

Abstract
Reactive oxygen species (ROS), a redox signal, are produced by various enzymatic reactions and chemical processes, which are essential for many physiological functions and act as second messengers. However, accumulating evidence has implicated the pathogenesis of several human diseases including neurodegenerative disorders related to increased oxidative stress. Under pathological conditions, increasing ROS production can regulate the expression of diverse inflammatory mediators during brain injury. Elevated levels of several proinflammatory factors including cytokines, peptides, pathogenic structures, and peroxidants in the central nervous system (CNS) have been detected in patients with neurodegenerative diseases such as Alzheimer's disease (AD). These proinflammatory factors act as potent stimuli in brain inflammation through upregulation of diverse inflammatory genes, including matrix metalloproteinases (MMPs), cytosolic phospholipase A2 (cPLA2), cyclooxygenase-2 (COX-2), and adhesion molecules. To date, the intracellular signaling mechanisms underlying the expression of target proteins regulated by these factors are elusive. In this review, we discuss the mechanisms underlying the intracellular signaling pathways, especially ROS, involved in the expression of several inflammatory proteins induced by proinflammatory factors in brain resident cells. Understanding redox signaling transduction mechanisms involved in the expression of target proteins and genes may provide useful therapeutic strategies for brain injury, inflammation, and neurodegenerative diseases.
1. Introduction
In general, inflammation is a protective response to various cell and tissue injuries. The purpose of this process is to destroy and remove the detrimental agents and injured tissues, thereby benefiting tissue repair. When this helpful response is uncontrolled, the effect initiates excessive cell and tissue damages that result in destruction of normal tissue and chronic inflammation [1–3]. Moreover, the brain inflammatory diseases, including Alzheimer's disease (AD) and Parkinson's disease (PD), are characterized by “redox state” imbalance and chronic inflammation, a major cause of cell damage and death. Reactive oxygen species (ROS) are widely recognized as key mediators of cell survival, proliferation, differentiation, and apoptosis [4, 5]. Excessive production of ROS (termed “oxidative stress”) by mitochondria and NADPH oxidase (Nox) is usually thought to be responsible for tissue injury associated with a range of brain injury, inflammation, and degenerative diseases such as AD [5–8]. Moreover, many of the well-known inflammatory target proteins, including matrix metalloproteinase-9 (MMP-9), cytosolic phospholipase A2 (cPLA2), cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), and adhesion molecules, are associated with oxidative stress (ROS generation) induced by proinflammatory factors such as cytokines, peptides, infections, and peroxidants [3, 5, 9]. Brain cells, especially neuroglial cells, are susceptible to the injurious effects of oxidative stress. Several studies have shown that brain cells like microglia and astrocytes induce and release diverse inflammatory mediators in response to oxidative stress [9–11]. In addition, ROS act as a critical signaling molecule to trigger inflammatory responses in central nervous systems (CNS) through the activation of the redox-sensitive transcription factors, including nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) [5, 9]. Thus, this review will focus on many general aspects of oxidative stress regulation and summarize the current progresses regarding the occurrence and effects of redox signals on CNS and their involvement in the expression of inflammatory target proteins in response to proinflammatory factors during brain inflammation. Moreover, the pharmacological interventions which protect against oxidative stress-induced neuroinflammation and neurodegenerative diseases will be discussed.
2. Role of Neuroglial Cells in CNS Physiological and Pathological Events
CNS consists of neurons and glial cells. Among glial cells, astrocytes constitute nearly 40% of the total CNS cell population in the adult human brain, and they maintain homeostasis in normal CNS. Astrocytes have also been proposed to exert a wide range of functions including guidance of the development and migration of neurons during brain development, production of growth factors, maintenance of the integrity of the blood-brain barrier (BBB), and participating in the immune and repairing responses to disease and brain injury [12, 13]. Microglial cells represent resident brain macrophages and can be transformed into activated immunocompetent antigen-presenting cells during the pathological process. An increased number of activated microglial cells have consistently been reported in PD, which may have a deleterious effect on dopaminergic neurons [14]. Astrocytes, as well as microglia, display an array of receptors involved in innate immunity, including Toll-like receptors (TLRs), nucleotide-binding oligomerization domains, double-stranded RNA dependent protein kinase, mannose receptor, and components of the complement system [10]. One common feature of a variety of neurodegenerative disorders is the presence of a large number of activated glial cells including astrocytes and microglia that involve the changes of morphology and expression of many inflammation-related proteins. Gliosis, especially astrogliosis, is characterized by astrocytic proliferation, extensive hypertrophy of the cell body, and functional changes, when stimulated with various factors including lipopolysaccharide (LPS), interleukin-1β (IL-1β), and tumor necrosis factor-(TNF-α) [15, 16].
Moreover, the cell-cell interactions between glial cells and neurons may be important in the regulation of brain inflammation and neurodegeneration. Many recent reports implicate that inflammation contributes to a wide variety of brain pathologies, apparently killing neurons via glia [10, 11, 17]. Thus, the activated glial cells, especially microglia and astrocytes, are thought to play a critical role in the pathogenesis and progression of neurodegeneration (Figure 1). Previously, many reports have shown that microglial cells may be a major inflammatory cell of the brain [14]. The activated microglia produce several inflammatory mediators including COX-2/prostaglandins (PGs), iNOS/nitric oxide (NO), or cytokines as well as neurotoxic substances, which are thought to be responsible for brain injuries and diseases including trauma, AD, and neural death due to the exposure of LPS, interferon-γ, or β-amyloid [18, 19]. Although most studies have demonstrated that microglial cells play an important role in neuroinflammation and neurodegeneration, accumulating evidence has also demonstrated the characteristic changes of astrocytes in neurodegenerative diseases such as dementia [10, 11, 20]. Recently, we have demonstrated the upregulation of several inflammatory mediators including MMP-9, cPLA2, COX-2, iNOS, and oxidative stress by various proinflammatory factors such as cytokines (e.g., IL-1β), peptides (e.g., bradykinin (BK) or endothelin-1 (ET-1)), infections (e.g., bacteria or virus), and peroxidants (e.g., oxidized low-density lipoprotein (oxLDL)) in rat brain astrocytes [21–29]. More recent data indicated that multiple factors including ROS, MMP-9, and heme oxygenase-1 (HO-1)/carbon monoxide (CO) from BK-challenged brain astrocytes may contribute to the neuronal cell apoptosis [30]. Together these results implicate that activated neuroglial cells, especially astrocytes, play a key role in the pathogenesis of the CNS inflammation leading to neurodegenerative diseases (Figure 1).
Figure 1.
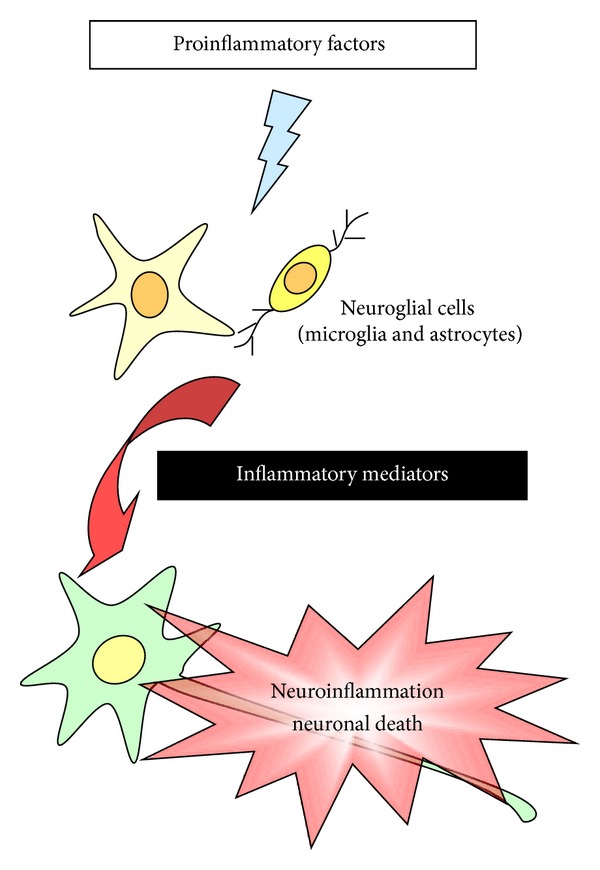
Schematic presentation of the interaction of the brain cells, including neurons and glial cells. In the central nervous system (CNS), proinflammatory factors induce the expression of various inflammatory mediators in neuroglial cells, particularly microglia and astrocytes. These induced inflammatory mediators from glial cells may cause the neuroinflammation or neuronal death, and then leading to neurodegenerative disorders.
3. Role of Oxidative Stress (Redox Signaling) in the Brain Inflammation and Neurodegenerative Diseases
In CNS inflammation, various proinflammatory factors may cause the development of an oxidative stress and antioxidants imbalance, which induces redox signal-dependent expression of genes for inflammatory mediators or protective antioxidants (Figure 2). The oxidative stress (i.e., ROS and reactive nitrogen species (RNS)) is produced by various enzymatic reactions and chemical processes or directly inhaled. ROS that are particularly responsible in oxidative stress include superoxide anion (O2 ∙−), hydrogen peroxide (H2O2), and hydroxyl radical (∙OH). Furthermore, the RNS include nitric oxide (NO) and peroxynitrite (ONOO−). These oxidative stresses (i.e., ROS/RNS) are essential for many physiological functions at low concentrations [2–6] and killing invading microorganisms [31]. However, several lines of evidence have suggested that the pathogenesis of human diseases is attributed to increased oxidative stress [2, 31]. Moreover, oxidative stress has been shown to mediate the pathogenesis of neurodegenerative diseases, including PD [6], AD [32], and cerebrovascular disorders such as stroke [31]. There are several major sources of ROS/RNS generation in the cells, including Nox, Xanthine oxidase (Xox), P450 enzymes, COX, and NOS (Figure 2), which contribute to several physiological and pathological functions including brain inflammation and neurodegeneration [8]. The physiological role of ROS/RNS (along with O2 ∙− and NO) also extends to the control of vascular tone in the brain, which is tightly modulated by the metabolic activity within neurons [6, 33]. Particularly in the brain, even small redox imbalances can be deleterious. Recently, accumulating evidence attributes the cellular damage in the CNS degenerative disorders to oxidative stress [5–9], suggesting that oxidative stress is an early event in AD [32]. Oxidative stress may be responsible for brain inflammatory disorders, which cause deleterious effects during CNS pathogenesis [34]. Furthermore, several reports have shown that ROS levels are increased with age in several major organs including brain [32]. Abnormally elevated ROS is implicated in age-related long-term potentiation (LTP) impairment [35]. ROS further induce expression and activation of proinflammatory factors or inflammatory mediators during brain injury and inflammation. Under various pathological conditions, excessive amounts of ROS can damage DNA, lipids, proteins, and carbohydrates leading to impairing cellular functions and enhancing inflammatory reactions [34, 36]. In brains of AD patients, cellular and animal models of AD, the elevated levels of these oxidative stress-modified molecules are also detected [32]. Recently, increasing evidence attributes the cellular damage in neurodegenerative disorders such as AD and PD to oxidative stress that leads to generation of ROS associated with brain inflammatory disorders [2, 6]. Thus, these results indicate that oxidative stress (i.e., ROS production) plays an important role in CNS inflammation and neurodegenerative disorders (Figure 4).
Figure 2.
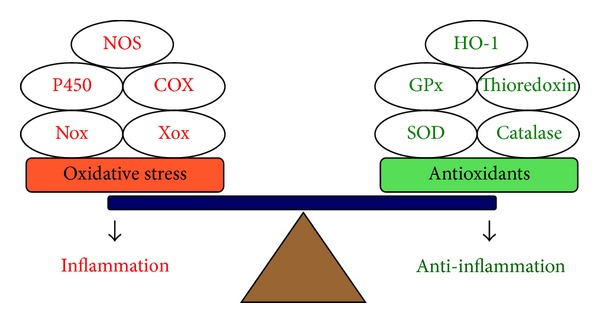
Oxidative stress and antioxidants imbalance in inflammation. In inflammation, the balance appears to be tipped in favor of increased oxidative stress by various specialized enzymes, including Nox, Xox, P450, COX, or NOS, either because of excessive ROS release or inflammatory mediators leading to the amplification of the proinflammatory effects. In contrast, induction of several antioxidants, such as SOD, catalase, GPx, thioredoxin, or HO-1, may reduce ROS generation and attenuate the inflammatory response (anti-inflammation). Nox: NADPH oxidase; Xox: Xanthine oxidase; P450: P450 enzyme; COX: cyclooxygenase; NOS: nitric oxide synthase; SOD: superoxide dismutase; GPx: glutathione peroxidase; HO-1: heme oxygenase-1.
Figure 4.
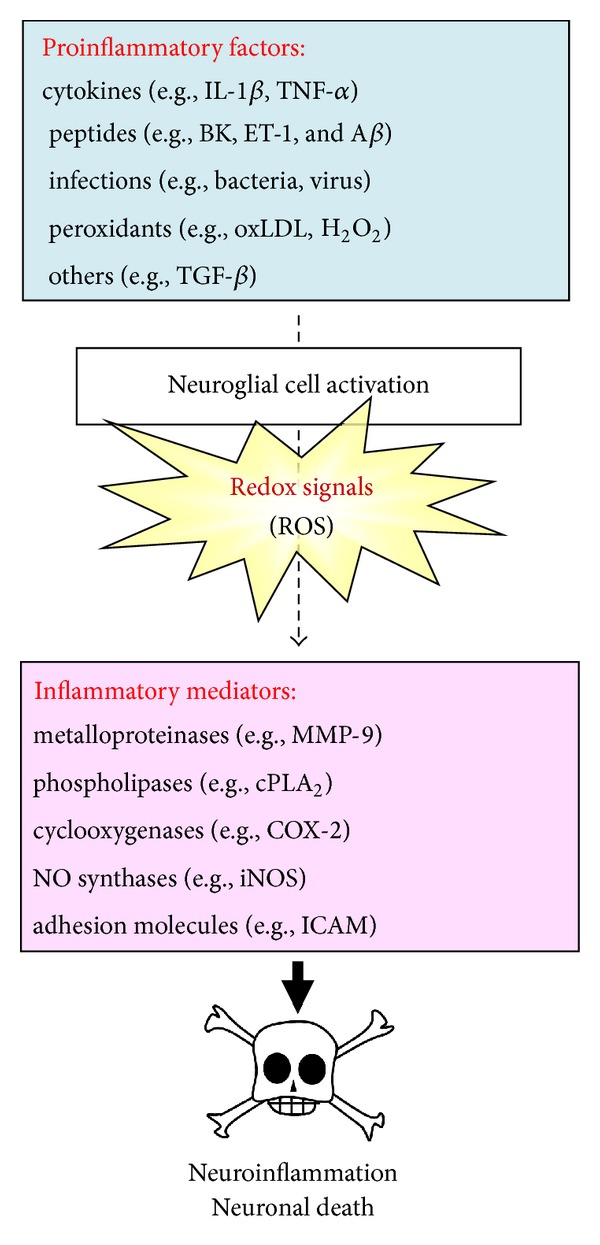
Schematic representation of the redox signals (ROS production) and their role in the development of neuroinflammation and neuronal death. Many of the well-known inflammatory target proteins, such as MMP-9, ICAM-1, VCAM-1, COX-2, and cPLA2, can be upregulated by various proinflammatory factors, including cytokines, peptides, bacterial or viral infection, peroxidants, via a ROS signal-dependent manner in neuroglial cells. These inflammatory mediators can cause neuroinflammation and neuronal death. IL-1β: interleukin-1β; TNF-α: tumor necrosis factor-α; BK: bradykinin; ET-1: endothelin-1; Aβ: β-amyloid; oxLDL: oxidized low-density lipoprotein; H2O2: hydrogen peroxide; TGF-β: transforming growth factor-β; MMP-9: matrix metalloproteinase-9; cPLA2: cytosolic phospholipase A2; COX-2: cyclooxygenase-2; iNOS: inducible nitric oxide synthase; ICAM: intercellular adhesion.
Oxidative stress activates several intracellular signaling cascades that may have a deleterious effect on the cellular homeostasis. The molecular mechanisms associated with ROS production (e.g., mitochondrial dysfunction and Nox activation) and its influences have been investigated in various models of chronic inflammation and neurodegenerative disorders [9]. Recently, there are extensive pieces of literature supporting a role of mitochondrial dysfunction and oxidative damage in the pathogenesis of AD [5, 37], and ROS are associated with neuroinflammatory and neurodegenerative processes [9, 17, 32]. Several proinflammatory factors (e.g., LPS and BK) have been shown to induce the expression and activation of various inflammatory mediators via a ROS-dependent manner in brain cells [25, 36]. In microglial cells, ROS, as a major signaling molecule, mediate microglial activation induced by proinflammatory mediators such as Aβ or LPS [38, 39]. However, the roles of oxidative stress that contribute to these events are not well characterized in brain cells including astrocytes. Our recent reports have demonstrated that ROS signals contribute to the expression of many inflammatory genes (e.g., MMP-9) by several proinflammatory factors, including BK [25], LTA [27], and TGF-β1 [40] in brain astrocytes. More recent result indicates that ROS generation from BK-challenged astrocytes contributes to neuronal apoptosis through a caspase-3-dependent manner [30]. Although oxidative stress is implicated as a causative factor in neurodegenerative disorders, the signaling pathways linking ROS production with neuronal cell death are not well characterized [6]. Hence, there are several targets and signals that need to be identified and explored for the development of therapeutic strategies in the future.
4. Redox Signaling and Proinflammatory Factors in Brain Inflammation and Neurodegenerative Diseases
The senile and neuritic plaque of AD are accompanied by inflammatory responses in activated glial cells (i.e., astrocytes and microglia). In CNS, several cytokines and inflammatory mediators produced by activated glia have the potential to initiate or exacerbate the progression of neuropathology [41]. Moreover, traumatic injury to CNS results in the production of inflammatory cytokines via intrinsic (brain cells) and extrinsic means (by infiltrating macrophages and other leukocytes). The expression of many inflammatory mediators including cytokines, MMPs, cPLA2, COX-2, and iNOS has been shown to be regulated by various extracellular stimuli such as proinflammatory cytokines (e.g., IL-1β and TNF-α), peptides (e.g., BK, ET-1, and Aβ), infections (e.g., bacteria and virus), peroxidants (e.g., oxLDL and H2O2), and other stresses (e.g., TGF-β) in neuronal and neuroglial cells [4–9, 42] (Figure 4).
4.1. Cytokines
IL-1β and TNF-α are two of the inflammatory cytokines significantly elevated in neurodegenerative diseases such as AD, and they play a central role in initiating and regulating the cytokine cascades during inflammatory responses [43]. IL-1β is a pleiotropic cytokine and classified as a dominant injury biomarker. Furthermore, several studies have shown that the level of IL-1β is elevated in the cerebrospinal fluid (CSF) of patients with AD, traumatic brain injury [44], and stroke [45]. Thus, IL-1β plays an important role in both acute and chronic neurodegenerative diseases. The effects of IL-1β on ROS generation have been reported to be associated with brain inflammatory disorders, cancers, and myocardial remodeling [46, 47]. ROS generation by IL-1β leads to the expression of several inflammatory genes like MMP-9 which may increase BBB permeability, recruit immune cells infiltrating through BBB into the tissues, and subsequently result in brain inflammation and edema during brain injury [6, 34]. ROS may also act as an inflammatory signaling factor mediated microglial activation induced by IL-1β [39]. Moreover, in culture of glia/neuron, IL-1β induces neurotoxicity through the release of free radicals [48]. In addition, TNF-α is also produced in response to oxidative stress and Aβ. In brain, TNF-α is produced by microglia and its overproduction has been linked with neuronal cell death [49]. These studies indicate that cytokines, especially IL-1β and TNF-α, contribute to the CNS inflammation and neurodegenerative diseases through redox signalings.
4.2. Peptides
AD is defined by progressive impairments in memory and cognition and by the presence of extracellular neuritic plaques (Aβ) and intracellular neurofibrillary tangles (tau protein) [5, 32]. Among these molecules, Aβ is an insoluble fibrous protein and aggregates sharing specific structural traits. It arises from at least 18 inappropriately folded versions of proteins and polypeptides present naturally in the body. The misfolded structures alter their proper configuration such that they erroneously interact with other cell components forming insoluble fibrils. Aβ has been associated with the pathology of more than 20 human diseases including AD. Abnormal accumulation of amyloid fibrils in brain may play a role in neurodegenerative disorders. Although Aβ peptide is neurotoxic species implicated in the pathogenesis of AD, mechanisms through which intracellular Aβ impairs cellular properties and produces neuronal dysfunction remain unclear. Accumulating evidence has indicated that Aβ can stimulate the production of free radicals [50]. Interestingly, intracellular Aβ is present in mitochondria from brains of transgenic mice with targeted neuronal overexpression of mutant human amyloid precursor protein and AD patients. Importantly, mitochondria-associated Aβ, principally Aβ 1–42, was detected as early as 4 months, before extensive extracellular Aβ deposits [51]. Moreover, activation of Nox by Aβ 1–42 results in ROS production in rat primary culture of microglial cells [52]. In mouse models of plaque formation, oxidative stress occurs prior to Aβ deposition in a Tg2576 APP transgenic mice [53]. Moreover, increased levels of oxidative damage occur in individuals with mild cognitive impairment (MCI), which is often believed to be one of the earliest stages of AD [54]. Additionally, glial HO-1 expression in the MCI temporal cortex and hippocampus is also significantly greater than that of the nondemented group [55]. These results support Aβ-induced redox signaling serving as an early event that leads to the development of the CNS pathological features such as AD. Moreover, glial cells may play a key role in the events.
In addition to Aβ peptide, BK and related peptides are produced and released during trauma, stroke, and neurogenic inflammation [56]. All these pathological processes may be involved in tissue remodeling, which were regulated by MMPs. Moreover, astrocytes possess receptors for numerous transmitters such as glutamate and BK [57]. These peptides mediate several inflammatory responses including increasing vasodilatation and vascular permeability, promotion of fluid secretion and ion transport, and eliciting itching and pain at the sites exposed to noxious stimuli. Thus, the elevated level of BK plays a key role in the initiation of inflammatory responses in target tissues, including CNS. It is well established that BK interacts with two BK receptor subtypes, including BK B1 and B2 [58]. Astrocytes are known to express B2-type BK receptors and this type of receptors is found only on astrocytes type 1 [57]. The B2 BK receptor is a heterotrimeric G-protein-coupled receptor (GPCR) that can be coupled to intracellular signaling molecules via interaction with Gq protein [59]. Activation of BK receptors stimulates intracellular signaling molecules, including Ca2+, PKCs, and MAPKs, in several cell types including astrocytes [57–59]. Activation of these signaling pathways may lead to cell survival, proliferation, differentiation, and the expression of several inflammatory genes such as iNOS and MMP-9 [36, 60]. During brain injury, BK has been shown to induce the expression of several inflammatory genes by increasing ROS production [6, 34]. Moreover, Nox is expressed in astrocytes and contributes to ROS generation [61, 62]. In brain astrocytes, BK induces the expression of several inflammatory genes like MMP-9 by ROS-dependent signaling pathways [25]. Moreover, ROS released from BK-challenged brain astrocytes cause neuronal cell apoptosis [30]. These pieces of literature suggest that BK plays an important role in brain inflammation and neurodegenerative disorders.
Endothelial cells are known to produce vasotone mediators such as endothelins (ETs) and NO to maintain hemodynamic responses. The ETs are 21-amino acid vasoconstricting peptides produced primarily in the endothelium, which play a key role in vascular homeostasis and have been implicated in brain inflammatory diseases. Among the ET family, the bioactivity of ET-1 is mediated through potent vasoconstrictor and proinflammatory action in vascular diseases, including the heart, circulation system, and brain [63–66]. Two types of ET receptors, ET type A (ETA) and type B (ETB), are responsible for ET-1-triggered biological effects, which are mediated via G-protein-dependent processes [63–65]. In CNS, ET-1 also plays a substantial role in the normal development and CNS diseases. Both endothelial cells and astrocytes are potential sources of ET-1 release in response to hypoxic/ischemic injury of the brain [66]. On astrocytes, the ETB receptors are predominantly expressed and modulate postinjury responses of astrocytes in CNS [67]. Circumstantial evidence has further demonstrated that overexpression of ET-1 has deleterious effects on astrocytes in ischemic brain [68]. Similarly, ET-1 causes hypertrophy of ETB/GFAP-immunoreactive astrocytes, a typical characteristic of astrogliosis, in the normal optic nerve, leading to glial scar formation following CNS injury [68]. Endothelial ET-1 induces cytokine production such as IL-1β released by astrocytes, which directly contributes to BBB breakdown during CNS inflammation [69]. These findings further imply the involvement of ET-1 in the CNS inflammation and diseases.
4.3. Infections
Bacterial infections have been shown to be involved in brain inflammation [70]. A well-known endotoxin from Gram-negative bacteria, LPS, regulates the expression of inflammatory proteins associated with inflammatory diseases. Many studies have also shown that ROS are the major signaling molecule which mediates microglial activation induced by inflammatory mediators, including LPS [71]. However, the signaling mechanisms of which activated brain cells in response to Gram-positive bacterial infection remain undefined. Gram-positive bacterial infections of CNS occur in bacterial meningitis and brain abscess, being localized to the membranes surrounding the brain and in its parenchyma [72]. Lipoteichoic acid (LTA), an amphiphilic polymer, is embedded in-cell wall of Gram-positive bacteria [73]. The Gram-positive bacterium Streptococcus pneumoniae is the most common cause of acute bacterial meningitis worldwide [74], revealing a close relationship between LTA challenges and CNS diseases. For the initiation of LTA signaling, TLRs are believed to be responsible for LTA recognition challenged by Gram-positive bacteria such as Staphylococcus aureus and Streptococcus pneumoniae [75]. Upon binding to TLR heterodimers (i.e., TLR2/TLR1 or TLR2/TLR6 complex), LTA exerts a sequential activation of members of IL-1 receptor-associated kinase (IRAK) family and tumor necrosis factor receptor-associated factor 6 (TRAF6), mediated by a TLR adaptor protein MyD88. Ultimately, TLR signalings activate MAPK family and NF-κB, leading to modulation of gene expression of cytokines and other inflammatory proteins [76]. Among the diverse cell types in CNS, glial cells such as astrocytes and microglia are regarded as targets in Gram-positive bacterial infection [77–79]. Several lines of evidence suggest a causal relationship between LTA challenges and the CNS diseases, which involves glial activation and TLR2 signalings [77–79]. TLR signalings in astrocytes have been shown to be involved in inflammatory responses in CNS [80], accompanied with upregulation of genes with inflammatory and proapoptotic effects [81]. The pathogenic progression involves glial activation and TLR2 signalings stimulated by LTA, which are linked to inflammatory neurodegeneration [82]. Additionally, LTA exhibits detrimental effects on brain cellular functions, including induction of apoptosis, production of oxidative stresses, and disruption of BBB following group B Streptococcus or Staphylococcus aureus challenge in CNS [82]. Although the effects of LTA on ROS generation have been reported in several cell types such as renal diseases [83], LTA-induced brain cell responses through the ROS signals are not well characterized. Recent report indicates that LTA-induced MMP-9 expression is mediated through Nox2-derived ROS generation in brain astrocytes [27]. These data suggest that targeting LTA and its specific signaling components could yield useful therapeutic targets for CNS inflammatory diseases upon infection with Gram-positive bacteria.
Moreover, increasing evidence has shown that viral infections such as Japanese encephalitis virus (JEV) and Enterovirus 71 (EV71) may contribute to several inflammatory responses in CNS [28]. Neurotropic viruses can cause massive neuronal dysfunction and destruction that lead to neurological diseases. EV71, a single-positive-stranded RNA virus, belongs to the Enterovirus B genus of the Picornaviridae family [84]. EV71 and Coxsackievirus A16 (CVA16) are the major causative agents of hand-foot-and-mouth disease (HFMD) that is usually mild exanthematous infection and self-limiting in the young children. However, EV71, but not CVA16, can progress to severe neurological diseases including fatal encephalitis, aseptic meningitis, and fatal neurogenic pulmonary edema [85]. Children under 5 years old of age group are susceptible to these infections and may develop permanent neurological sequelae or even succumb to such disorders [86]. In 1998, an EV71 outbreak infected more than 130,000 children resulted in 78 fatalities. Since then, EV71 infection has recurred every year in Taiwan and EV71 outbreaks have been periodically reported throughout the world, representing a major public health concern particularly in the Asia-Pacific regions including Taiwan, Malaysia, Singapore, Japan, and China [85, 87]. The emerging evidence suggests that ROS affect the interaction between host and viral pathogens. Recently, EV71 has been shown to induce oxidative stress-dependent viral replication in human neuroblastoma SK-N-SH cell line [88]. Similarly, JEV is a single-stranded, positive-sense RNA virus belonging to the family Flaviviridae. JEV is transmitted between animals and humans by culex mosquitoes [89]. After the bite of an infected mosquito, JEV amplifies peripherally producing transient viremia before entering into CNS [89]. The principal target cells for JEV are localized in CNS, including neurons and astrocytes [90]. Several lines of evidence suggest that JEV frequently causes severe encephalitis in the world, especially in Eastern and Southeastern Asia. The infection with JEV is characterized by clinical manifesting with fever, headache, vomiting, signs of meningeal irritation, and altered consciousness leading to high mortality [89, 90]. The generation of ROS plays an important role in diverse cellular functions including signal transduction, oxygen sensing and host defense during infection by viruses such as JEV [91]. In CNS, JEV infection has been shown to upregulate MMP-9 gene expression through ROS-dependent pathways in brain astrocytes [28]. These findings concerning JEV-induced expression of inflammatory genes in brain astrocytes imply that JEV might play a critical role in the brain inflammation and neurodegenerative diseases.
4.4. Peroxidants
Oxidative stress may cause production of several peroxidants such as oxidized lipoprotein. Clinical reports reveal that the patients with AD exhibit an increased oxidation of lipoproteins potentially toxic to neurons in CNS [92]. Among these, the oxidized low-density lipoprotein (oxLDL) is a well-known predominantly risk factor of atherosclerosis, which has been reported to participate in the progression of the CNS diseases. In CNS, oxLDL exhibits detrimental effects on brain cell functions, including induction of apoptosis, disruption of capillary homeostasis, and alteration of inflammatory protein activity in various brain cells [93]. Furthermore, in patients with cerebral infarction, oxLDL is present in brain parenchyma and stimulates astrocytes to secrete interleukin-6 [94] and may serve as an indicator to reflect the level of oxidative stress [95]. In brain astrocytes, oxLDL can induce MMP-9 expression and cell migration, which plays a critical role in the progression of inflammatory diseases and remodeling processes in target tissues, including CNS [29, 96]. These findings suggest that peroxidants like oxLDL might play a key role in the progression of the CNS diseases and also that targeting these peroxidants-stimulated signaling components may provide useful therapeutic strategies for brain inflammation and neurodegenerative diseases.
4.5. Others
In addition to these well-known factors, there are many factors that may also contribute to neuroinflammatory responses. Among these, TGF-β has been implicated to participate in the responses. TGF-β binds to two serine/threonine kinase receptors which consist of TGF-βRI and TGF-βRII. During ligand binding, TGF-βRII phosphorylates TGF-βRI and activates Smad-dependent intracellular signaling pathways and thus leads to expression of several genes [97, 98]. In addition to activation of Smad-dependent pathways, TGF-β can affect several signal transduction pathways in a Smad-independent manner, such as MAPKs [97, 98]. In human gingival and skin fibroblasts, both p38 MAPK and Smad3 cooperate in regulating TGF-β-induced MMP-13 expression, whereas ERK1/2 cooperates with Smad3 in regulating connective tissue growth factor expression [99]. Recently, increasing evidence has attributed the cellular damage in neurodegenerative disorders to oxidative stress leading to generation of ROS that are responsible for brain inflammation and neurodegenerative disorders [6, 34]. TGF-β can stimulate ROS production, which participates in the expression of diverse inflammatory genes such as MMPs in the processes of several human inflammatory diseases [100]. In brain astrocytes, TGF-β1 has been shown to induce inflammatory protein expression via a ROS-dependent manner [40]. These results suggest that TGF-β1 may play a key role in the process of brain inflammation and neurodegenerative diseases.
5. Role of Redox Signaling in the Regulation of Inflammatory Mediators
Neuroinflammation is an active defensive process against diverse insults, metabolic and traumatic injuries, infection, and neurodegenerative diseases. Although neuroinflammation serves as a neuroprotective mechanism associated with repair and recovery, it can also cause brain damage [101]. However, if inflammation in the brain is chronic or inappropriately controlled, it may become detrimental to neurons, thus representing one of the various pathological insults induced by various proinflammatory factors and by inflammatory mediators in CNS [101]. Experimental and clinical studies have shown that various inflammatory mediators are present in brain, CSF, and blood in brain injury. In particular, the histological analysis of human brain from individuals with brain disorder such as AD or epilepsy of various etiologies strongly suggests the existence of a chronic inflammatory state in the brain almost invariably associated with neuronal loss or reactive gliosis [102]. In experimental models of rodent brain seizures, a variety of inflammatory mediator mRNAs and protein levels are rapidly increased after the induction of seizures, including MMPs (e.g., MMP-9, especially), multiple forms of PLA2 (e.g., cPLA2), COX-2, NOS (e.g., iNOS), and adhesion molecules (e.g., ICAM-1 and VCAM-1) [102, 103]. After expression of these inflammatory mediators, several CNS damaging factors will be produced such as cytokines shedding by MMPs, arachidonic acid (AA)/PGE2 releasing by cPLA2/COX-2 system, and NO generation by NOS [102, 103]. Herein, we reviewed the role and mechanism of these inflammatory mediators in the brain inflammation and neurodegeneration and whether oxidative stress plays a crucial role in these events.
5.1. Matrix Metalloproteinases
MMPs are a large family of zinc-dependent endopeptidases, which play an important role in the turnover of extracellular matrix (ECM) and pathophysiological processes [104]. To date, 24 MMPs have been identified in mammals. Among these MMPs, some are membrane-type MMPs which are anchored to the cell surface and others are secreted into the extracellular space. In general, MMPs are released as inactive proform MMPs and activated by proteolytic cleavage of the N-terminal domain. In gelatinase subfamily of MMPs (i.e., MMP-2 and MMP-9), the catalytic domain that contains the Zn2+ binding site and repeats of fibronectin motifs allowing the ability to bind their major substrate gelatin. MMP-9 (gelatinase B; 92 kDa) is usually low and its expression can be induced by various proinflammatory factors such as cytokines. The other class of gelatinase, MMP-2 (gelatinase A; 72 kDa), is constitutively expressed in several cell types and usually not inducible. In CNS, MMPs, especially, MMP-9 are implicated in several important physiological events, including morphogenesis, wounding healing, and neurite outgrowth [105]. Moreover, upregulation of MMP-9 may contribute to the pathogenesis of several CNS diseases such as stroke, AD, multiple sclerosis, and malignant glioma [105]. Several proinflammatory factors including cytokines, endotoxins, and oxidative stress have been shown to upregulate MMP-9 in astrocytes in vitro [106, 107], implying that MMP-9 activity may be regulated by diverse factors in CNS during neuroinflammation. Moreover, many proinflammatory mediators like cytokines and BK induce the expression of MMP-9 during brain injury by increasing ROS production [25, 62]. Recently, upregulated MMP-9 and ROS generation from brain astrocytes have been reported to contribute to neuronal cell death in vitro [30]. These studies suggest that upregulation and activation of MMP-9 by proinflammatory factors are mediated through oxidative stress (ROS production) during brain injury and inflammation (Figure 4). Therefore, the inhibition of MMP-9-mediated inflammatory pathways may provide therapeutic strategies to brain inflammation and neurodegenerative diseases.
5.2. Cytosolic Phospholipase A2
There are three forms of phospholipase A2 (PLA2) superfamily including the secretory PLA2, type IV PLA2, also known as cPLA2, and calcium-independent PLA2 in mammalian cells [108–110]. The secretary PLA2 (sPLA2) is expressed in a variety of cell types and it has no preference for AA at sn-2 position, requires millimolar amounts of Ca2+ for activity and is sensitive to sulfhydryl reducing agents, such as dithiothreitol (DTT), and is resistant to heat or acid conditions [109]. The calcium-independent PLA2 (iPLA2) does not require Ca2+ for catalytic activity. The iPLA2 prefers plasmalogen substrates and does not appear to have a preference for the type of fatty acid at the sn-2 position. The third class is the novel and high molecular weight (85 kDa) cPLA2. The cPLA2 catalyzes the hydrolysis of the sn-2 position of membrane glycerophospholipids, leading to production of free fatty acids and lysophospholipids. This reaction is of particular importance if the esterified fatty acid is AA, which is converted by downstream metabolic enzymes to various bioactive lipophilic compounds called eicosanoids, including PGs and leukotrienes (LTs) [110]. PLA2 could be the initial and rate-limiting enzyme in this conversion. The increase in cPLA2 activation and expression following external stimuli, including proinflammatory cytokines, growth factors, and microbial toxin, is often observed in several systems [111]. Among these enzymes, cPLA2 is the only one that plays a key role in mediating agonist-induced AA release for eicosanoid production in various cell types [112]. Several studies have indicated that cPLA2 is constitutively expressed in the cytosol of most resting brain cells and tissues. In brain, cPLA2 has been shown to co-localize with glial fibrillary acidic protein (GFAP), a principal marker for brain astrocytes [113]. Moreover, under brain inflammatory and neurodegenerative conditions such as AD, there is an increase in immunoreactivity to cPLA2 in astrocytes from the cortex of patients [114, 115]. A variety of proinflammatory factors including IL-1β, TNF-α, or BK may exert as modulators of cPLA2 activity and/or expression in various cell types including astrocytes [23, 111]. Upregulation and activation of cPLA2 leading to PGE2 production have been implicated in a number of neurodegenerative diseases [111, 114, 115]. Recently, PGE2 production and cPLA2 activation have also been shown to regulate the CREB-dependent iNOS expression in microglia [116] or cPLA2 expression in amnion fibroblasts [117]. However, a series of highly reactive PGs, free fatty acids, lysophospolipids, eicosanoids, platelet-activating factor, and ROS, all generated by enhanced PLA2 activity and AA release, participate in cellular injury, particularly in neurodegeneration [118]. Thus, cPLA2 seems to function as a crucial upstream regulator of the production of eicosanoids during brain inflammation and is correlated to the process of neurodegenerative diseases (Figure 4). The inhibition of cPLA2-mediated pathways may provide a therapeutic strategy to brain inflammation and neurodegenerative diseases.
5.3. Cyclooxygenase-2
COX, known as a prostaglandin-endoperoxide synthase, is a rate-limiting key enzyme in the synthesis of PGs. In this process, PLA2 catalyzes the release of AA from membrane phospholipids, while COX catalyzes the conversion of AA into PGs [119]. Significant advances have been made in understanding the role of COX in certain biologic processes, including inflammation, angiogenesis, development, and several homeostasis [119]. COX exists in two isoforms: COX-1, which is expressed constitutively under normal conditions in most tissues, mediates regulating normal physiological responses, and controls renal homeostasis, and the inducible COX-2, is not detectable in most normal tissues or resting cells, but its expression can be induced rapidly by a variety of stimuli including cytokines, bacterial or viral infections, and other mediators to produce PGs during inflammation [120]. In addition, COX-2 gene promoter which contains multiple regulatory elements has been shown to be regulated by different transcription factors, including NF-κB, AP-1, and cyclic AMP-response element binding protein (CREB) in various cell types [121]. Previous studies showed that COX-2 immunoreactivity is a characteristic finding in the synovial macrophage of patients with arthritis as well as in other forms of inflammation. Moreover, several lines of evidence have confirmed COX-2 as a major therapeutic target for the treatment of inflammatory disorders such as arthritis [119, 122]. Recently, the mice with homozygous deletion of the COX-2 gene suppress endotoxin-induced inflammation [123]. In brain, expression of COX-2 leads to increased production of prostanoids which are potent inflammatory mediators, and upregulated COX-2 expression has been reported in neurodegenerative disorders [124]. Moreover, upregulation of COX-2 and PGE2 release by viral infection such as EV71 have been reported in brain astrocytes and human neuroblastoma cells via diverse signaling pathways [125, 126]. Upregulation of COX-2/PGE2 by ET-1 via MAPK-dependent NF-κB pathway in brain microvascular endothelial cells [127]. A recent report also indicates that the ROS-induced COX-2 expression can be found in ALS [128]. However, the expression of COX-2 appears to be strongly induced and activated during AD, indicating the importance of inflammatory gene pathways as a response to brain injury [118]. Thus, COX-2 may play an important role in the development of brain inflammation and neurodegenerative diseases.
5.4. Nitric Oxide Synthase
NO is a free radical that displays diverse bioactivity in various organ systems, including CNS. Depending on the concentration, excess NO levels are implicated in the pathogenesis of CNS diseases including ischemia, trauma, neuroinflammatory, and neurodegenerative diseases [129–131]. Production of NO from L-arginine is catalyzed by NOS. The level of iNOS in healthy brain is undetectable. Accumulating evidence supports the role of iNOS in the pathogenesis of CNS disorders. In CNS, upregulation of iNOS in various cell types, including astrocytes and microglia, is proposed to be the leading source of NO production during neuroinflammation [132]. Furthermore, knockout strategies of iNOS gene protect against focal cerebral ischemia and LPS challenges [133, 134]. iNOS is induced by a variety of stimuli, such as viral and bacterial infections, cytokines, cell-cell contact, and neurotoxins [131]. The consequent product NO reacts with superoxide to form peroxynitrite (ONOO−), the most toxic derivative of NO (Figure 3). As for the involvement of NO derivatives in neuropathology, many studies have revealed that the reference of iNOS/NO/ONOO− plays an important role in neurodegenerative disorders [131]. However, following inflammatory insults, reactive astrocytes express iNOS, which causes the neuronal damage associated with cerebral ischemia and/or demyelinating diseases [132]. In CNS, appearance of iNOS in astrocytes is related to several neurodegenerative diseases such as ALS [130] and multiple sclerosis (MS) [129]. These findings imply that astrocytes are the leading regulators in neurodegenerative diseases. Moreover, activation of astrocytes has been reported to involve in the expression of inflammatory genes. It has been well established that the regulation of iNOS expression is mediated via tyrosine kinases such as JAK, MAPKs, ROS, and various transcription factors including STAT-1, NF-κB, and AP-1 in astrocytes [131]. Increasing evidence suggests that activation of signal transduction pathways like c-Src, PI3K/Akt, and MAPK cascades contributes to activation of astrocytes and microglia, leading to expression of inflammatory proteins and advanced damage in neurodegenerative diseases [25, 26, 135].
Figure 3.
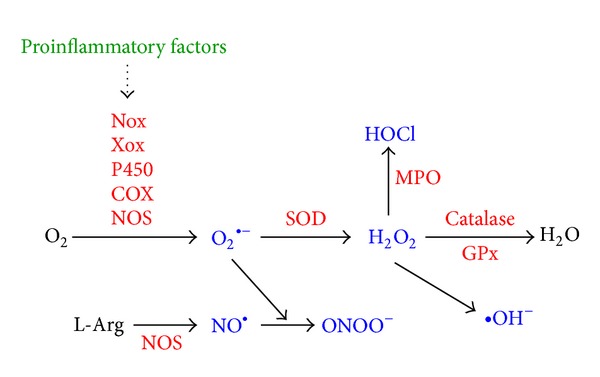
Major pathways of reactive oxygen (nitrogen) species generation and metabolism. Several proinflammatory factors can stimulate O2 ∙− generation through activation of several specialized enzymes, such as the Nox, Xox, P450, COX, or NOS. SOD then converts the O2 ∙− to H2O2, which is then converted into the highly reactive ∙OH or has to be rapidly removed from the system that is generally achieved by catalase or peroxidases, such as the GPx. Further, O2 ∙− can be either converted into ROO∙ or can react with NO to yield ONOO−. NO is mostly generated by L-Arg via NOS. H2O2 can be converted to HOCl by the action of MPO. myeloperoxidase. O2: molecular oxygen; H2O: water; O2 ∙−: superoxide radical anion; ∙OH: hydroxyl radical; ROO∙: peroxyl radical; H2O2: hydrogen peroxide; ONOO−: peroxynitrite; NO: nitric oxide; L-Arg: L-arginine; HOCl: hypochlorous acid.
5.5. Adhesion Molecules
Cell adhesion molecules play an important role in inflammatory responses. Leukocytes continuously circulate throughout the body in order to come in contact with antigens sequestered within tissues. To enter tissues, circulating leukocytes migrate from the blood between vascular endothelial cells and into the tissue [136]. During this migration, leukocytes initially bind to endothelial cells via low-affinity adhesion molecules. The low-affinity adhesion in combination with the force of the blood flow results in rolling leukocytes on endothelial cells. Subsequently, adhesion molecule affinity is upregulated and leukocytes firmly adhere to the endothelium [136]. Finally, bound leukocytes migrate between the endothelial cells and into the tissue. The vascular cell adhesion molecule 1 (VCAM-1) is one of the inducible cell transmembrane glycoproteins of the immunoglobulin supergene family expressed on several cell types and plays an important role in a number of inflammatory and immune responses [137]. It was first identified as an adhesion molecule induced on endothelial cells by proinflammatory cytokines or LPS [138]. VCAM-1 expression is induced on endothelial cells during inflammatory bowel disease, atherosclerosis, and infections [139]. Upregulation of VCAM-1 expression on cytokine-triggered vascular endothelial cells enhances the targeted transmigration of PMNs into extravascular space of inflammation [137]. In brain, proinflammatory cytokine-mediated expression of cell surface adhesion molecules plays a key role in endothelial cell injury, leading to vascular inflammation and the development of many cerebrovascular diseases [140]. Moreover, astrocytes can be induced by viral infections to express the adhesion molecules. Upregulation of adhesion molecules such as ICAM-1 (intercellular adhesion molecule 1) and VCAM-1 in astrocytes is required for monocyte-astrocyte interaction which increases infiltration of monocytes into the CNS observed in the patients with HIV-1 dementia [141]. HIV-1 Tat enhances monocyte adhesion by upregulation of ICAM-1 and VCAM-1 genes via a ROS-dependent NF-κB activation in astrocytes [141]. Understanding the role of ROS in proinflammatory factor-mediated adhesion molecule expression and subsequently increased adhesion of monocyte to brain cells provides an occasion for the development of anti-inflammatory compounds that may be useful as therapeutic strategies for the CNS inflammation and ROS-associated neurotoxicity.
5.6. Stress Protective Proteins
In contrast with inflammatory proteins, recent reports indicate that the ROS can also induce several stress protective proteins, such as HO-1 and heat-shock proteins (HSP70 in particular), which may exert protective effects from the deleterious effects of inflammation [142]. Abnormal protein folding has been shown as a cause of various diseases like neurodegenerative diseases in association with inflammatory mechanisms. In the events, the HSPs play a crucial role in preventing protein misfolding and inhibiting apoptotic activity and represent a class of proteins potentially involved in PD pathogenesis [143]. Recent studies have shown that HSPs are colocalized in protein aggregates in AD, PD, and other neurodegenerative disorders [144, 145]. Many experimental findings have demonstrated that selective overexpression of HSP70 prevents the disease progression in various animal models and cellular models [145]. Furthermore, HSP70 dysfunction activates intracellular signaling like NF-κB that can also promote neurodegeneration [146]. Thus, the expression of HSP70 may prove diagnostic and prognostic values in inflammatory conditions and therapeutical applications are being considered on the basis of these reports.
6. Redox Signal-Mediated Signaling Transduction
Recently, increasing evidence has demonstrated that oxidative stress (ROS generation) also plays a key signaling molecule in regulation of various inflammatory mediators in several cell types. Although many cells from brain tissue can produce various inflammatory mediators [42, 105], the intracellular signaling mechanisms responsible for the regulation of diverse inflammation-relating mediators expression induced by proinflammatory factors in brain cells like astrocytes are not completely characterized. Next, we review some signaling molecules in several inflammatory target protein expressions induced by proinflammatory factors in brain cells.
6.1. Mitogen-Activated Protein Kinases
Many proinflammatory cytokines and chemokines transducer signals are mediated via activation of MAPKs pathways. There is growing evidence that members of the MAPK family may play a central role in neurodegeneration [147]. MAPKs are important components of signaling modules activated by neurotransmitters, cytokines, and growth factors, as well as chemical and mechanical stressors. In mammals, three groups of MAPKs have been identified: the extracellular signal-regulated protein kinases (ERKs), the c-Jun NH2-terminal kinases (JNKs), and the p38 MAPK. ERK is activated by diverse stimuli, including growth factors and cytokines [147]. The p38 MAPK is activated by cellular stresses, including cytokines, LPS, growth factors, and UV radiation. The JNK is activated by many of the same stimuli that activate p38 MAPK, such as cellular stresses and various cytokines. Moreover, abnormal MAPK regulation might be implicated in CNS injury and inflammation [148]. Several mediators such as BK have been reported to act as an important proinflammatory factors through activation of MAPK cascades in different cell types [21–26]. In brain cells, the activation of ERK1/2 is mainly associated with proliferation, differentiation, and development in response to nerve growth factors. In contrast, the JNK and p38 MAPK signaling pathways are activated by various environmental stress and inflammatory factors that have been shown to promote neuronal cell death [149]. Moreover, the JNK and p38 MAPK signaling cascades can also be strongly activated by stress-induced ROS production or a mild oxidative shift of the redox state [28]. Both JNK and p38 MAPK are recognized as contributors to neurodegeneration by their ability to mediate intracellular stress events in transgenic mouse models of AD [19]. The p38 MAPK activation and COX-2 and PGE2 induction are served as contributors to neuronal damage in AD in response to oxidative stress [150].
In nonneural cells like astrocytes, many studies have found that Aβ peptide can activate astrocytes, including morphological alterations, cytokine induction, NO release [151], and chemokine and matrix-degrading proteinases production [152]. These findings further indicate that induction of several inflammatory mediators by the Aβ-stimulated activation of MAPKs in glial cells may be involved in AD progression. Moreover, our recent reports in astrocytes have demonstrated that the proinflammatory factors including TGF-β and BK can induce many inflammatory mediators such as MMP-9 expression through the ROS-dependent MAPK cascades [40]. These results suggest that upregulation of inflammatory mediators via ROS-mediated activation of MAPKs in astrocytes might play a key role during the CNS inflammation and neurodegeneration. Moreover, these results also implicate that the distinct groups of MAPKs are activated by a ROS-dependent manner which contribute to the expression of various inflammatory genes and are dependent on the external stimuli during brain inflammation. Thus, ROS may mediate MAPKs activation and expression of inflammatory genes in response to proinflammatory mediators in the CNS inflammatory disorders (Figure 5).
Figure 5.
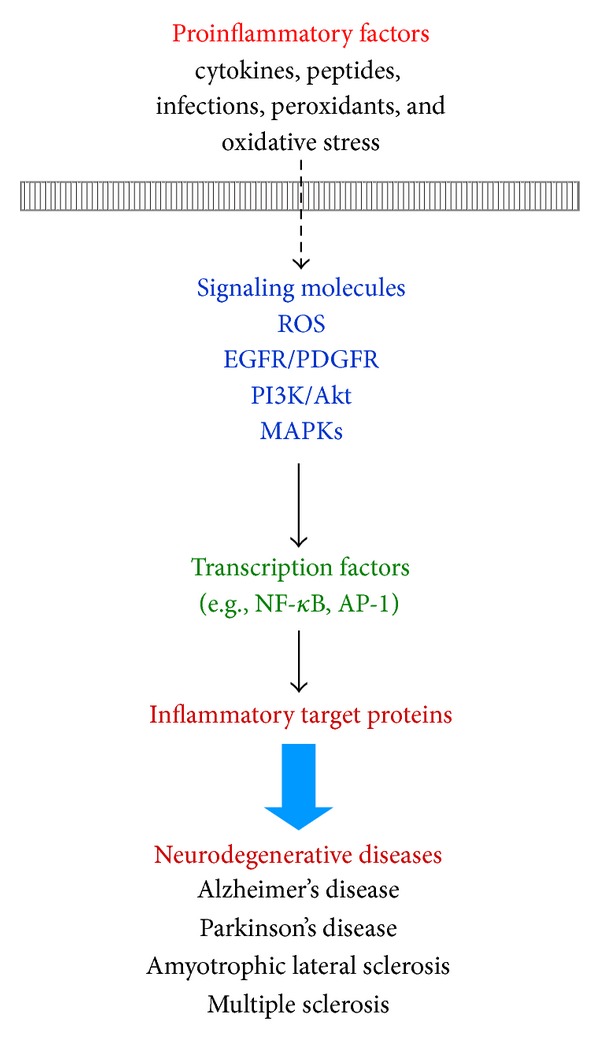
Proposed mechanisms of proinflammatory factors-stimulated activation of various signaling molecules and transcription factors leading to the expression of inflammatory target genes in brain resident cells. The intracellular signaling molecules include ROS, EGFR/PDFER, PI3K/Akt, and MAPKs. Oxidative stress may regulate these signaling pathways leading to activation of transcription factors such as NF-κB and AP-1 and recruitment of coactivator p300 in the transcription initiation complex. Ultimately, upregulation of diverse inflammatory target proteins can cause the pathogenesis of several neurodegenerative diseases. EGFR: epidermal growth factor receptor; PDGFR: platelet-derived growth factor receptor; PI3K: phosphoinositide-3′-kinase; MAPKs: mitogen-activated protein kinases; NF-κB: Nuclear factor-κB; AP-1: activator protein-1.
6.2. Transactivation of Receptor Tyrosine Kinases
Cross-communication between different signaling systems allows the integration of the great diversity of stimuli that a cell receives under varying physiological situations. The most direct mechanism is receptor heterodimerization that is well described for members of the epidermal growth factor receptor (EGFR) family [153]. In addition to growth factor receptor tyrosine kinases (RTKs) cross-talk, also completely unrelated cell surface receptors are able to communicate and influence each other, which play a key role in the transmission of information from outside the cell into the cytoplasm and nucleus. A variety of cytokines and growth factors that act as respective receptors have been reported to induce production of ROS in nonimmune cells. The prototype for such a pathway is the GPCR-induced transactivation of EGFR signal [154]. Treatment of cells with GPCR agonists induces phosphorylation of the EGFR by metalloprotease-dependent release of EGF-like ligands such as HB-EGF, thereby coupling GPCRs to EGFR characteristic downstream signaling pathways such as MAPKs or PI3K/Akt pathway [155]. In addition to the EGFR, other RTKs have been shown to be activated in response to GPCR stimulation, comprising the Trk receptor [156] and platelet-derived growth factor receptor (PDGFR) [157]. Previous studies have shown that in developing carcinoma cells, the early effects of COX-2-derived PGE2 and lysophosphatidic acid are in part mediated by the EGFR or PDGER, and this transactivation is responsible for subsequent downstream effects including the stimulation of cell migration and invasion [158]. However, receptor cross-talk can also occur in a ligand-independent manner involving for instance, non-RTKs such as c-Src [159]. Production of ROS results from the activation of signaling through the EGF and PDGF receptors [160]. In addition, ROS have been shown to stimulate c-Src-dependent transactivation of PDGFRα [161]. Accumulating evidence has shown that PKC-dependent activation of Nox is essential for PDGF-stimulated ROS generation, which is important for PDGF-induced MAPKs activation [162]. In the adult CNS, the EGFR pathway is highly upregulated and activated in astrocytes following neuronal injury [163]. Activation of the EGFR pathway triggers quiescent astrocytes to become reactive astrocytes that appear to be destructive to neurons in the adult CNS [163]. Regulation of RTKs such as EGFR in astrocytes may be a new therapeutic strategy for the treatment of neural disorders. These studies suggest that growth factor RTKs may play a pivotal role in mediating inflammatory genes regulation through ROS signal in several diseases including the CNS disorders (Figure 5).
6.3. Phosphoinositide-3′-Kinase (PI3K)/Akt Cascade
The phosphoinositide-3′-kinase (PI3K)/Akt cascade, the common downstream signal of EGF and PDGF receptors, is a cell survival pathway and regulated by various growth factor receptor-dependent mechanisms. Recent studies suggested that numerous components of the PI3K/Akt pathway play a crucial role in the expression and activation of inflammatory mediators, inflammatory cell recruitment, immune cell function, and tissue remodeling in chronic inflammatory diseases. In astrocytes, we demonstrated that ET-1 induced iNOS expression and NO production through PI3K/Akt cascade [26]. Moreover, PI3K/Akt cascade contributes to the expression of various inflammatory mediators induced by several proinflammatory factors in brain cells including astrocytes [125, 127]. Selective PI3K inhibitors such as wortmannin and LY294002 have been developed that reduce inflammation and some characteristics of disease in experimental animal models. In addition, ROS induction is often accompanied by the activation of PI3K/Akt cascade. For example, LY294002 has been shown to reduce chemokine-induced ROS generation in phagocytes [164], which was further confirmed by studies using PI3K knockout mice. Many studies have indicated the ROS generation induced by cytokines, PDGF, or VEGF in several cell types, which is reduced by inhibition of PI3K activity, suggesting that PI3K is involved in the ROS production induced by cytokines and growth factors. In addition to the role of PI3K/Akt cascade in ROS production, several reports support that the opposite hierarchical relationship exists between ROS and PI3K/Akt cascade. PI3K/Akt was activated in response to the exogenous treatment of H2O2 in several cell types [165]. Moreover, ROS have been shown to regulate phosphorylation of Akt [166] and then induce the expression of inflammatory genes associated with inflammation in various cell types. Taken together, these results implicate that ROS-dependent PI3K/Akt cascade or PI3K/Akt-mediated ROS signal may be critical for regulating the expression of inflammatory proteins in the brain inflammation and neurodegenerative disorders (Figure 5).
6.4. Transcription Factors
The progressive increase of oxidative stress during injuries not only causes oxidative damage to cellular macromolecules, but also modulates the pattern of gene expression through functional alterations of transcription factors. Here we focus on the roles of many transcription factors (e.g., NF-κB and AP-1), which are well known to be modulated during oxidative stress associated with physiological and pathological events [32]. The transcription factors such as NF-κB and AP-1 play a key role in the regulation of several gene expressions including proinflammatory cytokines, adhesion molecules, chemokines, growth factors, and inducible enzymes (e.g., MMPs, cPLA2, COX-2, and iNOS) during inflammation, immunity, cell proliferation, stress response, and apoptosis [167–169]. One important and widely investigated transcription factor which is NF-κB is a major participant in signaling pathways governing cellular responses to environmental (oxidative) stresses [168]. The nuclear translocation and activation of NF-κB in response to various stimuli, such as proinflammatory cytokines, LPS, and oxidative challenge (ROS production), are sequentially organized at the molecular level [168]. Moreover, NF-κB act as a positive regulator in the expression of many inflammatory genes such as COX-2 involved in chronic inflammatory diseases [169]. Cytokines such as IL-1β and TNF-α have been shown to activate NF-κB leading to upregulation of various NF-κB-dependent genes in several cell types [168]. It is of interest that many of the genes regulated by these MAPK pathways are dependent on NF-κB for transcription and lead to expression of inflammatory genes such as MMP-9 at the transcriptional level [169, 170]. In astrocytes, various stimuli can induce the expression of several inflammatory mediators, including MMP-9, cPLA2, COX-2, and iNOS, through ROS-mediated activation of NF-κB manner [40, 62].
In addition, activator protein-1 (AP-1) is a sequence-specific transcriptional activator mainly composed of members of the Fos, Jun, and ATF-2 families. These proteins associate to form a variety of homodimers or heterodimers that bind to an AP-1 binding element within the promoter region of inflammatory genes such as COX-2 and MMP-9. It is a well-known redox-regulated transcription factor for the expression of several AP-1-dependent genes induced by diverse stress signals such as ROS generation associated with physiological and pathological events [25, 62, 170]. Several reports indicate that AP-1 is also involved in the pathogenesis of brain inflammation (Figure 5). Many studies have demonstrated that ROS signals (e.g., O2 ∙− and H2O2) contribute to the expression or activation of AP-1 proteins (e.g., c-Fos) [62]. Recently, Kim et al. demonstrated that apocynin (a Nox inhibitor) shows potential antioxidant activities and inhibitory effects on the activation of redox-sensitive transcription factors, such as AP-1, induced by proinflammatory stimuli such as TNF-α [171]. The reports indicate that CSE induces cPLA2 expression through the production of ROS and subsequent activation of the MAPK pathway and AP-1 in human tracheal smooth muscle cells [172]. In astrocytes, we have demonstrated that AP-1 participates in the expression of several genes, including MMP-9 and HO-1, by BK through ROS-dependent manner [25, 62]. These results implicate that ROS play a central role in regulating AP-1 activation or expression and lead to inflammatory genes expression in brain inflammation and neurodegenerative disorders (Figure 5).
6.5. Transcription Coactivators
The transcription coactivator p300/CREB binding protein (CBP) is vital for the coactivation of several transcription factors such as NF-κB and AP-1 in the transcription machinery, which has a significant role in the activation of transcription factor-mediated gene expression for proinflammatory factors [173–175]. The p300 protein is a key regulator of RNA polymerase II-mediated transcription. Several studies indicate that p300 participates in the expression of inflammatory genes induced by cytokines and growth factors. Furthermore, the transcriptional cofactor p300/CBP is an important component of the transcriptional machinery that participates in regulation at the levels of both chromatin modification and transcription initiation [173–175]. Previous studies have indicated that the promoter of several gene transcriptions, chromatin remodeling, and histone modification is regulated by p300/CBP [175]. However, in astrocytes, the p300 is vital for the coactivation of several transcription factors such as AP-1 in the transcription machinery, which has a significant role in the activation of AP-1-mediated gene expression for proinflammatory mediators [173]. Previous results have indicated that p300 plays an important role in BK-, IL-1β-, and oxLDL-induced MMP-9 expression in astrocytes [21, 22, 96]. Recently, a study has shown that ROS-dependent p300 activation leads to cPLA2 expression by cigarette smoke extract in human tracheal smooth muscle cells [172]. Consistently, we have demonstrated that LTA induces p300/AP-1-dependent MMP-9 expression via ROS-mediated pathway in astrocytes [27]. Moreover, oxidative stress activates NF-κB resulting in the expression of proinflammatory mediators through the activation of intrinsic HAT activity on coactivator molecules. Oxidative stress also inhibits HDAC activity and in doing so enhances the expression of inflammatory genes which leads to a chronic inflammatory response. Oxidative stress can also increase complex formation between the coactivator p300 and the p65 subunit of NF-κB suggesting a further role of oxidative stress in chromatin remodeling [1]. Together, these studies indicate that the oxidative stress-stimulated coactivator p300 may play a critical role in the expression of inflammatory genes during brain inflammation and neurodegenerative disorders.
7. Conclusions
Glial cells maintain brain plasticity and protect the brain for functional recovery from injuries. Reactivation of glial cells may promote neuroinflammation and neurodegeneration (Figure 1) and, ultimately, the retraction of neuronal synapses, which leads to cognitive deficits [10]. Moreover, redox signaling is a critical event in several inflammatory diseases such as AD that precedes the formation of these disease pathologies. To date, although numerous effects have been made to develop therapies based on antioxidants in the past years, the actual benefits to the patients have been very limited. It is likely due to lack of potency, late administration, and poor penetration into the brain cells [7, 32]. Alternative strategies including searching for factors that initiate endogenous antioxidants are necessary to improve the efficacy of treatment (Figure 2). Moreover, increased oxidative stresses (ROS) by various proinflammatory factors, such as cytokines, peptides, bacterial or viral infections, peroxidants, and other stress, serve as intracellular signals in gene regulation and signaling transduction, in addition to their deleterious effects on cellular components. Thus, understanding how oxidative stress produces and modulates expression of several genes that might help to develop effectively therapeutic strategies for CNS diseases. First, the focus of this review is on glial cells and their effects on the CNS disorders. Moreover, this review summarized the interplay between oxidative stress and neuroinflammation via ROS production which contributes to neurodegeneration, thereby enhancing disease progression based on data collected from brain cells, particularly astrocytes, in in vitro and in vivo studies (Figure 1). Perhaps modifying the activity of glial cells to reduce their neurotoxic properties and enhance their neuroprotective effects may offer potential targets for therapeutic interventions in neurodegenerative diseases. Oxidative stress-induced signaling transduction pathways, including ROS, transactivation of EGFR or PDGFR, PI3K/Akt, MAPKs, NF-κB, and AP-1, that are associated with the CNS disorders were discussed (Figure 4). Moreover, the review highlighted current progress on the association of oxidative stress with the expression of various inflammatory genes, including MMP-9, cPLA2, COX-2, iNOS, and adhesion molecules and redox signal-sensitive transcription factors that may contribute to the development of the CNS inflammation and neurodegenerative diseases (Figure 5). Possible therapeutic strategies to target redox-sensitive signaling molecules, transcription factors, or cofactors are implicated based on the updated view of ROS-mediated regulation of inflammatory target genes in brain inflammation and neurodegenerative disorders.
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
This work was supported by National Science Council, Taiwan, Grant nos. NSC102-2321-B-182-011, NSC101-2320-B-182-039-MY3, and NSC102-2320-B-255-005-MY3; Chang Gung Medical Research Foundation, Grant nos. CMRPD1C0101, CMRPD1B0382, CMRPD1C0561, CMRPF1C0191, and CMRPF1A0063; and the Ministry of Education, Taiwan; Grant nos. EMRPD1C0261 and EMRPD1C0271.
Abbreviations
- ROS:
Reactive oxygen species
- CNS:
Central nervous system
- AD:
Alzheimer's disease
- PD:
Parkinson's disease
- MMPs:
Matrix metalloproteinases
- cPLA2:
Cytosolic phospholipase A2
- COX-2:
Cyclooxygenase-2
- Nox2:
NADPH oxidase 2
- iNOS:
Inducible nitric oxide synthase
- LPS:
Lipopolysaccharide
- IL-1β:
Interleukin-1
- TNF-α:
Tumor necrosis factor-α
- BBB:
Blood-brain barrier
- TLRs:
Toll-like receptors
- PGs:
Prostaglandins
- NO:
Nitric oxide
- Aβ:
β-Amyloid
- BK:
Bradykinin
- ET-1:
Endothelin-1
- oxLDL:
Oxidized low-density lipoprotein
- HO-1:
Heme oxygenase-1
- CO:
Carbon monoxide
- RNS:
Reactive nitrogen species
- Xox:
Xanthine oxidase
- GPCR:
G-Protein-coupled receptor
- LTA:
Lipoteichoic acid
- JEV:
Japanese encephalitis virus
- EV71:
Enterovirus 71
- AA:
Arachidonic acid
- VCAM-1:
Vascular cell adhesion molecule 1
- MAPKs:
Mitogen-activated protein kinases
- ERKs:
Extracellular signal-regulated protein kinases
- JNKs:
c-Jun NH2-terminal kinases
- EGFR:
Epidermal growth factor receptor
- RTKs:
Receptor tyrosine kinases
- PDGFR:
Platelet-derived growth factor receptor
- PI3K:
Phosphoinositide-3′-kinase
- NF-κB:
Nuclear factor-κB
- AP-1:
Activator protein 1
- CREB:
Cyclic AMP-response element binding protein
- CBP:
CREB binding protein.
References
- 1.Rahman I, Marwick J, Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-κB and pro-inflammatory gene expression. Biochemical Pharmacology. 2004;68(6):1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 2.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry and Cell Biology. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochemical Pharmacology. 2012;84(5):581–590. doi: 10.1016/j.bcp.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Dröge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 5.von Bernhardi R, Eugenín J. Alzheimer’s disease: redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxidants and Redox Signaling. 2012;16(9):974–1031. doi: 10.1089/ars.2011.4082. [DOI] [PubMed] [Google Scholar]
- 6.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of Neurochemistry. 2006;97(6):1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 7.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Current Neuropharmacology. 2009;7(1):65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melo A, Monteiro L, Lima RMF, de Oliveira DM, de Cerqueira MD, El-Bachá RS. Oxidative stress in neurodegenerative diseases: mechanisms and therapeutic perspectives. Oxidative Medicine and Cellular Longevity. 2011;2011:14 pages. doi: 10.1155/2011/467180.467180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiurchiù V, MacCarrone M. Chronic inflammatory disorders and their redox control: from molecular mechanisms to therapeutic opportunities. Antioxidants and Redox Signaling. 2011;15(9):2605–2641. doi: 10.1089/ars.2010.3547. [DOI] [PubMed] [Google Scholar]
- 10.Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer’s disease: Alzheimer’s review series. Journal of Cellular and Molecular Medicine. 2008;12(3):762–780. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuller S, Steele M, Münch G. Activated astroglia during chronic inflammation in Alzheimer’s disease-Do they neglect their neurosupportive roles? Mutation Research. 2010;690(1-2):40–49. doi: 10.1016/j.mrfmmm.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Kimelberg HK. Receptors on astrocytes—what possible functions? Neurochemistry International. 1995;26(1):27–40. doi: 10.1016/0197-0186(94)00118-e. [DOI] [PubMed] [Google Scholar]
- 13.Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathology. 1994;4(3):229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- 14.Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Experimental and Molecular Medicine. 2006;38(4):333–347. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- 15.Eddelston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54(1):15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends in Neurosciences. 1997;20(12):570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 17.Brown GC. Mechanisms of inflammatory neurodegeneration: INOS and NADPH oxidase. Biochemical Society Transactions. 2007;35(5):1119–1121. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- 18.Koistinaho M, Kettunen MI, Goldsteins G, et al. β-amyloid precursor protein transgenic mice that harbor diffuse Aβ deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1610–1615. doi: 10.1073/pnas.032670899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savage MJ, Lin Y-G, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-Terminal Kinase and p38 in an Alzheimer’s Disease Model Is Associated with Amyloid Deposition. The Journal of Neuroscience. 2002;22(9):3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mrak RE, Sheng JG, Griffin WST. Glial cytokines in Alzheimer’s disease: review and pathogenic implications. Human Pathology. 1995;26(8):816–823. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C-Y, Hsieh H-L, Sun C-C, Tseng C-P, Yang C-M. IL-1β induces proMMP-9 expression via c-Src-dependent PDGFR/PI3K/Akt/p300 cascade in rat brain astrocytes. Journal of Neurochemistry. 2008;105(4):1499–1512. doi: 10.1111/j.1471-4159.2008.05318.x. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh H-L, Wu C-Y, Yang C-M. Bradykinin induces matrix metalloproteinase-9 expression and cell migration through a PKC-δ-dependent ERK/Elk-1 pathway in astrocytes. Glia. 2008;56(6):619–632. doi: 10.1002/glia.20637. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh H-L, Wu C-Y, Hwang T-L, Yen M-H, Parker P, Yang C-M. BK-induced cytosolic phospholipase A2 expression via sequential PKC-δ, p42/p44 MARK, and NF-κB activation in rat brain astrocytes. Journal of Cellular Physiology. 2006;206(1):246–254. doi: 10.1002/jcp.20457. [DOI] [PubMed] [Google Scholar]
- 24.Hsieh H-L, Wang H-H, Wu C-Y, et al. BK-induced COX-2 expression via PKC-δ-dependent activation of p42/p44 MAPK and NF-κB in astrocytes. Cellular Signalling. 2007;19(2):330–340. doi: 10.1016/j.cellsig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Lin CC, Hsieh HL, Shih RH, et al. NADPH oxidase 2-derived reactive oxygen species signal contributes to bradykinin-induced matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Cell Communication and Signaling. 2012;10(1):p. 35. doi: 10.1186/1478-811X-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H-H, Hsieh H-L, Yang C-M. Nitric oxide production by endothelin-1 enhances astrocytic migration via the tyrosine nitration of matrix metalloproteinase-9. Journal of Cellular Physiology. 2011;226(9):2244–2256. doi: 10.1002/jcp.22560. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh HL, Lin CC, Shih RH, Hsiao LD, Yang CM. NADPH oxidase-mediated redox signal contributes to lipoteichoic acid-induced MMP-9 upregulation in brain astrocytes. Journal of Neuroinflammation. 2012;9:p. 110. doi: 10.1186/1742-2094-9-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tung W-H, Tsai H-W, Lee I-T, et al. Japanese encephalitis virus induces matrix metalloproteinase-9 in rat brain astrocytes via NF-ΚB signalling dependent on MAPKs and reactive oxygen species. British Journal of Pharmacology. 2010;161(7):1566–1583. doi: 10.1111/j.1476-5381.2010.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H-H, Hsieh H-L, Wu C-Y, Sun C-C, Yang C-M. Oxidized low-density lipoprotein induces matrix metalloproteinase-9 expression via a p42/p44 and JNK-dependent AP-1 pathway in brain astrocytes. Glia. 2009;57(1):24–38. doi: 10.1002/glia.20732. [DOI] [PubMed] [Google Scholar]
- 30.Yang CM, Hsieh HL, Lin CC, et al. Multiple factors from bradykinin-challenged astrocytes contribute to the neuronal apoptosis: involvement of astroglial ROS, MMP-9, and HO-1/CO system. Molecular Neurobiology. 2013;47(3):1020–1033. doi: 10.1007/s12035-013-8402-1. [DOI] [PubMed] [Google Scholar]
- 31.Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends in Molecular Medicine. 2008;14(11):495–502. doi: 10.1016/j.molmed.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Q, Gibson GE. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Disease and Associated Disorders. 2007;21(4):276–291. doi: 10.1097/WAD.0b013e31815721c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demchenko IT, Oury TD, Crapo JD, Piantadosi CA. Regulation of the brain’s vascular responses to oxygen. Circulation Research. 2002;91(11):1031–1037. doi: 10.1161/01.res.0000043500.03647.81. [DOI] [PubMed] [Google Scholar]
- 34.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. Journal of Cerebral Blood Flow and Metabolism. 2001;21(1):2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Serrano F, Klann E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Research Reviews. 2004;3(4):431–443. doi: 10.1016/j.arr.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Kamata H, Hirata H. Redox regulation of cellular signalling. Cellular Signalling. 1999;11(1):1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 37.Federico A, Cardaioli E, da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. Journal of the Neurological Sciences. 2012;322(1-2):254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 38.Kang J, Park EJ, Jou I, Kim J-H, Joe E-H. Reactive oxygen species mediate Aβ(25-35)-induced activation of BV-2 microglia. NeuroReport. 2001;12(7):1449–1452. doi: 10.1097/00001756-200105250-00030. [DOI] [PubMed] [Google Scholar]
- 39.Qin L, Liu Y, Wang T, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. The Journal of Biological Chemistry. 2004;279(2):1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- 40.Hsieh H-L, Wang H-H, Wu W-B, Chu P-J, Yang C-M. Transforming growth factor-β1 induces matrix metalloproteinase-9 and cell migration in astrocytes: roles of ROS-dependent ERK- and JNK-NF-κB pathways. Journal of Neuroinflammation. 2010;7, article 88 doi: 10.1186/1742-2094-7-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Research Reviews. 1995;21(2):195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39(3):279–291. doi: 10.1002/glia.10108. [DOI] [PubMed] [Google Scholar]
- 43.Fillit H, Ding W, Buee L, et al. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neuroscience Letters. 1991;129(2):318–320. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- 44.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nature Reviews Immunology. 2005;5(8):629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 45.Fassbender K, Rossol S, Kammer T, et al. Proinflammatory cytokines in serum of patients with acute cerebral ischemia: kinetics of secretion and relation to the extent of brain damage and outcome of disease. Journal of the Neurological Sciences. 1994;122(2):135–139. doi: 10.1016/0022-510x(94)90289-5. [DOI] [PubMed] [Google Scholar]
- 46.Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Research Bulletin. 2012;87(1):10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siwik DA, Colucci WS. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Failure Reviews. 2004;9(1):43–51. doi: 10.1023/B:HREV.0000011393.40674.13. [DOI] [PubMed] [Google Scholar]
- 48.Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. Journal of Neurochemistry. 2006;98(1):258–266. doi: 10.1111/j.1471-4159.2006.03872.x. [DOI] [PubMed] [Google Scholar]
- 49.Greig NH, Mattson MP, Perry T, et al. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-α inhibitors, and GLP-1 receptor agonists. Annals of the New York Academy of Sciences. 2004;1035:290–315. doi: 10.1196/annals.1332.018. [DOI] [PubMed] [Google Scholar]
- 50.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid β-peptide. Trends in Molecular Medicine. 2001;7(12):548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 51.Caspersen C, Wang N, Yao J, et al. Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. The FASEB Journal. 2005;19(14):2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 52.Della Bianca V, Dusi S, Bianchini E, Dal Prà I, Rossi F. β-amyloid activates the O− 2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. The Journal of Biological Chemistry. 1999;274(22):15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 53.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. The Journal of Neuroscience. 2001;21(21):8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ding Q, Dimayuga E, Keller JN. Oxidative damage, protein synthesis, and protein degradation in Alzheimer’s disease. Current Alzheimer Research. 2007;4(1):73–79. doi: 10.2174/156720507779939788. [DOI] [PubMed] [Google Scholar]
- 55.Schipper HM, Bennett DA, Liberman A, et al. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiology of Aging. 2006;27(2):252–261. doi: 10.1016/j.neurobiolaging.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 56.Kamiya T, Katayama Y, Kashiwagi F, Terashi A. The role of bradykinin in mediating ischemic brain edema in rats. Stroke. 1993;24(4):571–576. doi: 10.1161/01.str.24.4.571. [DOI] [PubMed] [Google Scholar]
- 57.Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiological Reviews. 1998;78(1):99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- 58.Regoli D, Rhaleb N-E, Dion S, Drapeau G. New selective bradykinin receptor antagonists and bradykinin B2 receptor characterization. Trends in Pharmacological Sciences. 1990;11(4):156–161. doi: 10.1016/0165-6147(90)90067-I. [DOI] [PubMed] [Google Scholar]
- 59.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacological Reviews. 1992;44(1):1–80. [PubMed] [Google Scholar]
- 60.Lin C-W, Shen S-C, Chien C-C, Yang L-Y, Shia L-T, Chen Y-C. 12-O-tetradecanoylphorbol-13-acetate-induced invasion/migration of glioblastoma cells through activating PKCα/ERK/NF-κB-dependent MMP-9 expression. Journal of Cellular Physiology. 2010;225(2):472–481. doi: 10.1002/jcp.22226. [DOI] [PubMed] [Google Scholar]
- 61.Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR. Expression and modulation of an NADPH oxidase in mammalian astrocytes. The Journal of Neuroscience. 2005;25(40):9176–9184. doi: 10.1523/JNEUROSCI.1632-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsieh H-L, Wang H-H, Wu C-Y, Yang C-M. Reactive oxygen species-dependent c-fos/activator protein 1 induction upregulates heme oxygenase-1 expression by bradykinin in brain astrocytes. Antioxidants and Redox Signaling. 2010;13(12):1829–1844. doi: 10.1089/ars.2009.2957. [DOI] [PubMed] [Google Scholar]
- 63.Levin ER. Endothelins. The New England Journal of Medicine. 1995;333(6):356–363. doi: 10.1056/NEJM199508103330607. [DOI] [PubMed] [Google Scholar]
- 64.Schinelli S. Pharmacology and physiopathology of the brain endothelin system: an overview. Current Medicinal Chemistry. 2006;13(6):627–638. doi: 10.2174/092986706776055652. [DOI] [PubMed] [Google Scholar]
- 65.Böhm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovascular Research. 2007;76(1):8–18. doi: 10.1016/j.cardiores.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 66.Hasselblatt M, Lewczuk P, Löffler B-M, et al. Role of the astrocytic ETB receptor in the regulation of extracellular endothelin-1 during hypoxia. Glia. 2001;34(1):18–26. doi: 10.1002/glia.1036. [DOI] [PubMed] [Google Scholar]
- 67.Rogers SD, Peters CM, Pomonis JD, Hagiwara H, Ghilardi JR, Mantyh PW. Endothelin B receptors are expressed by astrocytes and regulate astrocyte hypertrophy in the normal and injured CNS. Glia. 2003;41(2):180–190. doi: 10.1002/glia.10173. [DOI] [PubMed] [Google Scholar]
- 68.Lo ACY, Chen AYS, Hung VKL, et al. Endothelin-1 overexpression leads to further water accumulation and brain edema after middle cerebral artery occlusion via aquaporin 4 expression in astrocytic end-feet. Journal of Cerebral Blood Flow and Metabolism. 2005;25(8):998–1011. doi: 10.1038/sj.jcbfm.9600108. [DOI] [PubMed] [Google Scholar]
- 69.Didier N, Romero IA, Créminon C, Wijkhuisen A, Grassi J, Mabondzo A. Secretion of interleukin-1β by astrocytes mediates endothelin-1 and tumour necrosis factor-α effects on human brain microvascular endothelial cell permeability. Journal of Neurochemistry. 2003;86(1):246–254. doi: 10.1046/j.1471-4159.2003.01829.x. [DOI] [PubMed] [Google Scholar]
- 70.Lee SJ, Lee S. Toll-like receptors and inflammation in the CNS. Current Drug Targets Inflammation & Allergy. 2002;1(2):181–191. doi: 10.2174/1568010023344698. [DOI] [PubMed] [Google Scholar]
- 71.Kim S-Y, Lee J-G, Cho W-S, et al. Role of NADPH oxidase-2 in lipopolysaccharide-induced matrix metalloproteinase expression and cell migration. Immunology and Cell Biology. 2010;88(2):197–204. doi: 10.1038/icb.2009.87. [DOI] [PubMed] [Google Scholar]
- 72.Konat GW, Kielian T, Marriott I. The role of Toll-like receptors in CNS response to microbial challenge. Journal of Neurochemistry. 2006;99(1):1–12. doi: 10.1111/j.1471-4159.2006.04076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sutcliffe IC, Shaw N. Atypical lipoteichoic acids of gram-positive bacteria. Journal of Bacteriology. 1991;173(22):7065–7069. doi: 10.1128/jb.173.22.7065-7069.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sáez-Llorens X, McCracken GH., Jr. Bacterial meningitis in children. The Lancet. 2003;361(9375):2139–2148. doi: 10.1016/S0140-6736(03)13693-8. [DOI] [PubMed] [Google Scholar]
- 75.Mullaly SC, Kubes P. The role of TLR2 in vivo following challenge with Staphylococcus aureus and prototypic ligands. The Journal of Immunology. 2006;177(11):8154–8163. doi: 10.4049/jimmunol.177.11.8154. [DOI] [PubMed] [Google Scholar]
- 76.Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N. Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. Journal of Endocrinology. 2007;193(3):323–330. doi: 10.1677/JOE-07-0067. [DOI] [PubMed] [Google Scholar]
- 77.Kinsner A, Pilotto V, Deininger S, et al. Inflammatory neurodegeneration induced by lipoteichoic acid from Staphylococcus aureus is mediated by glia activation, nitrosative and oxidative stress, and caspase activation. Journal of Neurochemistry. 2005;95(4):1132–1143. doi: 10.1111/j.1471-4159.2005.03422.x. [DOI] [PubMed] [Google Scholar]
- 78.Lehnardt S, Henneke P, Lien E, et al. A mechanism for neurodegeneration induced by group B Streptococci through activation of the TLR2/MyD88 pathway in microglia. The Journal of Immunology. 2006;177(1):583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 79.Carpentier PA, Duncan DS, Miller SD. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain, Behavior, and Immunity. 2008;22(2):140–147. doi: 10.1016/j.bbi.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bsibsi M, Bajramovic JJ, van Duijvenvoorden E, et al. Identification of soluble CD14 as an endogenous agonist for toll-like receptor 2 on human astrocytes by genome-scale functional screening of glial cell derived proteins. Glia. 2007;55(5):473–482. doi: 10.1002/glia.20473. [DOI] [PubMed] [Google Scholar]
- 81.Jack CS, Arbour N, Manusow J, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. The Journal of Immunology. 2005;175(7):4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 82.Neher JJ, Brown GC. Neurodegeneration in models of Gram-positive bacterial infections of the central nervous system. Biochemical Society Transactions. 2007;35(5):1166–1167. doi: 10.1042/BST0351166. [DOI] [PubMed] [Google Scholar]
- 83.Chatterjee PK, Zacharowski K, Cuzzocrea S, et al. Lipoteichoic acid from Staphylococcus aureus reduces renal ischemia/reperfusion injury. Kidney International. 2002;62(4):1249–1263. doi: 10.1111/j.1523-1755.2002.kid580.x. [DOI] [PubMed] [Google Scholar]
- 84.Palacios G, Oberste MS. Enteroviruses as agents of emerging infectious diseases. Journal of NeuroVirology. 2005;11(5):424–433. doi: 10.1080/13550280591002531. [DOI] [PubMed] [Google Scholar]
- 85.McMinn PC. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiology Reviews. 2002;26(1):91–107. doi: 10.1111/j.1574-6976.2002.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 86.Huang C-C, Liu C-C, Chang Y-C, Chen C-Y, Wang S-T, Yeh T-F. Neurologic complications in children with enterovirus 71 infection. The New England Journal of Medicine. 1999;341(13):936–942. doi: 10.1056/NEJM199909233411302. [DOI] [PubMed] [Google Scholar]
- 87.Ho M, Chen E-R, Hsu K-H, et al. An epidemic of enterovirus 71 infection in Taiwan. The New England Journal of Medicine. 1999;341(13):929–935. doi: 10.1056/NEJM199909233411301. [DOI] [PubMed] [Google Scholar]
- 88.Tung W-H, Hsieh H-L, Lee I-T, Yang C-M. Enterovirus 71 induces integrin β1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication. Journal of Cellular Physiology. 2011;226(12):3316–3329. doi: 10.1002/jcp.22677. [DOI] [PubMed] [Google Scholar]
- 89.Misra UK, Kalita J. Overview: Japanese encephalitis. Progress in Neurobiology. 2010;91(2):108–120. doi: 10.1016/j.pneurobio.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 90.Raung S-L, Chen S-Y, Liao S-L, Chen J-H, Chen C-J. Tyrosine kinase inhibitors attenuate Japanese encephalitis virus-induced neurotoxicity. Biochemical and Biophysical Research Communications. 2005;327(2):399–406. doi: 10.1016/j.bbrc.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 91.Mishra MK, Koli P, Bhowmick S, Basu A. Neuroprotection conferred by astrocytes is insufficient to protect animals from succumbing to Japanese encephalitis. Neurochemistry International. 2007;50(5):764–773. doi: 10.1016/j.neuint.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 92.Montine TJ, Montine KS, Swift LL. Central nervous system lipoproteins in Alzheimer’s disease. American Journal of Pathology. 1997;151(6):1571–1575. [PMC free article] [PubMed] [Google Scholar]
- 93.Keller JN, Hanni KB, Markesbery WR. Oxidized low-density lipoprotein induces neuronal death: implications for calcium, reactive oxygen species, and caspases. Journal of Neurochemistry. 1999;72(6):2601–2609. doi: 10.1046/j.1471-4159.1999.0722601.x. [DOI] [PubMed] [Google Scholar]
- 94.Shie F-S, Neely MD, Maezawa I, et al. Oxidized low-density lipoprotein is present in astrocytes surrounding cerebral infarcts and stimulates astrocyte interleukin-6 secretion. American Journal of Pathology. 2004;164(4):1173–1181. doi: 10.1016/S0002-9440(10)63205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uno M, Harada M, Takimoto O, et al. Elevation of plasma oxidized LDL in acute stroke patients is associated with ischemic lesions depicted by DWI and predictive of infarct enlargement. Neurological Research. 2005;27(1):94–102. doi: 10.1179/016164105X18395. [DOI] [PubMed] [Google Scholar]
- 96.Wang H-H, Hsieh H-L, Wu C-Y, Yang C-M. Oxidized low-density lipoprotein-induced matrix metalloproteinase-9 expression via PKC-δ/p42/p44 MAPK/Elk-1 cascade in brain astrocytes. Neurotoxicity Research. 2010;17(1):50–65. doi: 10.1007/s12640-009-9077-2. [DOI] [PubMed] [Google Scholar]
- 97.Ten Dijke P, Hill CS. New insights into TGF-β-Smad signalling. Trends in Biochemical Sciences. 2004;29(5):265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 98.Massagué J. How cells read TGF-β signals. Nature Reviews Molecular Cell Biology. 2000;1(3):169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 99.Leivonen S-K, Chantry A, Häkkinen L, Han J, Kähäri V-M. Smad3 mediates transforming growth factor-β-induced collagenase-3 (matrix metalloproteinase-13) expression in human gingival fibroblasts: evidence for cross-talk between Smad3 and p38 signaling pathways. The Journal of Biological Chemistry. 2002;277(48):46338–46346. doi: 10.1074/jbc.M206535200. [DOI] [PubMed] [Google Scholar]
- 100.Koli K, Myllärniemi M, Keski-Oja J, Kinnula VL. Transforming growth factor-β activation in the lung: focus on fibrosis and reactive oxygen species. Antioxidants and Redox Signaling. 2008;10(2):333–342. doi: 10.1089/ars.2007.1914. [DOI] [PubMed] [Google Scholar]
- 101.Zipp F, Aktas O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends in Neurosciences. 2006;29(9):518–527. doi: 10.1016/j.tins.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 102.Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46(11):1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 103.Simi A, Tsakiri N, Wang P, Rothwell NJ. Interleukin-1 and inflammatory neurodegeneration. Biochemical Society Transactions. 2007;35(5):1122–1126. doi: 10.1042/BST0351122. [DOI] [PubMed] [Google Scholar]
- 104.Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinases and diseases of the CNS. Trends in Neurosciences. 1998;21(2):75–80. doi: 10.1016/s0166-2236(97)01169-7. [DOI] [PubMed] [Google Scholar]
- 105.Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nature Reviews Neuroscience. 2001;2(7):502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gottschall PE, Yu X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. Journal of Neurochemistry. 1995;64(4):1513–1520. doi: 10.1046/j.1471-4159.1995.64041513.x. [DOI] [PubMed] [Google Scholar]
- 107.Lee WJ, Shin CY, Yoo BK, et al. Induction of matrix metalloproteinase-9 (MMP-9) in lipopolysaccharide-stimulated primary astrocytes is mediated by extracellular signal-regulated protein kinase 1/2 (Erk1/2) Glia. 2003;41(1):15–24. doi: 10.1002/glia.10131. [DOI] [PubMed] [Google Scholar]
- 108.Hernández M, Nieto ML, Sánchez Crespo M. Cytosolic phospholipase A2 and the distinct transcriptional programs of astrocytoma cells. Trends in Neurosciences. 2000;23(6):259–264. doi: 10.1016/s0166-2236(00)01563-0. [DOI] [PubMed] [Google Scholar]
- 109.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins and Other Lipid Mediators. 2002;68-69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 110.Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clinical Immunology. 2006;119(3):229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 111.Xu J, Chalimoniuk M, Shu Y, et al. Prostaglandin E2 production in astrocytes: regulation by cytokines, extracellular ATP, and oxidative agents. Prostaglandins Leukotrienes and Essential Fatty Acids. 2003;69(6):437–448. doi: 10.1016/j.plefa.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 112.Leslie CC. Properties and regulation of cytosolic phospholipase A2. The Journal of Biological Chemistry. 1997;272(27):16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 113.Sun GY, Xu J, Jensen MD, et al. Phospholipase A2 in astrocytes: responses to oxidative stress, inflammation, and G protein-coupled receptor agonists. Molecular Neurobiology. 2005;31(1–3):27–41. doi: 10.1385/MN:31:1-3:027. [DOI] [PubMed] [Google Scholar]
- 114.Stephenson D, Rash K, Smalstig B, et al. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia. 1999;27(2):110–128. doi: 10.1002/(sici)1098-1136(199908)27:2<110::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 115.Gentile MT, Reccia MG, Sorrentino PP, et al. Role of cytosolic calcium-dependent phospholipase A2 in Alzheimer's disease pathogenesis. Molecular Neurobiology. 2012;45(3):596–604. doi: 10.1007/s12035-012-8279-4. [DOI] [PubMed] [Google Scholar]
- 116.Szaingurten-Solodkin I, Hadad N, Levy R. Regulatory role of cytosolic phospholipase A2α in NADPH oxidase activity and in inducible nitric oxide synthase induction by aggregated Aβ1-42 in microglia. Glia. 2009;57(16):1727–1740. doi: 10.1002/glia.20886. [DOI] [PubMed] [Google Scholar]
- 117.Guo C, Li J, Myatt L, Zhu X, Sun K. Induction of Gαs contributes to the paradoxical stimulation of cytosolic phospholipase A2α expression by cortisol in human amnion fibroblasts. Molecular Endocrinology. 2010;24(5):1052–1061. doi: 10.1210/me.2009-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bazan NG, Colangelo V, Lukiw WJ. Prostaglandins and other lipid mediators in Alzheimer’s disease. Prostaglandins and Other Lipid Mediators. 2002;68-69:197–210. doi: 10.1016/s0090-6980(02)00031-x. [DOI] [PubMed] [Google Scholar]
- 119.Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene. 1999;18(55):7908–7916. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- 120.Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1 β-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410(6827):471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 121.Tanabe T, Tohnai N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins and Other Lipid Mediators. 2002;68-69:95–114. doi: 10.1016/s0090-6980(02)00024-2. [DOI] [PubMed] [Google Scholar]
- 122.Korotkova M, Westman M, Gheorghe KR, et al. Effects of antirheumatic treatments on the prostaglandin E2 biosynthetic pathway. Arthritis and Rheumatism. 2005;52(11):3439–3447. doi: 10.1002/art.21390. [DOI] [PubMed] [Google Scholar]
- 123.Ejima K, Layne MD, Carvajal IM, et al. Cyclooxygenase-2-deficient mice are resistant to endotoxin-induced inflammation and death. The FASEB Journal. 2003;17(10):1325–1327. doi: 10.1096/fj.02-1078fje. [DOI] [PubMed] [Google Scholar]
- 124.Tocco G, Freire-Moar J, Schreiber SS, Sakhi SH, Aisen PS, Pasinetti GM. Maturational regulation and regional induction of cyclooxygenase-2 in rat brain: implications for Alzheimer’s disease. Experimental Neurology. 1997;144(2):339–349. doi: 10.1006/exnr.1997.6429. [DOI] [PubMed] [Google Scholar]
- 125.Tung W-H, Lee I-T, Hsieh H-L, Yang C-M. EV71 induces COX-2 expression via c-Src/PDGFR/PI3K/Akt/p42/p44 MAPK/AP-1 and NF-κB in rat brain astrocytes. Journal of Cellular Physiology. 2010;224(2):376–386. doi: 10.1002/jcp.22133. [DOI] [PubMed] [Google Scholar]
- 126.Tung W-H, Hsieh H-L, Lee I-T, Yang C-M. Enterovirus 71 modulates a COX-2/PGE2/cAMP-dependent viral replication in human neuroblastoma cells: role of the c-Src/EGFR/p42/p44 MAPK/CREB signaling pathway. Journal of Cellular Biochemistry. 2011;112(2):559–570. doi: 10.1002/jcb.22946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hsieh HL, Lin CC, Chan HJ, Yang CM, Yang CM. c-Src-dependent EGF receptor transactivation contributes to ET-1-induced COX-2 expression in brain microvascular endothelial cells. Journal of Neuroinflammation. 2012;9:p. 152. doi: 10.1186/1742-2094-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim DS, Kim JY, Han Y. Curcuminoids in neurodegenerative diseases. Recent Patents on CNS Drug Discovery. 2012;7(3):184–204. doi: 10.2174/157488912803252032. [DOI] [PubMed] [Google Scholar]
- 129.Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. The Lancet Neurology. 2002;1(4):232–241. doi: 10.1016/s1474-4422(02)00102-3. [DOI] [PubMed] [Google Scholar]
- 130.Barbeito LH, Pehar M, Cassina P, et al. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Research Reviews. 2004;47(1–3):263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 131.Saha RN, Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxidants and Redox Signaling. 2006;8(5-6):929–947. doi: 10.1089/ars.2006.8.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Galea E, Feinstein DL, Reis DJ. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):10945–10949. doi: 10.1073/pnas.89.22.10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Parmentier-Batteur S, Bohme GA, Lerouet D, et al. Antisense oligodeoxynucleotide to inducible nitric oxide synthase protects against transient focal cerebral ischemia-induced brain injury. Journal of Cerebral Blood Flow and Metabolism. 2001;21(1):15–21. doi: 10.1097/00004647-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 134.Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(28):9936–9941. doi: 10.1073/pnas.0502552102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Choi S-H, Joe EH, Kim SU, Jin BK. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. The Journal of Neuroscience. 2003;23(13):5877–5886. doi: 10.1523/JNEUROSCI.23-13-05877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76(2):301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 137.Cook-Mills JM. VCAM-1 signals during lymphocyte migration: role of reactive oxygen species. Molecular Immunology. 2002;39(9):499–508. doi: 10.1016/s0161-5890(02)00206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Osborn L, Hession C, Tizard R, et al. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. 1989;59(6):1203–1211. doi: 10.1016/0092-8674(89)90775-7. [DOI] [PubMed] [Google Scholar]
- 139.Michalska M, Machtoub L, Manthey HD, et al. Visualization of vascular inflammation in the atherosclerotic mouse by ultrasmall superparamagnetic iron oxide vascular cell adhesion molecule-1-specific nanoparticles. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32(10):2350–2357. doi: 10.1161/ATVBAHA.112.255224. [DOI] [PubMed] [Google Scholar]
- 140.Tang C, Xue H-L, Bai C-L, Fu R. Regulation of adhesion molecules expression in TNF-α-stimulated brain microvascular endothelial cells by tanshinone IIA: involvement of NF-κB and ROS generation. Phytotherapy Research. 2011;25(3):376–380. doi: 10.1002/ptr.3278. [DOI] [PubMed] [Google Scholar]
- 141.Song HY, Ryu J, Ju SM, et al. Extracellular HIV-1 Tat enhances monocyte adhesion by up-regulation of ICAM-1 and VCAM-1 gene expression via ROS-dependent NF-κB activation in astrocytes. Experimental and Molecular Medicine. 2007;39(1):27–37. doi: 10.1038/emm.2007.4. [DOI] [PubMed] [Google Scholar]
- 142.Jacquier-Sarlin MR, Fuller K, Dinh-Xuan AT, Richard M-J, Polla BS. Protective effects of hsp70 in inflammation. Experientia. 1994;50(11-12):1031–1038. doi: 10.1007/BF01923458. [DOI] [PubMed] [Google Scholar]
- 143.Aridon P, Geraci F, Turturici G, D’amelio M, Savettieri G, Sconzo G. Protective role of heat shock proteins in Parkinson’s disease. Neurodegenerative Diseases. 2011;8(4):155–168. doi: 10.1159/000321548. [DOI] [PubMed] [Google Scholar]
- 144.Luo W, Sun W, Taldone T, Rodina A, Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Molecular Neurodegeneration. 2010;5(1, article 24) doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Current Topics in Medicinal Chemistry. 2009;9(15):1337–1351. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yamashima T. Hsp70.1 and related lysosomal factors for necrotic neuronal death. Journal of Neurochemistry. 2012;120(4):477–494. doi: 10.1111/j.1471-4159.2011.07596.x. [DOI] [PubMed] [Google Scholar]
- 147.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiological Reviews. 2001;81(2):807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 148.Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. Journal of Cerebral Blood Flow and Metabolism. 2002;22(6):631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- 149.Harper SJ, Lograsso P. Signalling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cellular Signalling. 2001;13(5):299–310. doi: 10.1016/s0898-6568(01)00148-6. [DOI] [PubMed] [Google Scholar]
- 150.Hensley K, Floyd RA, Zheng N-Y, et al. p38 Kinase is activated in the Alzheimer’s disease brain. Journal of Neurochemistry. 1999;72(5):2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- 151.Hu J, Akama KT, Krafft GA, Chromy BA, van Eldik LJ. Amyloid-β peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Research. 1998;785(2):195–206. doi: 10.1016/s0006-8993(97)01318-8. [DOI] [PubMed] [Google Scholar]
- 152.Deb S, Zhang JW, Gottschall PE. β-amyloid induces the production of active, matrix-degrading proteases in cultured rat astrocytes. Brain Research. 2003;970(1-2):205–213. doi: 10.1016/s0006-8993(03)02344-8. [DOI] [PubMed] [Google Scholar]
- 153.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 154.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379(6565):557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 155.Prenzel N, Zwick E, Daub H, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402(6764):884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 156.Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(6):3555–3560. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Herrlich A, Daub H, Knebel A, et al. Ligand-independent activation of platelet-derived growth factor receptor is a necessary intermediate in lysophosphatidic, acid-stimulated mitogenic activity in L cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(15):8985–8990. doi: 10.1073/pnas.95.15.8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. The Journal of Biological Chemistry. 2003;278(37):35451–35457. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 159.Tanimoto T, Jin Z-G, Berk BC. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS) The Journal of Biological Chemistry. 2002;277(45):42997–43001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- 160.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. The FASEB Journal. 1999;13(1):9–22. [PubMed] [Google Scholar]
- 161.Lei H, Kazlauskas A. Growth factors outside of the platelet-derived growth factor (PDGF) family employ reactive oxygen species/Src family kinases to activate PDGF receptor α and thereby promote proliferation and survival of cells. The Journal of Biological Chemistry. 2009;284(10):6329–6336. doi: 10.1074/jbc.M808426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen KC-W, Zhou Y, Xing K, Krysan K, Lou MF. Platelet derived growth factor (PDGF)-induced reactive oxygen species in the lens epithelial cells: the redox signaling. Experimental Eye Research. 2004;78(6):1057–1067. doi: 10.1016/j.exer.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 163.Liu B, Neufeld AH. Activation of epidermal growth factor receptors in astrocytes: from development to neural injury. Journal of Neuroscience Research. 2007;85(16):3523–3529. doi: 10.1002/jnr.21364. [DOI] [PubMed] [Google Scholar]
- 164.Ptasznik A, Prossnitz ER, Yoshikawa D, Smrcka A, Traynor-Kaplan AE, Bokoch GM. A tyrosine kinase signaling pathway accounts for the majority of phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulated human neutrophils. The Journal of Biological Chemistry. 1996;271(41):25204–25207. doi: 10.1074/jbc.271.41.25204. [DOI] [PubMed] [Google Scholar]
- 165.Angeloni C, Motori E, Fabbri D, et al. H2O2 preconditioning modulates phase II enzymes through p38 MAPK and PI3K/Akt activation. American Journal of Physiology—Heart and Circulatory Physiology. 2011;300(6):H2196–H2205. doi: 10.1152/ajpheart.00934.2010. [DOI] [PubMed] [Google Scholar]
- 166.Pan J, Chang Q, Wang X, et al. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chemical Research in Toxicology. 2010;23(3):568–577. doi: 10.1021/tx9003193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jr Baldwin AS. The NF-kB and IkB proteins: new discoveries and insights. Annual Review of Immunology. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 168.Haddad JJ. Oxygen-sensitive pro-inflammatory cytokines, apoptosis signaling and redox-responsive transcription factors in development and pathophysiology. Cytokines, Cellular and Molecular Therapy. 2002;7(1):1–14. doi: 10.1080/13684730216401. [DOI] [PubMed] [Google Scholar]
- 169.Barnes PJ, Karin M. Nuclear factor-κB—a pivotal transcription factor in chronic inflammatory diseases. The New England Journal of Medicine. 1997;336(15):1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 170.Eberhardt W, Huwiler A, Beck K-F, Walpen S, Pfeilschifter J. Amplification of IL-1β-induced matrix metalloproteinase-9 expression by superoxide in rat glomerular mesangial cells is mediated by increased activities of NF-κB and activating protein-1 and involves activation of the mitogen-activated protein kinase pathways. The Journal of Immunology. 2000;165(10):5788–5797. doi: 10.4049/jimmunol.165.10.5788. [DOI] [PubMed] [Google Scholar]
- 171.Kim SY, Moon K-A, Jo H-Y, et al. Anti-inflammatory effects of apocynin, an inhibitor of NADPH oxidase, in airway inflammation. Immunology and Cell Biology. 2012;90(4):441–448. doi: 10.1038/icb.2011.60. [DOI] [PubMed] [Google Scholar]
- 172.Cheng S-E, Lin C-C, Lee I-T, Hsu C-K, Kou YR, Yang C-M. Cigarette smoke extract regulates cytosolic phospholipase A2 expression via NADPH oxidase/MAPKs/AP-1 and p300 in human tracheal smooth muscle cells. Journal of Cellular Biochemistry. 2011;112(2):589–599. doi: 10.1002/jcb.22949. [DOI] [PubMed] [Google Scholar]
- 173.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. Journal of Cell Science. 2001;114(13):2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 174.Asahara H, Tartare-Deckert S, Nakagawa T, et al. Dual roles of p300 in chromatin assembly and transcriptional activation in cooperation with nucleosome assembly protein 1 in vitro. Molecular and Cellular Biology. 2002;22(9):2974–2983. doi: 10.1128/MCB.22.9.2974-2983.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ma H, Nguyen C, Lee K-S, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/β-catenin-mediated survivin gene expression. Oncogene. 2005;24(22):3619–3631. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]