

Abstract
Inflammasomes are large cytoplasmic multi-protein complexes that activate caspase-1 in response to diverse intracellular danger signals. Inflammasome components termed NOD-like receptor proteins (NLRs) act as sensors for pathogen associated molecular patterns, stress or danger stimuli. We discovered that arsenicals, including arsenic trioxide and sodium arsenite, inhibited activation of the NLRP1, NLRP3, and NAIP5/NLRC4 inflammasomes by their respective activating signals, anthrax lethal toxin, nigericin, and flagellin. These compounds prevented the autoproteolytic activation of caspase-1 and the processing and secretion of IL-1β from macrophages. Inhibition was independent of protein synthesis induction, proteasome-mediated protein breakdown, or kinase signaling pathways. Arsenic trioxide and sodium arsenite did not directly modify or inhibit the activity of pre-activated recombinant caspase-1. Rather, they induced a cellular state inhibitory to both the autoproteolytic and substrate cleavage activities of caspase-1 which was reversed by the reactive oxygen species (ROS) scavenger N-acetyl-cysteine but not by reducing agents or nitric oxide pathway inhibitors. Arsenicals provided protection against NLRP1-dependent anthrax lethal toxin-mediated death and prevented NLRP3-dependent neutrophil recruitment in a monosodium-urate crystal inflammatory murine peritonitis model. These findings suggest a novel role in inhibition of the innate immune response for arsenical compounds which have been used as therapeutics for a few hundred years.
INTRODUCTION
Inflammasomes are large cytoplasmic multi-protein complexes that form in response to intracellular danger signals. These diverse danger signals include pathogen-derived stimuli such as bacterial toxins, flagellin, dsDNA; self-derived molecules such as uric acid, amyloid crystals, cholesterol, and ATP; and materials of environmental origin such as alum, asbestos and UV radiation (for reviews see (1, 2)). The NLR (nucleotide-binding domain, leucine-rich repeat containing/NOD-like receptor) proteins which act as the sensor components of inflammasomes are activated by several mechanisms. For example, anthrax lethal toxin (LT), a bipartite toxin made of a receptor binding moiety (protective antigen, PA) and a protease (lethal factor, LF), activates rodent NLRP1 inflammasomes by cleaving them in an N-terminal domain (3, 4). Flagellin activates the NAIP5/NLRC4 inflammasome by direct binding (5, 6). The exact mechanisms by which many disparate signals activate the “promiscuous” NLRP3 inflammasome are unknown (2). The end result of activation of all inflammasome sensors is the recruitment of caspase-1 to the sensor complex, followed by its autoproteolytic activation. Activated caspase-1 then rapidly processes the pro-inflammatory cytokines, IL-1β and IL-18, to mature forms, allowing their secretion. These cytokines, which are the first line of defense for the innate immune response, initiate a cascade of other immunological responses. Inflammasome activation is often accompanied by a caspase-1 dependent rapid cell death known as pyroptosis (for reviews see (1, 2)).
Not surprisingly, inflammasomes and the innate immune response play a key role in many infections (7). However, the pro-inflammatory response initiated by inflammasomes has also been implicated in metabolic disorders such as diabetes and inflammatory diseases such as gout and arthritis (8). Furthermore, polymorphisms in the inflammasome NLR sensors are associated with diseases including vitiligo, rheumatoid arthritis, and Alzheimer’s (1). The chronic inflammation etiologically associated with numerous cancers, most notably gastric, hepatic, and colorectal, has also been linked to activation of these sensors (9). Thus, the role played by inflammasome-initiated inflammation in human disease has led to much interest in developing therapeutics targeting inflammasomes or caspase-1.
In this report, we show that activation of multiple inflammasomes is inhibited by arsenical compounds. Sodium arsenite (NaAsO2) and arsenic trioxide (As2O3), known by its trade name Trisenox an FDA-approved drug with established clinical efficacy in treating a number of hematological cancers including acute promyelocytic leukemia and multiple myeloma (10), inhibit LT-induced inflammasome-dependent macrophage pyroptosis when used at clinically relevant doses. These compounds not only inhibit NLRP1 inflammasome activation by LT, but also the NAIP5/NLRC4 and NLRP3 inflammasome responses to their effectors. We found that arsenical compounds inhibit both caspase-1 self-activating autoproteolytic activity as well as pre-activated recombinant caspase-1. The inhibition does not occur through direct modification or inhibition of caspase-1 enzymatic function, but rather through induction of a cytoplasmic environment in intact cells which is inhibitory to its activity. Our findings suggest a novel role for arsenical compounds as inflammasome inhibitors, with possible off-target utility for treatment of inflammatory conditions, as well as a possible explanation of the mechanism for As2O3 efficacy in cytokine-dependent hematological cancers.
MATERIALS AND METHODS
Reagents
Arsenic trioxide (As2O3) and arsenic (III) chloride were purchased from Alfa Aesar (Ward Hill, MA). Other arsenicals included sodium arsenate (MP Biomedicals, Solon, OH) and arsenic (V) oxide (Strem Chemicals, Newburyport, MA). Cacodylic acid, cycloheximide, actinomycin D, puromycin, buthionine sulfoximine, N-acetyl-cysteine (NAC), uric acid, and propidium iodide (PI) were from Sigma-Aldrich (St Louis, MO). Sodium fluoride, sodium orthovanadate, and sodium arsenite were obtained from Fisher Scientific (Pittsburg, PA). Staurosporine was from Biotium (Hayward, CA). Nigericin, anti-Mek1 NT antibody (444942), lactacystin, NG-monomethyl-L-arginine (L-NMMA), and ultrapure lipopolysaccharide (LPS) were purchased from Calbiochem (San Diego, CA). Anti-Mek3NT antibody (sc-959), anti-actin (sc-1616), and anti-caspase1 p10 antibody (sc-514) were from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa Fluor 488-conjugated anti-Ly6 antibody was purchased from Biolegend (San Diego, CA). 2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (Carboxy PTIO), S-ethylisothiourea, and Ng-Nitro-L-arginine-methyl ester (L-NAME) were obtained from Enzo Life Sciences (Farmingdale, NY). Anti-IL-1β antibody (AF-401-NA) was purchased from R&D systems (Minneapolis, MN). Secondary antibodies used in these studies were anti-goat infrared dye (800CW) (Rockland Immunochemicals, Gilbertsville, PA) and anti-rabbit infrared dye (800CW) (Licor Biosciences, Lincoln, NE). Tris (carboxyethyl) phosphine hydrochloride was purchased from Affymetrix (Santa Clara, CA). Monosodium urate (MSU) crystals were prepared by crystallization of uric acid as described (11). Protective antigen (PA) and lethal factor (LF) were purified from Bacillus anthracis as described previously (12). LFn-Fla, a toxin also delivered by PA, is a fusion of the first 254 amino acids of LF to full-length flagellin from Legionella pneumophila (kind gift of Dr. Russell Vance, University of California at Berkeley, Berkeley, CA) (13). FlaTox is a combination of LFn-Fla and PA. Concentrations of LT correspond to the concentration of each toxin component (i.e. 1 μg/mL LT is 1 μg/mL PA + 1 μg/mL LF). Concentrations of FlaTox correspond to the concentration of LFn-Fla. Concentration of PA was always twice that of LFn-Fla in FlaTox experiments (i.e. 1 μg/mL of FlaTox is 2 μg/mL PA + 1 μg/mL LFn-Fla).
Cell culture
RAW264.7 cells and L929 mouse fibroblast cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 10 mM HEPES, and 50 μg/mL gentamicin (all purchased from Life Technologies, Grand Island, NY). Mouse bone marrow cells were cultured in complete DMEM (as above) supplemented with 30% L929 cell-conditioned supernatant and grown 7–9 days to allow time for differentiation to bone-marrow derived macrophages (BMDMs).
Animal studies
All mouse strains used for bone marrow, including mice deficient in promyelocytic leukemia protein (PML) were purchased from Jackson Laboratories (Bar Harbor, ME). Fischer CDF rats were purchased from Charles River (Wilmington, MA). Rats were given As2O3 (7 mg/kg, i.v.) or NaAsO2 (5 mg/kg, i.v.) 30 min prior to LT (12 μg, i.v.) and monitored continuously for malaise or death. For peritonitis studies As2O3 was injected i.p. at 1.25 mg/kg, followed by MSU crystals (i.p., 0.5 mg in 250 μl PBS/mouse). Peritoneal lavages were performed at 2 h with PBS and infiltrating cells counted after hypotonic erythrocyte lysis. For studies on the effects of As2O3 on circulating neutrophils, Ly6-staining of peripheral blood leukocytes was performed followed by flow cytometry analyses on a BD LSRII flow cytometer (BD Biosciences, San Jose, CA). All animal experiments were performed in strict accordance with guidelines from the NIH and the Animal Welfare Act, approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Cytotoxicity assays
RAW264.7 and BALB/cJ BMDM cells were grown in 96-well plates to 90% confluence and pre-treated with various drugs or vehicle at a range of doses or times (as described in figure legends). Cells were then treated with LT, FlaTox, or medium. Cell viability was assessed by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma-Aldrich, St Louis, MO) as previously described (14). In select flow cytometry experiments, propidium iodide was used for viability analysis on a BD LSRII flow cytometer (BD Biosciences, San Jose, CA).
MEK, caspase-1, and IL-1β cleavage
RAW264.7 and BALB/cJ BMDM cells were treated with LPS (1 μg/mL) for 2 h, with or without drugs (at doses and timing indicated in figure legends), prior to addition of inflammasome activators (LT, FlaTox or nigericin, at indicated doses). Cells were then lysed and processed for Western blotting using primary antibodies as previously described (14) in conjunction with IR-dye conjugated secondary antibodies and visualization with the Odyssey infrared imaging system (Licor Biosciences, Lincoln, NE).
In vitro caspase-1 assay
LPS (50 ng/ml for 8 h, or 1 μg/mL for 2 h) was used to induce IL-1β as a substrate for recombinant caspase-1. Sucrose buffer (250 mM sucrose, 10 mM Hepes) lysates of LPS-treated Balb/cJ BMDMs or RAW264.7 cells were incubated with 1 U active recombinant mouse caspase-1 per 50 μL of lysate (MBL International, Woburn, MA) in the presence or absence of arsenical drug, Boc-Asp-(OBzl)-chloromethylketone (Boc-D-CMK, Anaspec, San Jose, CA), or reducing agent for 3 h at 37°C. In other experiments cells were first pre-treated with arsenical drugs for 0.5–8 h (as indicated in figure legends) or NAC for 16 h prior to preparation of lysates. Caspase-1 mediated cleavage of IL-1β was analyzed by Western blotting as previously described (14). Because As2O3 as a potent NF-κB inhibitor prevents LPS-mediated upregulation of IL-1β if applied before LPS, all LPS priming was performed prior to As2O3 application.
Evaluation of caspase-1 sequestration in a high molecular weight complex
Sucrose buffer (250 mM sucrose, 10 mM Hepes) lysates of As2O3 treated or heat shocked (42°C) RAW264.7 cells were centrifuged at 10,000 × g for 10 min at 4°C. The supernatant and pellet were analyzed for caspase-1 by Western blotting as previously described (14).
Evaluation of glutathiolated proteins
Balb/cJ BMDMs were loaded with 250 μM BioGee glutathiolation detection reagent (Life Technologies, Grand Island, NY) for 1 h, as described (15). Cells were then treated with 50 μM of As2O3 for 1 h. Sucrose buffer lysates were prepared and biotin-labeled proteins were precipitated with streptavidin linked agarose (EMD Millipore, Darmstadt, Germany). Precipitated proteins were analyzed for caspase-1 by Western blotting as described (14).
RESULTS
Sodium arsenite and arsenic trioxide protect against LT-induced macrophage death
A screen of NF-κB inhibitors for protection against anthrax LT-induced macrophage pyroptosis identified sodium arsenite (NaAsO2) as a potent inhibitor. Both BALB/cJ BMDMs (Fig. 1A) and RAW264.7 (Supplemental Fig. 1A) were protected by NaAsO2 over a range of concentrations. We investigated other arsenic-containing compounds and found that arsenic trioxide (As2O3), an FDA-approved drug for the treatment of acute promyelocytic leukemia (APL), multiple myeloma and other myelodysplastic syndromes, also protected against LT intoxication (Fig. 1A and Supplemental Fig. 1A). Both drugs were non-toxic at protective doses (Fig. 1A and Supplemental Fig. 1A). Longer incubation times significantly lowered the protective concentration range of As2O3 on BMDMs (Fig. 1B) with doses as low as 3 uM providing 50–60% protection when cells were pre-treated for 16 h. In contrast, RAW264.7 cells did not show an added benefit with longer compound incubation times, requiring a four-fold higher dose to achieve 60% protection (Supplemental Fig. 1B). Another derivative, arsenic (III) chloride had similar protection to As2O3, while sodium arsenate required a ten-fold higher dose for protection (Supplemental Fig. 1C). Arsenical compounds arsenic (V) oxide and cacodylic acid were not protective (Supplemental Fig. 1C).
FIGURE 1.

NaAsO2 and As2O3 protect murine macrophages from LT-induced pyroptosis without affecting LT translocation or proteolytic activity. (A) Balb/cJ BMDMs were incubated with variable concentrations of NaAsO2 or As2O3 for 15 min before challenge with LT (1 μg/mL). Cell viability was assessed by MTT staining after 1.5 h of toxin treatment. Percent viability was assessed compared to untreated cells. (B) Balb c/J BMDMs were treated with As2O3 for the indicated times at the indicated concentrations followed by challenge with LT (1 μg/mL). Cell viability was assessed as above. Balb/cJ BMDMs were incubated with (C) NaAsO2 (75 μM) or (D) As2O3 (50 μM) for 15 min prior to challenge with LT (1 μg/mL) for various periods of time. Western blotting of cell lysates was performed with antibodies against the N-terminus of MEK1 and MEK3.
Sodium arsenite and arsenic trioxide do not inhibit LF translocation or proteolytic activity
To test if As2O3 protects against LT toxicity by inhibiting cytosolic translocation of LF or its proteolytic activity, the cleavage of the toxin’s cytoplasmic mitogen-activated protein kinase kinase (MEK) substrates was monitored in macrophages (16, 17). MEK1 and MEK3 were fully cleaved by 60 min in macrophages treated with LT regardless of whether NaAsO2 (Fig. 1C) or As2O3 (Fig. 1D) were added, demonstrating that arsenical compounds do not affect LT binding, uptake, translocation, or protease activity. Furthermore, the compounds were fully protective if applied 40 min after LT treatment (Supplemental Fig. 1D), a time point at which MEK substrates were fully cleaved (Fig. 1C, 1D). These results indicated that protection occurred by targeting late events downstream of MEK cleavage.
Sodium arsenite and arsenic trioxide inhibit multiple inflammasomes
LT-mediated macrophage death requires NLRP1-mediated activation of caspase-1 (18) which normally begins at 50–60 min after toxin treatment (19). Both NaAsO2 (Fig. 2A, left) and As2O3 (Fig. 2A, right) prevented LT-induced caspase-1 autoproteolysis (upper panels) and subsequent IL-1β processing (middle panels) and secretion (lower panels). Furthermore, addition of the compounds to cells 60 min after LT treatment, at a time when the bulk of the cell’s caspase-1 was processed, did not protect against cell death (Supplemental Fig. 1D. These results indicate that the compounds inhibit the LT-induced NLRP1-mediated activation of caspase-1. To determine if inhibition by arsenical compounds was specific to the NLRP1 inflammasome, the effect of the compounds on activation of the NLRP3 inflammasome by the ionophore nigericin was assessed. As2O3 and NaAsO2 inhibited nigericin-mediated caspase-1 processing and IL-1β maturation (Fig. 2B). Furthermore FlaTox-mediated caspase-1 activation and IL-1β maturation which occurs through the NAIP5/NLRC4 inflammasome was also inhibited by both compounds (Fig. 2C). A slight loss of inhibition was seen over intoxication times approaching 90 min (Fig. 2C). As2O3 also protected against the caspase-1 dependent cell death induced by FlaTox (Supplemental Fig. 2A). These results demonstrate that the compounds inhibit the caspase-1 activation induced by inflammasomes which have completely different activating danger signals and disparate mechanisms of activation.
FIGURE 2.

NaAsO2 and As2O3 prevent inflammasome-mediated caspase-1 activation in macrophages. Balb/cJ BMDMs were primed with LPS (1 μg/mL, 2 h), then pretreated with NaAsO2 (75 μM) or As2O3, (50 μM) for 15 min, and then treated with (A) LT (1 μg/mL), (B) nigericin (50 μM), or (C) FlaTox (1 μg/mL) for various periods of time. Western blotting of culture supernatants for secreted IL-1β (bottom panel) or of lysates for the p10 subunit of caspase-1 and intracellular forms of IL-1β (upper two panels) was performed with appropriate antibodies.
Arsenic trioxide does not directly inhibit caspase-1 enzymatic activity
To determine whether As2O3 inhibits caspase-1 enzymatic activity, its effect on the in vitro proteolytic function of pre-activated purified recombinant murine caspase-1 was tested. Pro-IL-1β present in lysates of LPS-treated macrophages was used as substrate. As2O3 failed to inhibit pro-IL-1β processing over a wide range of concentrations while the potent caspase-1 inhibitor Boc-D-CMK fully blocked processing (Fig. 3A). Similar results were found with NaAsO2 (Supplemental Fig. 3A). Interestingly, lysates made from cells pre-treated with As2O3 at a dose of 7 μM for 8 h (Fig. 3B) or 50 μM for 1 h (Fig. 3C) inhibited caspase-1 enzymatic activity. Furthermore, mixing lysates from As2O3 treated cells with those from untreated cells yielded an intermediate level of caspase-1 inhibition (Fig. 3C). As2O3 at these doses and treatment times does not upregulate pro-IL-1β or cause IL-1β maturation on its own (data not shown). These results demonstrate that As2O3-mediated inflammasome inhibition does not involve the drug’s direct inhibition of caspase-1 proteolytic activity. Rather, conditions induced by As2O3 in intact cells are sufficient to inhibit pre-activated recombinant caspase-1.
FIGURE 3.

As2O3 inhibits caspase-1 through indirect mechanisms. (A) Sucrose lysates from Balb/cJ BMDMs pre-treated with LPS (1 μg/mL, 2 h) were incubated (37°C, 3 h) with active recombinant caspase-1 (1 U/50 μL) in the presence or absence of a range of As2O3 concentrations or positive control caspase-1 inhibitor Boc-D-CMK (400 μM). IL-1β cleavage was monitored by Western blot. (B) Balb/cJ BMDMs pre-treated with LPS (50 ng/mL, 8 h) were incubated with 7 μM As2O3 for 8 h followed by sucrose lysis. Lysates were mixed with active recombinant caspase-1 (1 U/50 μL) and incubated at 37°C for 3 h. IL-1β cleavage was assessed by Western blot. (C) RAW264.7 cells were LPS treated (1 μg/mL, 2 h) followed by As2O3 treatment (50 μM, 1 h). Sucrose lysates from As2O3 pre-treated cells were mixed in various ratios with sucrose lysates from untreated cells. Recombinant caspase-1 was added and IL-1β cleavage monitored as above.
Arsenic trioxide protection is not dependent on caspase-1 sequestration, protein synthesis, proteasome-mediated protein breakdown, or phosphorylation events
We previously showed that heat shock results in the inhibition of caspase-1 through sequestration in a high molecular weight complex that can be separated from the cytosol by centrifugation (20). Because arsenical compounds have been shown to upregulate heat shock proteins (21, 22), we investigated if the mechanism of arsenical inhibition of caspase-1 also involved this high molecular weight complex. Treatment of cells with As2O3 did not cause trapping of caspase-1 into a high molecular weight complex (Supplemental Fig. 3B).
We hypothesized that As2O3-mediated upregulation of cellular stress proteins or inhibitory complexes could contribute to caspase-1 inhibition. Therefore, we tested the effects of the transcription inhibitor actinomycin D and the translation inhibitor puromycin to determine if protein synthesis was required for As2O3 protection. Pretreatment of macrophages with a range of concentrations of puromycin or actinomycin D (Fig. 4A) did not reverse As2O3 protection, indicating that protein synthesis is not necessary for As2O3-mediated inflammasome inhibition. These results were supported by our previous finding that NaAsO2 and As2O3 could protect cells even when applied 40 min after LT intoxication, making a requirement for new protein synthesis unlikely. Similar results were found with NaAsO2 and with use of translation inhibitor cycloheximide (Supplemental Fig. 4A).
FIGURE 4.

Protein synthesis or breakdown are not required for protection but can be reversed by ROS scavengers. (A) RAW264.7 cells were treated with variable concentrations of puromycin or actinomycin D for 1 h before incubation with As2O3 (50 μM, 15 min). Cells were then challenged with LT (1 μg/mL) and cell viability assessed at 2 h. (B) Balb/cJ BMDMs were primed with 1 μg/mL LPS for 1.5 h and pretreated with 20 μM lactacystin for 1 h, followed by As2O3 (50 μM, 15 min). Cells were then treated with FlaTox (1 μg/mL, 1 h). Western blotting was performed with antibodies against IL-1β. (C) RAW264.7 cells were pretreated for 16 h with variable concentrations of NAC, or (inset) with 25mM NAC, followed by As2O3 (50 μM, 15 min) and LT (1 μg/mL, 2 h). Cell viability was assessed by MTT staining and determined relative to untreated cells. Alternatively, cell death was evaluated by propidium iodide staining (inset). (D) Balb/cJ BMDMs were pretreated overnight with 25 mM NAC and primed with 1 μg/mL LPS for 2 h, followed first by As2O3 (50 μM, 15 min) and then by LT (1 μg/mL, 2 h). Western blotting was performed with antibodies against IL-1β. (E) RAW264.7 cells were pretreated for 16 h with 25 mM NAC. Cells were then treated and processed as in Figure 3C. (F) RAW264.7 cells were LPS treated (1 μg/mL, 2 h) followed by As2O3 treatment (50 μm, 1 h). Sucrose lysates were spiked with varying concentrations of DTT. Recombinant caspase-1 was added and IL-1β cleavage monitored as above.
Because proteasome inhibition prevents activation of the NLRP1 inflammasome (19, 23) we tested the role of protein breakdown induced by the arsenical compounds in the inhibition of the NAIP5/NLRC4 inflammasome. Unlike NLRP1 activation, which requires proteasome activity, FlaTox activation of the NAIP5/NLRC4 inflammasome is not impacted by proteasome inhibition (Fig. 4B and Supplemental Fig. 2B). We found that treatment of cells with the proteasome inhibitor lactacystin did not reverse As2O3 protection against FlaTox or As2O3 inhibition of IL-1β processing (Fig. 4B and Supplemental Fig. 2B), indicating that As2O3 did not manifest its effects through proteasome-mediated breakdown of a protein or proteins.
Finally, neither phosphatase inhibitors sodium fluoride and sodium vanadate nor the kinase inhibitor staurosporine reversed the inhibition of caspase-1 enzymatic activity in lysates of As2O3 treated cells (Supplemental Fig. 3C). Similarly, pretreatment of cells with phosphatase or kinase inhibitors had no effect on As2O3-based protection from LT mediated pyroptosis (data not shown). These results indicate that it is unlikely As2O3-based protection involves phospho-signaling pathways.
Arsenic trioxide protection requires reactive oxygen species but does not involve the nitric oxide pathway
As2O3 and NaAsO2 have been shown to directly induce ROS and some of their effects occur through the modification of ROS-controlling cellular enzymes (24). We utilized the potent anti-oxidant and ROS scavenger N-acetyl-cysteine (NAC) to determine if the protective effects of As2O3 require induction of an altered oxidative state in cells. Pretreatment of RAW264.7 cells with NAC, in a dose range where this ROS scavenger did not impact NLRP1 inflammasome activation, reversed the protective effects of As2O3 on LT-treated macrophages (Fig. 4C). NAC treatment of Balb/cJ BMDMs also restored IL-1β processing in response to LT treatment (Fig. 4D). NAC treatment also reversed the caspase-1 inhibitory ability of lysates made from As2O3 treated cells (Fig. 4E).
It has been shown that a generally oxidative cellular environment can lead to inhibition of caspase-1 through reversible glutathiolation of catalytic cysteine residues (15), and we hypothesized that arsenic treatment may create such an oxidative environment. However, treatment with the ROS generator buthionine sulfoximine or with hydrogen peroxide (Supplemental Fig. 4B) did not protect cells from LT-induced pyroptosis. Additionally, spiking lysates from As2O3 treated cells with the reducing agents DTT (Fig. 4F) or Tris (carboxyethyl) phosphine hydrochloride (data not shown) did not reverse the inhibitory effect of arsenic treatment on caspase-1 enzymatic activity. Finally, utilizing the biotin-labeled glutathione analog BioGee coupled with immunoprecipitation of caspase-1 from As2O3 treated cells, we found that caspase-1 was not glutathiolated in response to arsenic treatment (data not shown).
Inhibitory nitrosylation of caspase-1 were also eliminated as possible modifications. Pretreatment of cells with various inhibitors of NOS or with a scavenger of nitric oxide did not reverse As2O3 protection (Supplemental Fig. 4C).
These results indicate that specific ROS changes induced by As2O3 (but not general increases in ROS) are required to inhibit inflammasome activation. This also suggests that an ROS scavenger such as NAC can have both activating and inhibitory effects on inflammasome activation.
Arsenic trioxide inhibition of inflammasome activation is independent of promyelocytic leukemia protein degradation
As2O3 is known to catalyze the degradation of the promyelocytic leukemia protein (PML) (25). A recent report described inhibition of the NLRP3 inflammasome by As2O3 (26) and attributed this inhibition to the drug’s degradation of PML, which was suggested to be essential to NLRP3 inflammasome activation. Evidence for the breakdown of PML as the mechanism of NLRP3 inflammasome inhibition by As2O3, however, was not directly shown in those studies. To determine if PML degradation is the mechanism of As2O3 mediated inflammasome inhibition, we assessed the effect of As2O3 on inflammasome activation in BMDMs deficient in PML. As2O3 inhibited IL-1β maturation in response to NLRP3 (Fig. 5A) and NAIP5/NLRC4 (Fig. 5B) stimuli in PML deficient macrophages. The effect of LT-induced NLRP1 activation was not assessed in the knockout mice which are on the LT-nonresponsive C57BL/6 background (18). These results demonstrate that PML degradation is not required, or responsible for, As2O3-mediated inflammasome inhibition.
FIGURE 5.
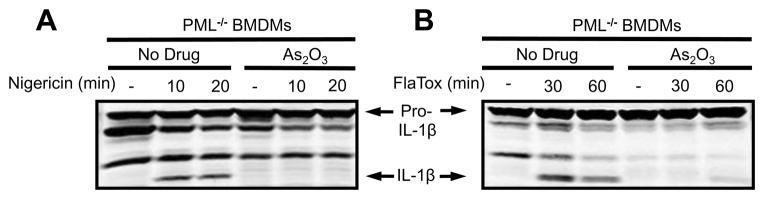
PML degradation is not the mechanism of As2O3-mediated inflammasome inhibition. PML−/ − BMDMs were primed with LPS (1 μg/mL, 2 h), then pretreated with As2O3, (50 μM) for 15 min, and then treated with (A) nigericin (50 μM) or (B) FlaTox (1 μg/mL) for various periods of time. Western blotting of lysates for intracellular forms of IL-1β was performed with appropriate antibodies.
Arsenic trioxide reduces inflammatory cell infiltrate in a murine peritonitis model
The anti-inflammatory properties of As2O3 were tested in a murine peritoneal inflammation model. As2O3 caused up to a two-fold reduction in the number of cells recruited to the peritoneum when administered i.p. (Fig. 6A) or i.v. (data not shown) 30 min prior to MSU crystals. This decrease was not due to As2O3-mediated death of peripheral neutrophils, since i.v. injection of As2O3 did not cause a significant decrease in the number of neutrophils in peripheral blood (data not shown). These results demonstrate that As2O3 can also inhibit the NLRP3 inflammasome in vivo.
FIGURE 6.

NaAsO2 and As2O3 inhibit inflammasomes in vivo. (A) Inflammatory cell recruitment was assessed after 2 h in the peritoneum of Balb/cJ mice injected i.p. with MSU (0.5 mg/250 μl/mouse). Mice had been injected with either PBS (i.p.) or As2O3 (1.25 mg/kg, i.p.). Error bars represent standard error of the mean. n = 5 animals/group. P = 0.0291. (B) Fischer rats pretreated with 7 mg/kg As2O3 i.v. (n=7) or 5 mg/kg NaAsO2 i.v. (n=7) were challenged with LT (12 μg, i.v.). Times to death (one symbol representing one animal) were compared to control animals that did not receive drug treatment (n=5). P-values comparing each treatment group to controls are <0.0075.
Sodium arsenite and arsenic trioxide extend survival time in LT-treated rats
LT induces rapid death of rats in 40–100 min through an unknown, but NLRP1-dependent, process (27). Pretreatment of toxin-sensitive Fischer rats with As2O3 or NaAsO2 prior to LT challenge extended mean time to death (Fig. 6B), demonstrating the ability of these drugs to alter the outcome of NLRP1/caspase-1 activation in vivo.
DISCUSSION
In this work we show that arsenical compounds inhibit activation of caspase-1 and IL-1β processing by the NLRP1, NLRP3 and NAIP5/NLRC4 inflammasome sensors. This inhibition of caspase-1 autoproteolytic activation and downstream cytokine processing was not due to inhibition of the uptake or activity of the inflammasome activating stimuli/toxins. It was also not a result of direct modification or inactivation of caspase-1 enzymatic activity. Instead inhibition occurred through induction of a cellular state, not involving protein synthesis, proteasome function or phosphorylation events, that was inhibitory to both caspase-1’s autoproteolytic processing and its enzymatic activity toward its cytokine substrates. A potent ROS scavenger, N-acetyl-cysteine could reverse the protection provided by the arsenical compounds, which have been previously shown to be potent inducers of ROS (24). Our findings suggest that arsenical compounds may be considered as a treatment in many conditions which involve inflammasome activation and uncontrolled proinflammatory responses, including gout, arthritis and various inherited disorders (28).
Arsenic has been used therapeutically for over 2,000 years in traditional Chinese medicine and in Western medicine since the time of Hippocrates. Its extensive use in the 18th and 19th centuries expanded to treatment of eczema, ulcers, malaria, plague, asthma, psoriasis, anemia, arthritis, epilepsy, Hodgkin’s disease, and leukemia (10, 29). Fowler’s solution, an alkaline solution of white arsenic, became a foundation of 19th century pharmacopeia and remained in use until 1950. Pharmacology texts in 1880 described the curative properties of arsenic as “almost magical,” and no other medication was said to remedy such an extensive assortment of ailments (30–32). Arsenic has a wide range of effects on cellular proteins and pathways. It has the ability to directly bind to thiol groups on proteins, potentially altering their function. A number of cellular proteins have been identified that are modified by arsenic, including the catalytic subunit of IκB kinase, resulting in inhibition of the NF-κB pathway and dampening inflammatory responses at the transcriptional level (33, 34). Modification of the antioxidant enzymes glutathione peroxidase and thioredoxin reductase (35, 36) results in potent ROS induction by arsenical compounds. In this work we present a new effect of arsenical compounds, as inhibitors of IL-1β processing and secretion.
Caspase-1 is the IL-1β activating/cleaving enzyme (ICE) responsible for the first line of cytokine responses by resident innate immune cells at all tissue sites. We found that As2O3 and NaAsO2 inhibit inflammasome–induced caspase-1 activation. The enzymatic activity of caspase-1, a cysteine protease, is dependent on catalytic cysteine residues that may be vulnerable to arsenic modification. Because arsenic compounds did not inhibit caspase-1 proteolytic activity in cell lysates, this possibility was eliminated. Lysates prepared from cells pre-treated with As2O3, however, inhibited cleavage of IL-1β by recombinant caspase-1, indicating a cellular condition inhibitory to its activity was induced in cells. Because generation of the autoproteolytic fragment p10 requires recruitment of pro-caspase-1 to the NLR platform, we hypothesized that this recruitment was possibly inhibited. One hypothesis was that arsenic compounds could induce binding of an endogenous caspase-1 inhibitory protein or cofactor within the cell. Heat shock inhibits caspase-1 through sequestration of the enzyme into a high molecular weight complex with unknown binding partner (20). Because arsenic treatment is known to induce a heat-shock like cellular state (21, 22), we tested if a similar large molecular complex was formed following treatment with As2O3. Arsenical treatment did not induce sequestration of caspase-1 into a high molecular weight complex. Arsenic treatment could also result in signaling events that lead to phosphorylation of various cellular proteins involved in interactions with or inhibition of caspase-1. We found that pan-kinase and phosphatase inhibitors did not impact As2O3 effects on caspase-1 activity.
Arsenic is a potent inducer of ROS (24). The antioxidant NAC reversed the protective effects of As2O3 and restored caspase-1 activity and IL-1β release, indicating that ROS generation could play a role in the protective effects of the drug. Paradoxically, some investigators have found that ROS production can be an activation signal for the NLRP3 inflammasome (37) and that ROS causes the induction of NLRP3 expression (38). Here we demonstrate that a known potent inducer of ROS can also inhibit NLRP3 inflammasome activation. Our findings support the hypothesis that ROS production is but one of a combination of cellular signals necessary for NLRP3 activation (39) and that certain ROS events can inhibit activation (40). Importantly, general increases in ROS do not inhibit all inflammasomes, as the pro-oxidants buthionine sulfoximine and hydrogen peroxide did not protect cells from LT or caspase-1 induced death. Therefore site-specific ROS regulation may be an important factor in arsenical compound mediated effects.
Reversible modification of redox-sensitive and catalytically necessary cysteine residues on caspase-1 under conditions of high ROS have been observed (15). Therefore, we hypothesized that redox dependent protein modifications could be the mechanism of arsenic based inhibition of caspase-1. Inhibitors of NOS or a nitric oxide scavenger, however, did not alter the effects of As2O3. Similarly, various reducing agents did not alter arsenic-based inhibition of caspase-1. Finally, we did not find glutathiolated caspase-1 in arsenic-treated cells. Therefore, we found no evidence of redox based caspase-1 modifications due to arsenic treatment. ROS increases can induce expression of numerous proteins, and it is known that arsenicals induces various cellular stress responses such as expression of heat shock proteins and the tumor suppressor protein p53 (24). We found that inhibitors of protein synthesis did not alter NaAsO2 or As2O3 protection against inflammasome activation, indicating that de novo protein synthesis was not required for arsenical compound effects.
Another effect of As2O3 is the breakdown of different cellular proteins, such as promyelocytic leukemia protein (PML) (25). While this manuscript was in preparation, Lo et. al. (26) described inhibition of the NLRP3 inflammasome by As2O3 and attributed this inhibition to degradation of PML, which they suggested to be essential to NLRP3 inflammasome formation. We demonstrate that PML-breakdown is not the mechanism of As2O3-mediated NLRP3 inflammasome inhibition. Furthermore, we show that the drug’s inhibitory effect is not specific to the NLRP3 inflammasome as we find potent inhibition of the NLRP1 and NAIP5/NLRC4 inflammasomes, which presumably do not require PML. As2O3 was effective at inhibiting IL-1β processing in PML-deficient macrophages in response to both NLRP3 and NAIP5/NLRC4 activating stimuli. Moreover, we demonstrate that proteasome activity, which catalyzes the breakdown of PML by As2O3 (25), is not necessary for inflammasome inhibition by this drug. Instead, our results show that, in addition to preventing the autoproteolytic activation of caspase-1 in intact cells, the drug can inhibit activity of pre-activated recombinant caspase-1.
As2O3 is an FDA-approved drug for treatment of refractory acute promyelocytic leukemia (APL), multiple myeloma, and other lymphoma conditions. Single agent therapy with As2O3 or combination therapy with all-trans retinoic acid leads to complete remission in over 80% of newly diagnosed APL patients (41). Recent clinical trials have demonstrated modest efficacy of As2O3 alone in treating advanced or refractory multiple myeloma (42). Trials of myelodysplastic syndrome patients treated with As2O3 alone or in combination with thalidomide have demonstrated hematological improvement and increases in progression free and overall survival compared to control patients (43). A number of mechanisms have been proposed to explain the drug’s efficacy in treatment of these cancers, including apoptosis, degradation of PML, induction of oxidative damage, and inhibition of angiogenesis and NF-κB signaling (for review see (44)). We hypothesize that the ability of As2O3 to inhibit caspase-1, and thus IL-1β inflammatory signaling, plays a major role in its anti-cancer effects. Many cancers cells, including multiple myeloma, breast cancer, and advanced melanoma cells actively secrete IL-1β, providing a proliferative advantage through either autocrine or paracrine signaling (45–47). For example, IL-1β secreted by myeloma cells has been shown to have paracrine effects on bone marrow stromal cells, inducing them to produce the IL-6 that is required for a proliferative cancer environment (45). IL-1β signaling has also been shown to play a role in tumor angiogenesis (48). As2O3 is already known to potently inhibit NF-κB signaling (33, 34) and thus this compound obstructs inflammation at two distinct steps; NF-κB dependent upregulation of pro-inflammatory cytokines, such as IL-1β, as well as maturation and release of the cytokine through inhibition of caspase-1 activation as shown in this work. Thus, As2O3-mediated disruption of IL-1β maturation may partially account for the successful treatment of multiple myeloma patients with As2O3 in recent clinical trials. The varied effects of As2O3 on cellular processes and its continued use as a therapeutic necessitate further study of its molecular mechanisms of action.
Because of the roles that inflammasomes and caspase-1 mediated inflammatory signaling play in various other human malignancies (8), the identification of inflammasome-inhibiting compounds is of clinical importance (49). We suggest that As2O3 and other arsenical compounds can also be utilized as potent anti-inflammatories for treatment of localized diseases, such as in topical treatments for skin conditions. These drugs have been used to treat animal models of asthma, coronary restenosis, colitis, and lupus (50–53). Gout, a condition in which uric acid crystals in joints activate the NLRP3 inflammasome to cause painful inflammation (54), is an example of a condition that could benefit from a caspase-1 inhibitory treatment. We demonstrated a two-fold reduction in MSU-induced cell recruitment upon treatment with As2O3, comparable to what was seen with allopurinol in other studies (55). One can imagine other possibilities for use of these compounds as anti-inflammatories in diseases where caspase-1 activation plays a prominent role.
Our studies demonstrate that arsenical compounds, including the FDA-approved anti-cancer therapeutic Trisenox, are potent inhibitors of caspase-1 and the innate immune response and thus may have potential in treatment for inflammatory disorders. The application of the drug’s anti-inflammatory actions in various inflammasome-related diseases is an area of future study by our laboratory.
Supplementary Material
Acknowledgments
We thank Dr. Russell Vance for providing the LFn-Fla protein. We thank the Flow Cytometry Section of the NIAID Research Technologies Branch for use of their facilities and expertise.
This research was supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
Abbreviations used in this article
- ROS
reactive oxygen species
- NAC
N-acetyl-cysteine
- NLRP1
nucleotide-binding oligomerization domain, leucine-rich repeat proteins 1
- NLRP3
nucleotide-binding oligomerization domain, leucine-rich repeat proteins 3
- NAIP5
nucleotide-binding oligomerization domain-like receptor family, apoptosis inhibitory protein 5
- NLRC4
nucleotide-binding oligomerization domain -like receptor family, caspase-1 recruitment domain-containing protein 4, APL, acute promyelocytic leukemia
- PML
promyelocytic leukemia protein
- BMDM
bone-marrow derived macrophages
- LT
lethal toxin (toxin consisting of lethal factor and protective antigen)
- FlaTox
toxin consisting of LFn-Fla and protective antigen
- LF
lethal factor
- PA
protective antigen
- LFn-Fla
fusion protein between LF N-terminus and flagellin
References
- 1.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 2.Lamkanfi M, V, Dixit M. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 3.Hellmich Ka, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, Liu S, Leppla SH, Moayeri M. Anthrax lethal factor cleaves mouse Nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PloS One. 2012;7:e49741. doi: 10.1371/journal.pone.0049741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012;8:e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 7.Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffman HM, Wanderer AA. Inflammasome and IL-1B-mediated disorders. Curr Allergy Asthma Rep. 2010;10:229–235. doi: 10.1007/s11882-010-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343–351. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
- 10.Emadi A, Gore SD. Arsenic trioxide - an old drug rediscovered. Blood Rev. 2010;24:191–199. doi: 10.1016/j.blre.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin WJ, Walton M, Harper J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. 2009;60:281–289. doi: 10.1002/art.24185. [DOI] [PubMed] [Google Scholar]
- 12.Park S, Leppla SH. Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr Purif. 2000;18:293–302. doi: 10.1006/prep.2000.1208. [DOI] [PubMed] [Google Scholar]
- 13.von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman ZL, Sirianni N, Mawhinney C, Lee MS, Leppla SH, Moayeri M, Johansen LM. Auranofin protects against anthrax lethal toxin-induced activation of the Nlrp1b inflammasome. Antimicrob Agents Chemother. 2011;55:1028–1035. doi: 10.1128/AAC.00772-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol. 2008;9:866–872. doi: 10.1038/ni.1633. [DOI] [PubMed] [Google Scholar]
- 16.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 17.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- 18.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 19.Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2008;10:332–343. doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin TC, Wickliffe KE, Leppla SH, Moayeri M. Heat shock inhibits caspase-1 activity while also preventing its inflammasome-mediated activation by anthrax lethal toxin. Cell Microbiol. 2008;10:2434–2446. doi: 10.1111/j.1462-5822.2008.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhagat L, V, Singh P, Dawra RK, Saluja AK. Sodium arsenite induces heat shock protein 70 expression and protects against secretagogue-induced trypsinogen and NF-kappaB activation. J Cell Physiol. 2008;215:37–46. doi: 10.1002/jcp.21286. [DOI] [PubMed] [Google Scholar]
- 22.Kim YH, Park EJ, Han ST, Park JW, Kwon TK. Arsenic trioxide induces Hsp70 expression via reactive oxygen species and JNK pathway in MDA231 cells. Life Sci. 2005;77:2783–2793. doi: 10.1016/j.lfs.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 23.Muehlbauer SM, Lima H, Goldman DL, Jacobson LS, Rivera J, Goldberg MF, Palladino MA, Casadevall A, Brojatsch J. Proteasome inhibitors prevent caspase-1-mediated disease in rodents challenged with anthrax lethal toxin. Am J Pathol. 2010;177:735–743. doi: 10.2353/ajpath.2010.090828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flora SJS. Arsenic-induced oxidative stress and its reversibility. Free Radic Biol Med. 2011;51:257–281. doi: 10.1016/j.freeradbiomed.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Lallemand-Breitenbach V, Zhu J, Chen Z, de Thé H. Curing APL through PML/RARA degradation by As2O3. Trends Mol Med. 2012;18:36–42. doi: 10.1016/j.molmed.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Lo YH, Huang YW, Wu YH, Tsai CS, Lin YC, Mo ST, Kuo WC, Chuang YT, Jiang ST, Shih HM, Lai MZ. Selective inhibition of the NLRP3 inflammasome by targeting to promyelocytic leukemia protein in mouse and human. Blood. 2013;121:3185–3194. doi: 10.1182/blood-2012-05-432104. [DOI] [PubMed] [Google Scholar]
- 27.Newman ZL, Printz MP, Liu S, Crown D, Breen L, Miller-Randolph S, Flodman P, Leppla SH, Moayeri M. Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog. 2010;6:e1000906. doi: 10.1371/journal.ppat.1000906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 29.Waxman S, Anderson KC. History of the development of arsenic derivatives in cancer therapy. Oncologist. 2001;6:3–10. doi: 10.1634/theoncologist.6-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- 30.Doyle D. Notoriety to respectability: a short history of arsenic prior to its present day use in haematology. Br J Haematol. 2009;145:309–317. doi: 10.1111/j.1365-2141.2009.07623.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhu J, Chen Z, Lallemand-Breitenbach V, de Thé H. How acute promyelocytic leukaemia revived arsenic. Nat Rev Cancer. 2002;2:705–713. doi: 10.1038/nrc887. [DOI] [PubMed] [Google Scholar]
- 32.Aronson S. Arsenic and old myths. R I Med. 1994;77:233–234. [PubMed] [Google Scholar]
- 33.Kapahi P, Takahashi T, Natoli G, Adams SR, Chen Y, Tsien RY, Karin M. Inhibition of NF-kappaB activation by arsenite through reaction with a critical cysteine in the activation loop of Ikappa B kinase. J Biol Chem. 2000;275:36062–36066. doi: 10.1074/jbc.M007204200. [DOI] [PubMed] [Google Scholar]
- 34.Roussel RR, Barchowsky A. Arsenic inhibits NF-kappaB-mediated gene transcription by blocking IkappaB kinase activity and IkappaBalpha phosphorylation and degradation. Arch Biochem Biophys. 2000;377:204–212. doi: 10.1006/abbi.2000.1770. [DOI] [PubMed] [Google Scholar]
- 35.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Styblo M, Serves SV, Cullen WR, Thomas DJ. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem Res Toxicol. 1997;10:27–33. doi: 10.1021/tx960139g. [DOI] [PubMed] [Google Scholar]
- 37.Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–619. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- 38.Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 40.Rubartelli A. Redox control of NLRP3 inflammasome activation in health and disease. J Leukoc Biol. 2012;92:951–958. doi: 10.1189/jlb.0512265. [DOI] [PubMed] [Google Scholar]
- 41.Lengfelder E, Hofmann WK, Nowak D. Impact of arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia. 2012;26:433–442. doi: 10.1038/leu.2011.245. [DOI] [PubMed] [Google Scholar]
- 42.Röllig C, Illmer T. The efficacy of arsenic trioxide for the treatment of relapsed and refractory multiple myeloma: a systematic review. Cancer Treat Rev. 2009;35:425–430. doi: 10.1016/j.ctrv.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Wei W, Zhou F, Zhang Y, Guo L, Shi H, Hou J. A combination of thalidomide and arsenic trioxide is effective and well tolerated in patients with myelodysplastic syndromes. Leuk Res. 2012;36:715–719. doi: 10.1016/j.leukres.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 44.Carney DA. Arsenic trioxide mechanisms of action--looking beyond acute promyelocytic leukemia. Leuk Lymphoma. 2008;49:1846–1851. doi: 10.1080/10428190802464745. [DOI] [PubMed] [Google Scholar]
- 45.Lust JA, Donovan KA. The role of interleukin-1 beta in the pathogenesis of multiple myeloma. Hematol Oncol Clin North Am. 1999;13:1117–1125. doi: 10.1016/s0889-8588(05)70115-5. [DOI] [PubMed] [Google Scholar]
- 46.Dunn JH, Ellis LZ, Fujita M. Inflammasomes as molecular mediators of inflammation and cancer: potential role in melanoma. Cancer Lett. 2012;314:24–33. doi: 10.1016/j.canlet.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Goldberg JE, Schwertfeger KL. Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Curr Drug Targets. 2010;11:1133–1146. doi: 10.2174/138945010792006799. [DOI] [PubMed] [Google Scholar]
- 48.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–329. doi: 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.López-Castejón G, Pelegrín P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs. 2012;21:995–1007. doi: 10.1517/13543784.2012.690032. [DOI] [PubMed] [Google Scholar]
- 50.Bobe P, Bonardelle D, Benihoud K, Opolon P, Chelbi-Alix MK. Arsenic trioxide: a promising novel therapeutic agent for lymphoproliferative and autoimmune syndromes in MRL/ lpr mice. Blood. 2006;108:3967–3975. doi: 10.1182/blood-2006-04-020610. [DOI] [PubMed] [Google Scholar]
- 51.Shen L, Gong F, Tian W, Li W, Zhang F, Qian J, Sun A, Zou Y, Yang W, Ge J. Anti-inflammatory effects of arsenic trioxide eluting stents in a porcine coronary model. Biomed Res Int. 2013;2013:1–9. doi: 10.1155/2013/937936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singer M, Trugnan G, Chelbi-Alix MK. Arsenic trioxide reduces 2,4,6-trinitrobenzene sulfonic acid-induced murine colitis via nuclear factor-kB down-regulation and caspase-3 activation. Innate Immun. 2011;17:365–374. doi: 10.1177/1753425910371668. [DOI] [PubMed] [Google Scholar]
- 53.Xu Z-p, Huo J-m, Sang Y-l, Kang J, Li X. Effects of arsenic trioxide (As2O3) on airway remodeling in a murine model of bronchial asthma. Can J Physiol Pharmacol. 2012;90:1576–1584. doi: 10.1139/y2012-127. [DOI] [PubMed] [Google Scholar]
- 54.Kingsbury SR, Conaghan PG, McDermott MF. The role of the NLRP3 inflammasome in gout. J Inflamm Res. 2011;4:39–49. doi: 10.2147/JIR.S11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.