

Abstract
pH-responsive megagates have been fabricated around mesoporous silica material SBA-15 in order to mechanize the mesopores. These megagates remain closed in neutral conditions, but open at pH 5. The capping components of the megagates were designed to be capable of controlling pores up to 6.5 nm in diameter. Selectivity of payloads with different sizes can be achieved through the use of different capping components. The operation of the megagates was demonstrated by time-resolved fluorescence spectroscopy which is capable of monitoring the release of both the payload and the cap. This study opens up new possibilities in the field of controllable release.
Introduction
Mesoporous silica materials have been studied widely in the fields of controlled release and drug delivery,1 on account of their high surface areas, porous structures, and ease of functionalization. One of the most important applications of these materials has been to employ them as scaffolds for the controlled release of cargo molecules.2 In order to achieve this end, the access to the pore opening needs to be controlled, preferably by means of a chemical method that can mechanize the pores so as to result in a stimulus-responsive release. A variety of methods have been developed to meet this criterion, such as macromolecule capping,3 nanocrystal capping,4 polymer coating,5 and nanomachine construction.6 These methods have all proven to be successful and many of them have resulted in materials with potential biomedical applications.2,3,6b These methods, however, have mostly been applied only to MCM-41-type materials in which the typical pore size is 2–3 nm. This choice of materials limits considerably the size range of molecules that can serve as payloads, thus restricting the future applications of the delivery systems. Some pioneering works have demonstrated the storage and release of larger cargos,7 such as proteins, polymers, and RNAs, from large pored mesoporous silica materials, where the simple electrostatic interaction between the cargos and the silica served as the dominant storage-release mechanism. It remains challenging to design a method to achieve on-demand release in a more precisely controlled manner.
In this paper, we describe the design and synthesis of a cargo size-selective pH-responsive megagate system that is able to mechanize mesopores with diameters of up to 6.5 nm. The megagates are designed to be fluorescent so that their operation may be monitored facilely. We have chosen a well-established standard polymer (dextran) as a model cargo for proof of principle. By using another cargo molecule with a smaller size—fluorescein disodium salt—we show that size-selective controlled release can be achieved upon tuning the capping components of the megagates.
Experimental
General methods
All reactions were performed under an argon atmosphere and in dry solvents, unless otherwise stated. Tetraethylorthosilicate (99%) and 1-butylaldehydetriethoxysilane (90%) were purchased from Gelest Inc. (PA). Propargyl bromide (80% in toluene), 2-[2-(2-chloroethoxy)-ethoxy]-ethanol (96%), 1-adamantanecarbonyl chloride (95%), copper(II) sulfate pentahydrate (98%), sodium L-ascorbate (98%), Pluronic P123, fluorescein disodium salt (98.5%), and fluorescein isothiocyanate-dextran (MW 3k), and all solvents were purchased from Sigma-Aldrich (St. Louis, MO). 6-(2-Aminoethyl)amino-6-deoxy-β-cyclodextrin,81,9 and 3 (ref. 10) were synthesized according to literature procedures. Analytical thin-layer chromatography (TLC) was performed on aluminum sheets, precoated with silica gel 60-F254 (Merck 5554). Column chromatography was carried out using silica gel 60F (230–400 mesh). 1H and 13C NMR spectra were recorded on Bruker ARX 500 MHz and DSX 500 MHz spectrometers, respectively, at ambient temperature, unless otherwise noted. Solid-state 13C CP/MAS Spectra was recorded on a Bruker DSX 300 MHz spectrometer. High-resolution MS data were recorded on an Applied Biosystems-MDS Sciex 4000 Q Trap with ESI sources. The chemical shifts are listed in ppm on the δ scale and coupling constants were recorded in Hertz (Hz). Chemical shifts are reported in ppm relative to the signals corresponding to the residue non-deuterated solvents (CDCl3: δ 7.26 ppm, CD3SOCD3: δ 2.47 ppm). The following abbreviations are used to explain the multiplicities: s, singlet; d, doublet; t, triplet; b, broad peaks; m, multiplet or overlapping peaks.
Synthesis of compound 2
SiCl4 (5.25 mL, 45.7 mmol) was added over 5 min to a solution of 1 (ref. 9) (2.0 g, 11.5 mmol) in EtOH (10 mL). After the reaction mixture had been stirred for a further 15 min, the precipitates were collected by vacuum filtration and washed with MeOH to yield 2 as a pink solid (1.1 g, 61%). 1H NMR (500 MHz, CDCl3): δ = 7.66 (s, 3H), 7.63 (d, J = 8.7 Hz, 6H), 7.09 (d, J = 8.7 4.76 (s,3H), 7.63 (d, J = 8.7Hz, 6H), 7.09 (d, J = 8.7Hz, 6H), 4.76 (d, J = 2.4Hz, 6H), 2.56 (t, J=2.2Hz, 3H). 13CNMR(125MHz,CDCl3): δ = 157.2, 141.7, 134.7, 128.4, 124.1, 115.2, 78.5, 75.7, 55.9.
Synthesis of compound 4
Anhydrous K2CO3 (0.77 g, 5.6 mmol) was added to a solution of 3 (ref. 10) (0.23g, 0.7 mmol) in anhydrous DMF (30mL). The solution was purged with argon for 30 min. Propargyl bromide (80% in PhMe, 700 μL, 7.9 mmol) was added to this solution. The mixture was heated at 80 °C under argon protection for 18 h. After cooling to room temperature, the mixture was poured into ice water (150 mL). The precipitate was collected by filtration, washed with EtOAc (60 mL) and Me2CO–H2O (70 mL, 3 : 4 w/v), and dried under vacuum to afford 4 as an off-white powder (0.3 g, 77%). 1H NMR (500 MHz, CD3SOCD3): δ = 8.12 (s, 6H), 5.08 (s, 12H), 3.60 (s, 6H).
Synthesis of compound 5
Sodium azide (3 g, 46.1 mmol) was added to a solution of 2-[2-(2-chloroethoxy)-ethoxy]ethanol (5.0 g, 29.7 mmol) in anhydrous DMF (50 mL). The mixture was heated at 80 °C for 18 h under argon. After cooling to room temperature, the mixture was concentrated by rotary evaporation, and the residue was dissolved in anhydrous CH2Cl2 and passed through Celite. The resultant solution was concentrated under vacuum and chromatographed (SiO2 and EtOAc) to yield 5 as a colorless oil (4.5 g, 85%). 1H NMR (500 MHz, CDCl3): δ = 3.36 (t, J = 4.9 Hz, 2H), 3.58 (t, J = 3.4 Hz, 2H), 3.63–3.65 (m, 6H), 3.70 (t, J = 3.7 Hz, 2H).
Synthesis of compound 6
Anhydrous K2CO3 (1 g, 7.2 mmol) was added to a solution of 5 (1.3 g, 7.4 mmol) in anhydrous CH2Cl2 (5 mL). A solution of 1-adamantanecarbonyl chloride (2.0 g, 10.0 mmol) in anhydrous CH2Cl2 (10 mL) was added to the reaction dropwise. The solution was stirred at room temperature for 24 h. The insoluble solid was separated from the solution by filtration. The filtrate was concentrated by rotary evaporation. The residue was then dissolved in EtOAc and washed with 5% NaHCO3 aqueous solution and brine. The organic layer was dried (Na2SO4), filtered, and concentrated by rotary evaporation. The crude was purified by column chromatography (SiO2 and hexanes–EtOAc 3 : 1) to yield 6 as a colorless oil (0.35 g, 20%). 1H NMR (500 MHz, CDCl3): δ = 1.71 (m, 6H), 1.90 (d, J = 4.9 Hz, 6H), 2.01 (s, 3H), 3.39 (t, J = 6.2 Hz, 2H), 3.66–3.71 (m, 8H), 4.21 (t, J = 6.2 Hz, 2H).
Synthesis of compound MG-1
CuSO4·5H2O (20 mg, 80 μmol) and sodium ascorbate (50 mg, 0.3 mmol) were added to a solution of 6 (0.4 g, 1.2 mmol) and 2 (0.15 g, 0.3 mmol) in DMF (5 mL). The mixture was stirred for 4 d at room temperature then concentrated by rotary evaporation, redissolved in EtOAc, and washed with brine. The organic layer was dried (MgSO4), filtered, and concentrated by rotary evaporation. The crude was purified by column chromatography (Al2O3, hexanes–EtOAc 1 : 2, then EtOAc, then EtOAc–MeOH 50 : 1) to yield MG-1 as a pale yellow solid (0.36 g, 80%). 1H NMR (500 MHz, CDCl3): δ = 1.65 (m, 18H), 1.83 (d, J = 3.5 Hz, 18H), 1.95 (s, 9H), 3.57–3.62 (m, 18H), 3.86 (t, J = 4.6 Hz, 6H), 4.16 (t, J = 4.8 Hz, 6H), 4.53 (t, J = 4.6 Hz, 6H), 5.24 (s, 6H), 7.08 (d, J = 8.6 Hz, 6H), 7.58 (d, J = 8.6 Hz, 6H), 7.62 (s, 3H), 7.84 (s, 3H). 13C NMR (125 MHz, CDCl3): δ = 177.3, 170.9, 157.9, 143.7, 141.6, 134.0, 128.2, 123.9, 123.7, 115.0, 70.4, 70.3, 69.3, 69.1, 50.2, 40.5, 38.6, 36.3, 27.8. ESI-MS: C84H105O15N9 calculated: [M]+ 1480.7919, Found: 1480.5782.
Synthesis of compound MG-2
CuSO4·5H2O (20 mg, 80 mmol) and sodium ascorbate (50 mg, 0.3 mmol) were added to a solution of 6 (0.35 g, 1.0 mmol) and 4 (0.1 g, 0.2 mmol) in DMF (5 mL). The mixture was stirred for 4 d at room temperature. The mixture was concentrated by rotary evaporation, redissolved in EtOAc, and washed with brine. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated by rotary evaporation. The crude was purified by column chromatography (Al2O3, hexanes–EtOAc 1 : 2, then EtOAc, then EtOAc–MeOH 50 : 1) to yield MG-2 as a pale yellow solid (0.13 g, 30%). 1H NMR (500 MHz, CDCl3): δ = 1.62–1.70 (m, 36H), 1.70 (d, J = 3.3 Hz, 36H), 1.96 (s, 18H), 3.50–3.58 (m, 36H), 3.88 (t, J = 6.5 Hz, 12H), 4.13 (t, J = 4.9 Hz, 12H), 4.55 (t, J = 6.5 Hz, 12H), 5.53 (s, 12H), 8.09 (s, 6H), = 8.14 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 177.4, 148.0, 143.8, 124.5, 123.8, 108.2, 70.5, 70.3, 69.4, 69.1,=50.2, 40.5, 38.7, 36.3, 27.8. ESI-MS: C138H186O30N18 calculated: [M]+ 2577.0696, Found: [M + H]+ 2578.0671.
Synthesis of SBA-15
Pluronic P123 (2.0 g) was dissolved in a mixture of H2O (15 mL) and 2 M HCl (60 mL). The mixture was heated to 40 °C until the solution became clear. Tetraethylorthosilicate (4.25 g, 21.6 mmol) was then added to the solution dropwise. The mixture was stirred at 40 °C for 24 h after which it was transferred to a Teflon container and heated under pressure at 80 °C for 24 h. The precipitant, SBA-15,11 was collected by filtration and dried in air.
Synthesis of SBA-15-aldehyde
As-synthesized SBA-15 (200 mg) was dried under vacuum and suspended in anhydrous PhMe (10 mL). 1-Butylaldehyde-triethoxysilane (30 μL, 0.1 mmol) was added to the reaction mixture. The suspension was heated under reflux over argon for 15 h. After cooling to room temperature, the precipitates were collected by filtration, washed with MeOH, and dried under vacuum. The P123 surfactant was extracted by EtOH in a Soxhlet extractor.
Synthesis of SBA-15-cyclodextrin
SBA-15-aldehyde (100 mg) was suspended in anhydrous DMF (10 mL). 6-(2-Aminoethyl)amino-6-deoxy-β-cyclodextrin8 (100 mg, 0.08 mmol) and anhydrous MgSO4 (100 mg, 0.8 mmol) were added into the solution. The mixture was heated at 60 °C for 24 h under argon. After cooling to room temperature, the resultant precipitates were collected by filtration and washed with H2O. The product was dried under vacuum.
Loading and capping for SBA-15-cyclodextrin
SBA-15-cyclodextrin (30 mg) was suspended in an aqueous solution of fluorescein disodium salt (FDS) (2 mL, 2 mg mL−1) or FITC-dextran (2 mL, 2 mg mL−1). The mixture was stirred at room temperature for 24 h. A solution of the capping agent, MG-1 or MG-2, (20 mg) in Me2SO (2 mL) was then added into the mixture which was stirred for 30 min and the precipitate was collected by centrifugation, washed extensively with Me2SO–H2O (1 : 1) and dried under vacuum.
Assessment of cargo release and cap dissociation
The release profiles were obtained by a time-resolved fluorescence spectroscopy method. The solid sample was placed at the corner of a quartz cuvette. The cuvette was then filled with PBS buffer (pH 7.4) and the solution was gently stirred to accelerate the dispersion of the released dye. A probe beam (448 nm, 20 mW for FDS and FITC-dextran; 257 nm, 20 mW for MG-1; 351 nm, 30 mW for MG-2) was directed into the solution to excite the dissolved cargo molecules or dissociated cap molecules. The luminescence spectrum was collected in 1 s intervals over the course of the experiment. The pH of the solution was adjusted to the desired value with 1 M HCl. The luminescence intensity at the emission maximum of the cargo or cap was plotted as a function of time to generate a release profile.
Results and discussion
In this investigation, we have employed the well-known mesoporous silica material, SBA-15,11 as the model platform. The synthesis of SBA-15 was based on established procedures. A Pluronic P123 triblock copolymer was used as the templating agent, resulting in 2D hexagonal-patterned pores. N2 adsorption–desorption analysis gave a BET surface area of 650 m2 g−1, and the average pore size was calculated to be around 6.5 nm (ESI†).
In our previous studies where MCM-41-type materials were employed, we designed6,12 a variety of nanovalve systems to mechanize the pores. The typical design of these nanovalves consisted of two components, a linear one (the stalk) that is grafted at the opening of the mesopores, and a bulky compound (the cap) that can associate with the stalk as a result of non-covalent bonding interactions and block the pores.6,12 Upon appropriate stimulation, the interactions between the stalk and the cap can be weakened, allowing the cap to depart from the binding site, thus opening up the pore. In order to control access to the bigger pores found in SBA-15, a much larger capping agent, namely a megagate molecule (MGM), becomes necessary.
It is not necessary, however, to cover the pores completely in order to prevent leakage when attempting to store large molecules. We envisioned that the use of a weblike cap large enough to extend across the pores would be sufficient to serve as a megagate. With this thought in mind, we designed MG-1, consisting of a 1,3,5-triphenylbenzene core linked to three adamantine moieties by triethylene glycol chains using Cucatalyzed azide–alkyne cycloaddition (CuAAC).13 The core was synthesized (Fig. 1a) by first of all propargylating 4′-hydroxyacetophenone to obtain the precursor 1. A subsequent SiCl4-induced cyclotrimerization yielded 2, a versatile trialkyne building block. The arms of the cap were obtained (Fig. 1c) by converting 2-[2-(2-chloroethoxy)-ethoxy]-ethanol to the azide 5 and then reacting 5 with 1-adamantanecarbonyl chloride under basic conditions to yield 6. A simple CuAAC between 2 and 6 provided (Fig. 1d) the desired compound, MG-1.
Fig. 1.

The synthesis of (a) 2, (b) 4, and (c) 6. The construction of (d) MG-1 and MG-2 by Cu-catalyzed click reactions.
MG-1 has a theoretical diameter of about 5.5 nm—calculated by a molecular mechanics method in SPARTAN 5.1—when fully extended. While this diameter is less than that of the average pore size (ca. 6.5 nm) in the nanoparticles, MG-1 is still a good candidate because (i) in the case of sufficiently large cargo molecules, blocking even just a small section of the pore orifice—much like a mathematical chord of a circle—will prevent leakage from the nanoparticle, (ii) this dimension does not include the size of the surface-attached cyclodextrin moiety, which is around 1.5 nm, and (iii) more importantly, more than one cap molecule may be associated with each pore.
The bare SBA-15 nanoparticles were functionalized with 1-butylaldehydetriethoxysilane to provide an anchoring point for the stalks. The amine-modified cyclodextrin stalk 7 was grafted (Fig. 2b) onto SBA-15 by imine bond formation. It is well-known14 that imine bonds are acid-sensitive, and will cleave upon lowering the pH, providing the mechanism for operation of the megagate.‡ The successful surface modification of the SBA-15 was confirmed by solid-state NMR spectroscopy (ESI†).
Fig. 2.
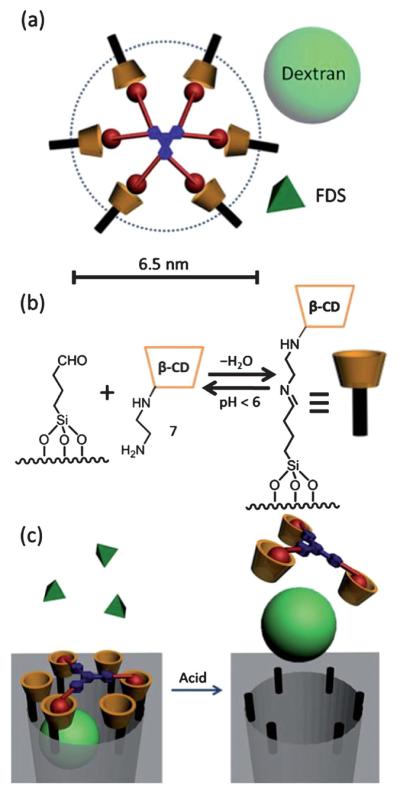
(a) The fully extended structure of a megagate incorporating MG-2 as the capping molecule. The dashed circle, the light green ball and the dark green pyramid show the relative sizes of the pore, the dextran cargo, and the fluorescein disodium salt (FDS), respectively. (b) The attachment of cyclodextrin onto the aldehyde-modified silica through imine formation. (c) The operation of a megagate capped by MG-1. The size of MG-1 allows FDS to escape freely from the pore, while dextran is trapped within the pore until the gate is opened by the addition of acid.
The overall operation of the megagate is illustrated in Fig. 2c. The modified SBA-15 is loaded with cargo, and then capped by a MGM. In aqueous solutions, the adamantane moieties bind strongly to the cyclodextrin tori as a result of hydrophobic interactions. Upon binding, the MGMs serve to prevent the loaded cargo from leaking out of the pores. When the pH of the solution is lowered, the imine bonds in the stalks are cleaved, allowing the cyclodextrin tori to dissociate from the surface. As a result, the MGMs, which are still bound to the cyclodextrin tori, are no longer blocking the pores, thus leading to the release of cargo from the nanoparticles.
In order to demonstrate the functioning of the megagates, a well-established12 time-resolved fluorescence spectroscopic method was employed. The SBA-15 was first of all loaded with the fluorescent cargo, that is, either FITC-dextran (M.W. 3k, hydrodynamic size about 4 nm (ref. 15)) or fluorescein disodium salt (FDS). MGMs were then used to cap the pores. After drying under vacuum, the capped SBA-15 was placed in a PBS buffer solution. The pH of the solution was then adjusted to 5 to initiate the release of the cargo. The released cargo in the supernatant was excited by a probe laser beam and the corresponding fluorescence spectrum was recorded by a CCD spectrometer to generate the release profile.
When MG-1 was employed as the capping agent (Fig. 3a), FITC-dextran was trapped successfully in the pores of SBA-15. There was no significant premature release, indicating that as predicted, MG-1 is large enough to block the pore orifices and prevent the FITC-dextran molecules from escaping. A release capacity of about 3.3% wt was determined by UV-Vis spectroscopy after the completion of the release. This relatively low value arises from the fact that the capping process occurs on a statistical basis. Only those pores blocked by extended MGMs will contribute to the effective release capacity. Cargos loaded in incompletely capped pores will be washed out during the subsequent washing process. This release capacity is enough, however, for certain biomedical applications.16 The prolonged time course of the release is a consequence of the low diffusion rate of the bulky FITC-dextran cargo. In a control experiment where there was no MG-1 capping agent present, a significantly smaller amount of cargo release was observed as molecules could not be trapped within the pores during the loading process. The near-negligible release detected may be attributed to residual surface-adsorbed molecules. This proves that megagates equipped with MG-1 caps operate as designed. By using a different probe beam, the dissociation process of the megagates from the silica surface to the solution can be monitored (Fig. 3b). The fast completion of this process correlates well with the proposed release mechanism.
Fig. 3.
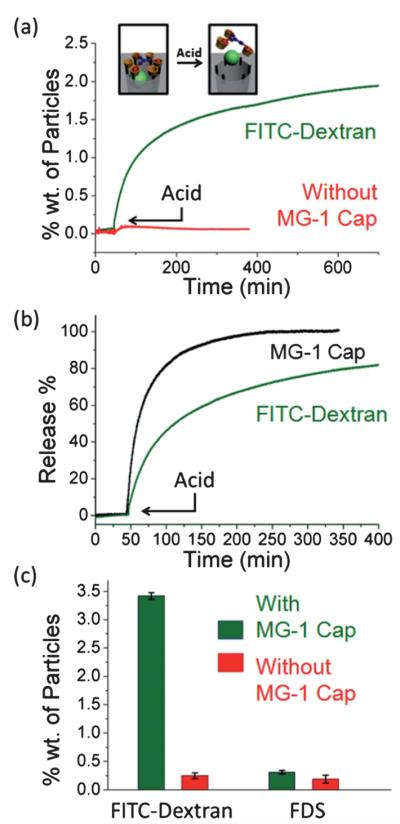
The release profile of FITC-dextran from SBA-15 capped with MG-1 (green line). The control experiment with no cap resulted in negligible release (red line). (b) The release profile of the MG-1 cap (black line). The release of the cargo (green line) was used as a reference. (c) Release capacities of nanoparticles loaded with different cargoes, with and without the MG-1 cap.
When a smaller cargo (FDS) was used in analogous experiments, however, MG-1 failed (Fig. 3c) to function as a cap, presumably because the gaps between the adamantane moieties were too big to prevent FDS from leaking out. In order to trap smaller molecules, a “tighter” cap is needed. Therefore, a cap with twice as many adamantane arms as MG-1 was designed. The synthesis (Fig. 1b) of MG-2 was facile. Hexahydroxytriphenylene (3) was propargylated to yield the hexaalkyne 4, which was then reacted with 6 under CuAAC conditions to yield (Fig. 1d) MG-2. With its hexameric structure, MG-2 exhibits much less space between adamantane moieties while maintaining a similar area of coverage as MG-1 (Fig. 2a). Indeed, MG-2 was able (Fig. 4a) to trap FDS molecules successfully. The release profile, which was again tracked by time-resolved fluorescence spectroscopy, was faster than that of FITC-dextran, an observation which can be explained by the higher mobility of the smaller cargo. The dissociation profile of MG-2 also correlates well with the release course (Fig. 4b) of the FDS. In order to compare the efficiency of MG-2 with that of MG-1, FITC-dextran was employed as cargo in the MG-2 system, and a release capacity of 3.3% wt was determined. This result is close to that from the MG-1 system, suggesting that in spite of the size-selective effect, the cap does not influence the loading or release of the cargo.
Fig. 4.

(a) The release profile of FDS from SBA-15 capped with MG-2 (green line). The control experiment with no cap resulted in negligible release (red line). (b) The release profile of the MG-2 cap (black line). The release of FDS was used as reference (green line).
Conclusions
We have developed a “megagate” methodology to mechanize the mesopores of SBA-15. Depending on the number of arms on the cap, the megagate is capable of storing, and then releasing, differently sized cargoes from SBA-15 upon pH stimulation. Further investigations towards biomedical applications are currently ongoing.
Supplementary Material
Acknowledgements
The research was funded by the US Public Health Service Grants, RO1 CA 133697, by US DOD DTRA 1-08-1-0041 and by the National Science Foundation (NSF) under grant CHE 0809384 and is based upon work supported by NSF equipment grant CHE 1048804 and by the National Center for Research Resources under grant S10RR024605. J. F. S. and D. C. also acknowledge support from the World Class University (WCU) Program (R-31-2008-000-10055-0) in Korea. D. C. thanks the NSF for a Graduate Research Fellowship.
Footnotes
Electronic supplementary information (ESI) available: 13C CP/MAS NMR spectrum of SBA-15-cyclodextrin and N2 absorption isotherm of SBA-15. See DOI: 10.1039/c2nr32170j
The surface density of hydroxyl groups on the silica nanoparticles and gels is 4.9 OH per nm2 (L. T. Zhuravlev, Langmuir, 1987, 3, 316–318). Based on this model, the average distance between two surface OH groups is 5.3 Å corresponding to ~38 OH groups around one mesopore (diameter 6.5 nm). Given that attaching one stalk to the silica surface requires at least two OH groups, the maximum number of aldehyde stalks around one pore is ~19. The diameter of the periphery of the secondary face of β-cyclodextrin is ~1.5 nm (J. Szejtli, Chem. Rev., 1998, 98, 1743–1754). The maximum number of cyclodextrins around one pore is about 13, which exceeds the number of binding sites required to complex with both MG-1 and MG-2.
Notes and references
- 1.(a) Vallet-Regi M, Rámila A, del Real RP, Pérez-Pariente J. Chem. Mater. 2001;13:308–311. [Google Scholar]; (b) Slowing II, Trewyn BG, Giri S, Lin VS-Y. Adv. Funct. Mater. 2007;17:1225–1236. [Google Scholar]; (c) Kim J, Kim HS, Lee N, Kim T, Kim H, Yu T, Song IC, Moon WK, Hyeon T. Angew. Chem., Int. Ed. 2008;47:8438–8441. doi: 10.1002/anie.200802469. [DOI] [PubMed] [Google Scholar]; (d) Liong M, Angelos S, Choi E, Stoddart JF, Zink JI. J. Mater. Chem. 2009;19:6251–6257. [Google Scholar]; (e) Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nat. Mater. 2011;10:389–397. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Trewyn BG, Slowing II, Giri S, Chen H-T, Lin VS-Y. Acc. Chem. Res. 2007;40:846–853. doi: 10.1021/ar600032u. [DOI] [PubMed] [Google Scholar]; (b) Li Z, Barnes JC, Bosoy A, Stoddart JF, Zink JI. Chem. Soc. Rev. 2012;41:2590–2605. doi: 10.1039/c1cs15246g. [DOI] [PubMed] [Google Scholar]; (c) Qian KK, Bogner RH. J. Pharm. Sci. 2012;101:444–463. doi: 10.1002/jps.22779. [DOI] [PubMed] [Google Scholar]; (d) Rosenholm JM, Mamaeva V, Sahlgren C, Linden M. Nanomedicine. 2012;7:111–120. doi: 10.2217/nnm.11.166. [DOI] [PubMed] [Google Scholar]
- 3.(a) Radu DR, Lai C-Y, Jeftinija K, Rowe EW, Jeftinija S, Lin VS-Y. J. Am. Chem. Soc. 2004;126:13216–13217. doi: 10.1021/ja046275m. [DOI] [PubMed] [Google Scholar]; (b) Schlossbauer A, Warncke S, Gramlich PME, Kecht J, Manetto A, Carell T, Bein T. Angew. Chem., Int. Ed. 2010;49:4734–4737. doi: 10.1002/anie.201000827. [DOI] [PubMed] [Google Scholar]
- 4.Lai C-Y, Trewyn BG, Jeftinija DM, Jeftinija K, Xu S, Jeftinija S, Lin VS-Y. J. Am. Chem. Soc. 2003;125:4451–4459. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- 5.(a) Xue JM, Shi M. J. Controlled Release. 2004;98:209–217. doi: 10.1016/j.jconrel.2004.04.023. [DOI] [PubMed] [Google Scholar]; (b) Zhu Y, Shi J, Shen W, Dong X, Feng J, Ruan M, Li Y. Angew. Chem., Int. Ed. 2005;44:5083–5087. doi: 10.1002/anie.200501500. [DOI] [PubMed] [Google Scholar]; (c) Meng H, Liong M, Xia T, Li Z, Ji Z, Zink JI, Nel AE. ACS Nano. 2010;4:4539–4550. doi: 10.1021/nn100690m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Argyo C, Cauda V, Engelke H, Räadler J, Bein G, Bein T. Chem.–Eur. J. 2012;18:428–432. doi: 10.1002/chem.201102926. [DOI] [PubMed] [Google Scholar]
- 6.(a) Coti K, Belowich ME, Liong M, Ambrogio MW, Lau YA, Khatib HA, Zink JI, Khashab NM, Stoddart JF. Nanoscale. 2009;1:16–39. doi: 10.1039/b9nr00162j. [DOI] [PubMed] [Google Scholar]; (b) Meng H, Xue M, Xia T, Zhao Y-L, Tamanoi F, Stoddart JF, Zink JI, Nel AE. J. Am. Chem. Soc. 2010;132:12690–12697. doi: 10.1021/ja104501a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ambrogio MW, Thomas CR, Zhao Y-L, Zink JI, Stoddart JF. Acc. Chem. Res. 2011;44:903–913. doi: 10.1021/ar200018x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang C, Li Z, Cao D, Zhao Y-L, Gaines JW, Bozdemir OA, Ambrogio MW, Frasconi M, Botros YY, Zink JI, Stoddart JF. Angew. Chem., Int. Ed. 2012;51:5460–5465. doi: 10.1002/anie.201107960. [DOI] [PubMed] [Google Scholar]
- 7.(a) Yang Q, Wang S, Fan P, Wang L, Di Y, Lin K, Xiao F-S. Chem. Mater. 2005;17:5999–6003. [Google Scholar]; (b) Slowing II, Trewyn BG, Lin VS-Y. J. Am. Chem. Soc. 2007;129:8845–8849. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]; (c) Gao F, Botella P, Corma A, Blesa J, Dong L. J. Phys. Chem. B. 2009;113:1796–1804. doi: 10.1021/jp807956r. [DOI] [PubMed] [Google Scholar]; (d) Kim T-W, Slowing II, Chung P-W, Lin VS-Y. ACS Nano. 2011;5:360–366. doi: 10.1021/nn101740e. [DOI] [PubMed] [Google Scholar]; (e) Kim M-H, Na H-K, Kim Y-K, Ryoo S-R, Cho HS, Lee KE, Jeon H, Ryoo R, Min D-H. ACS Nano. 2011;5:3568–3576. doi: 10.1021/nn103130q. [DOI] [PubMed] [Google Scholar]
- 8.May BL, Kean SD, Easton CJ, Lincoln SF. J. Chem. Soc., Perkin Trans. 1997;1:3157–3160. [Google Scholar]
- 9.Hans RH, Guantai EM, Chibale K, Lategan C, Smith PJ, Wan B, Franzblau SG, Gut J, Rosenthal PJ. Bioorg. Med. Chem. Lett. 2010;11:942–944. doi: 10.1016/j.bmcl.2009.12.062. [DOI] [PubMed] [Google Scholar]
- 10.Krebs FC, Schioedt NC, Batsberg W, Bechgaard K. Synthesis. 1997;11:1285–1290. [Google Scholar]
- 11.Zhao D, Feng J, Huo Q, Melosh N, Fredrickson GH, Chmelka BF, Stucky GD. Science. 1998;279:548–552. doi: 10.1126/science.279.5350.548. [DOI] [PubMed] [Google Scholar]
- 12.(a) Nguyen TD, Tseng H-R, Celestre PC, Flood AH, Liu Y, Stoddart JF, Zink JI. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10029–10034. doi: 10.1073/pnas.0504109102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Angelos S, Khashab NM, Yang YW, Trabolsi A, Khatib H, Stoddart JF, Zink JI. J. Am. Chem. Soc. 2009;131:12912–12914. doi: 10.1021/ja9010157. [DOI] [PubMed] [Google Scholar]; (c) Angelos S, Yang YW, Khashab NM, Stoddart JF, Zink JI. J. Am. Chem. Soc. 2009;131:11344–11346. doi: 10.1021/ja9042752. [DOI] [PubMed] [Google Scholar]; (d) Ferris DP, Zhao Y-L, Khashab NM, Khatib HA, Stoddart JF, Zink JI. J. Am. Chem. Soc. 2009;131:1686–1688. doi: 10.1021/ja807798g. [DOI] [PubMed] [Google Scholar]; (e) Du L, Liao S, Khatib HA, Stoddart JF, Zink JI. J. Am. Chem. Soc. 2009;131:15136–15142. doi: 10.1021/ja904982j. [DOI] [PubMed] [Google Scholar]; (f) Thomas CR, Ferris DP, Lee J-H, Choi E, Cho MH, Kim ES, Stoddart JF, Shin J-S, Cheon J, Zink JI. J. Am. Chem. Soc. 2010;132:10623–10625. doi: 10.1021/ja1022267. [DOI] [PubMed] [Google Scholar]
- 13.(a) Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (b) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 14.(a) Lee C-H, Cheng S-H, Huang I-P, Souris JS, Yang C-S, Mou C-Y, Lo L-W. Angew. Chem., Int. Ed. 2010;49:8214–8219. doi: 10.1002/anie.201002639. [DOI] [PubMed] [Google Scholar]; (b) Zhao Y-L, Li Z, Kabehie S, Botros YY, Stoddart JF, Zink JI. J. Am. Chem. Soc. 2010;132:13016–13025. doi: 10.1021/ja105371u. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi N, Kishimoto T, Nemoto T, Kadowaki T, Kasai H. Science. 2002;297:1349–1352. doi: 10.1126/science.1073806. [DOI] [PubMed] [Google Scholar]
- 16.Meng H, Xue M, Xia T, Ji Z, Tarn DY, Zink JI, Nel AE. ACS Nano. 2011;5:4131–4144. doi: 10.1021/nn200809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.