to a proteolytic degradation and contribute in cell dysfunctions. Even insulin per se as a peptide may be subject to proteolytic degradation or reduced secretion, contributing to a reduced glucose transport into cells. Degrading enzymes may influence the de-novo biosynthesis of the receptor, restoration into the cell membrane and its internalization, and they may degrade the membrane glucose transporters, mechanisms that require further studies.
Furthermore, entry of digestive enzymes into the wall of the intestine serves to generate proinflammatory mediators (26), which in combination with digestive enzymes may continue to damage cells in the circulation and undermine multiple physiological functions besides the insulin resistance.
It is evident there is a need for early intervention to minimize the enzymatic breakdown of the mucosal barrier and receptor dysfunction. Ideal in this respect is blockade of digestive enzymes in the lumen of the intestine as a preventive measure, an approach that may have clinical utility in elective surgery. Our evidence in experimental models of intestinal ischemia shows, however, that delayed treatment may also be possible in spite of the progression of microvascular apoptosis in shock as long as sufficient cell functions are still preserved and it is possible to return blood pressure to control states by return of shed blood volume (9). Experimental shock studies showed that the enteral blockade of digestive enzymes with TXA in polyethylene glycol electrolyte solution in non-ischemic controls has no noticeable effect on post-surgical animal behaviors (eating, drinking, grooming, body position or mobility) or body weight with observations over several days after treatment (9).
The objective of the current study was to investigate proteolytic mechanisms responsible for reduction of extracellular binding sites of the insulin receptor and subsequent insulin resistance in hemorrhagic shock. The evidence we obtained here is in line with similar evidence for cleavage of the extracellular domain of the insulin receptor in other models with insulin resistance (12). In the spontaneously hypertensive rat, a strain with multiple co-morbidities typical for the metabolic syndrome, other membrane receptors are cleaved as well and the loss of a particular receptor is associated with a deficient activity by the receptor. For example, cleavage of the β2 adrenergic receptor attenuates the arteriolar response and is associated with central pressure dysregulation, cleavage of the vascular endothelial growth factor receptor 2 promotes apoptosis of endothelial cells and induces microvascular rarefaction with loss of capillaries, cleavage of membrane adhesion molecules causes a defective leukocyte adhesion to the endothelium, or cleavage of the FPR receptor is associated with loss of chemotactic and mechanotransduction mechanism in circulating leukocytes (29). Thus, there is a possibility that other receptors may become dysfunctional in hemorrhagic shock upon generation of uncontrolled proteolytic activity and this is associated with loss of cell function or loss of a response to drug treatment.
In this context it is interesting to note that shock is accompanied by multiple cell dysfunctions, for example a depressed vasopressor response (2), reduced eNOS-derived NO production and failure of arginine transport (24) followed after some delay by enhanced NO production from iNOS in the irreversible phase of shock, e.g. from macrophages (20). The NO production is rate limited by the transport of arginine via cationic transporters (CAT-1, -2, -4 and their isoforms) (7). Elevated permeability in post-injury lung endothelium shows signs of VE-cadherin dysfunction (14), without necessarily new gene expression of endothelial junctional proteins CD31 (PECAM-1, which regulates permeability in shock (3)) or VE-cadherin attachment (31). The symptoms are organ specific and may have in common a proteolytic breakdown of membrane components that regulate their function.
Ectodomain receptor cleavage is expected to be associated with soluble receptor fragments in the plasma. These receptor fragments tend to be in low concentrations in plasma, and in chronic forms of human Type II diabetes correlate with blood glucose levels (16). In the current experimental model in the rat we have not detected soluble receptors. In the presence of protease activity in the plasma receptor fragments and their antibody binding sites may be further degraded, preventing detection by immunochemical techniques, such as ELISA. The issue remains to be explored in patients.
Acknowledgement
Supported by an unrestricted gift from Leading Biosciences Inc., San Diego, CA and by NIH grant GM 85072.
Footnotes
Competing Interest: G.W.S.-S. is scientific advisor to Leading Biosciences Inc.. F.A.D., and G.W.S.S. own equity in InflammaGen, a company by Leading Bioscience Inc., which develops therapy for shock patients.
REFERENCES
- 1.Barroso-Aranda J, Schmid-Schönbein GW, Zweifach BW, Engler RL. Granulocytes and the no-reflow phenomenon in irreversible hemorrhagic shock. Circ Res. 1988;63:437–447. doi: 10.1161/01.res.63.2.437. [DOI] [PubMed] [Google Scholar]
- 2.Brierre S, Kumari R, Deboisblanc BP. The endocrine system during sepsis. Am J Med Sci. 2004;328:238–247. doi: 10.1097/00000441-200410000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Carrithers M, Tandon S, Canosa S, Michaud M, Graesser D, Madri JA. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am J Pathol. 2005;166:185–196. doi: 10.1016/S0002-9440(10)62243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang M, Alsaigh T, Kistler EB, Schmid-Schönbein GW. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PloS one. 2012;7:e40087. doi: 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang M, Kistler EB, Schmid-Schönbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 2012;37:297–305. doi: 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaudry IH, Sayeed MM, Baue AE. Insulin resistance and its reversal by in vivo infusion of ATP in hermorrhagic shock. Can J Physiol Pharmacol. 1976;54:736–741. doi: 10.1139/y76-102. [DOI] [PubMed] [Google Scholar]
- 7.Chen SJ, Wu CC, Yen MH. Alterations of ex vivo vascular reactivity in intraperitoneal sepsis. J Cardiovasc Pharmacol. 1994;24:786–793. doi: 10.1097/00005344-199424050-00014. [DOI] [PubMed] [Google Scholar]
- 8.Das UN. Insulin: an endogenous cardioprotector. Current opinion in critical care. 2003;9:375–383. doi: 10.1097/00075198-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 9.DeLano FA, Hoyt DB, Schmid-Schönbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Science translational medicine. 2013;5:169ra111. doi: 10.1126/scitranslmed.3005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeLano FA, Parks DA, Ruedi JM, Babior BM, Schmid-Schönbein GW. Microvascular display of xanthine oxidase and NADPH oxidase in the spontaneously hypertensive rat. Microcirculation. 2006;13:551–566. doi: 10.1080/10739680600885152. [DOI] [PubMed] [Google Scholar]
- 11.Delano FA, Schmid-Schönbein GW. Proteinase Activity and Receptor Cleavage. Mechanism for Insulin Resistance in the Spontaneously Hypertensive Rat. Hypertension. 2008 doi: 10.1161/HYPERTENSIONAHA.107.104356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeLano FA, Zhang H, Tran EE, Zhang C, Schmid-Schönbein GW. New hypothesis for insulin resistance in hypertension due to receptor cleavage. Expert Review of Endocrinology and Metabolism. 2010;5:149–158. doi: 10.1586/eem.09.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzal F, DeLano FA, Young C, Rosario HS, Schmid-Schönbein GW. Pancreatic protease inhibition during shock attenuates cell activation and peripheral inflammation. J Vasc Res. 2002;39:320–329. doi: 10.1159/000065544. [DOI] [PubMed] [Google Scholar]
- 14.Gorbunov NV, Asher LV, Ayyagari V, Atkins JL. Inflammatory leukocytes and iron turnover in experimental hemorrhagic lung trauma. Exp Mol Pathol. 2006;80:11–25. doi: 10.1016/j.yexmp.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Granger DN, McCord JM, Parks DA, Hollwarth ME. Xanthine oxidase inhibitors attenuate ischemia-induced vascular permeability changes in the cat intestine. Gastroenterology. 1986;90:80–84. doi: 10.1016/0016-5085(86)90078-8. [DOI] [PubMed] [Google Scholar]
- 16.Group TSRS. Soluble insulin receptor ectodomain is elevated in the plasma of patients with diabetes. Diabetes. 2007;56:2028–2035. doi: 10.2337/db07-0394. [DOI] [PubMed] [Google Scholar]
- 17.Ji S, Guan R, Frank SJ, Messina JL. Insulin inhibits growth hormone signaling via the growth hormone receptor/JAK2/STAT5B pathway. J Biol Chem. 1999;274:13434–13442. doi: 10.1074/jbc.274.19.13434. [DOI] [PubMed] [Google Scholar]
- 18.Jiang S, Gavrikova TA, Sharifov OF, Messina JL. Role of tissue macrophages in the development of critical illness diabetes. Shock. 2012;37:70–76. doi: 10.1097/SHK.0b013e31823180a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang S, Messina JL. Role of inhibitory kappaB kinase and c-Jun NH2-terminal kinase in the development of hepatic insulin resistance in critical illness diabetes. Am J Physiol Gastrointest Liver Physiol. 2011;301:G454–G463. doi: 10.1152/ajpgi.00148.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly E, Shah NS, Morgan NN, Watkins SC, Peitzman AB, Billiar TR. Physiologic and molecular characterization of the role of nitric oxide in hemorrhagic shock: evidence that type II nitric oxide synthase does not regulate vascular decompensation. Shock. 1997;7:157–163. doi: 10.1097/00024382-199703000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Kistler EB, Hugli TE, Schmid-Schönbein GW. The pancreas as a source of cardiovascular cell activating factors. Microcirculation. 2000;7:183–192. [PubMed] [Google Scholar]
- 22.Lang CH, Dobrescu C. In vivo insulin resistance during nonlethal hypermetabolic sepsis. Circulatory shock. 1989;28:165–178. [PubMed] [Google Scholar]
- 23.Li L, Messina JL. Acute insulin resistance following injury. Trends in endocrinology and metabolism: TEM. 2009;20:429–435. doi: 10.1016/j.tem.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendes Ribeiro AC, Brunini TM. L-Arginine transport in disease. Curr Med Chem Cardiovasc Hematol Agents. 2004;2:123–131. doi: 10.2174/1568016043477288. [DOI] [PubMed] [Google Scholar]
- 25.Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Generation of in vivo activating factors in the ischemic intestine by pancreatic enzymes. Proc Natl Acad Sci U S A. 2000;97:1772–1777. doi: 10.1073/pnas.97.4.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penn AH, Hugli TE, Schmid-Schönbein GW. Pancreatic enzymes generate cytotoxic mediators in the intestine. Shock. 2007;27:296–304. doi: 10.1097/01.shk.0000235139.20775.7f. [DOI] [PubMed] [Google Scholar]
- 27.Ren J, Wu S. A burning issue: do sepsis and systemic inflammatory response syndrome (SIRS) directly contribute to cardiac dysfunction? Front Biosci. 2006;11:15–22. doi: 10.2741/1776. [DOI] [PubMed] [Google Scholar]
- 28.Ryan NT, George BC, Egdahl DH, Egdahl RH. Chronic tissue insulin resistance following hemorrhagic shock. Ann Surg. 1974;180:402–407. doi: 10.1097/00000658-197410000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmid-Schönbein GW. An emerging role of degrading proteinases in hypertension and the metabolic syndrome: autodigestion and receptor cleavage. Current hypertension reports. 2012;14:88–96. doi: 10.1007/s11906-011-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson LH, Kim HT, Ma Y, Kokorina NA, Messina JL. Acute, muscle-type specific insulin resistance following injury. Mol Med. 2008;14:715–723. doi: 10.2119/2008-00081.Thompson. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Meurs M, Wulfert FM, Knol AJ, De Haes A, Houwertjes M, Aarts LP, Molema G. Early organ-specific endothelial activation during hemorrhagic shock and resuscitation. Shock. 2008;29:291–299. doi: 10.1097/SHK.0b013e318145a7c1. [DOI] [PubMed] [Google Scholar]
- 32.Williams VL, Martin RE, Franklin JL, Hardy RW, Messina JL. Injury-induced insulin resistance in adipose tissue. Biochem Biophys Res Commun. 2012;421:442–448. doi: 10.1016/j.bbrc.2012.03.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Kim HT, Ma Y, Zhao L, Zhai L, Kokorina N, Wang P, Messina JL. Trauma and hemorrhage-induced acute hepatic insulin resistance: dominant role of tumor necrosis factor-alpha. Endocrinology. 2008;149:2369–2382. doi: 10.1210/en.2007-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhai L, Ballinger SW, Messina JL. Role of reactive oxygen species in injury-induced insulin resistance. Mol Endocrinol. 2011;25:492–502. doi: 10.1210/me.2010-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]