

Abstract

Chagas disease, caused by the protozoan parasite Trypanosoma cruzi (T. cruzi), is an increasing threat to global health. Available medicines were introduced over 40 years ago, have undesirable side effects, and give equivocal results of cure in the chronic stage of the disease. We report the development of two compounds, 6 and (S)-7, with PCR-confirmed curative activity in a mouse model of established T. cruzi infection after once daily oral dosing for 20 days at 20 mg/kg 6 and 10 mg/kg (S)-7. Compounds 6 and (S)-7 have potent in vitro activity, are noncytotoxic, show no adverse effects in vivo following repeat dosing, are prepared by a short synthetic route, and have druglike properties suitable for preclinical development.
Introduction
Chagas disease is a major human health issue. The disease is found primarily in endemic areas of 21 Latin American countries,1 with Bolivia currently experiencing the highest disease burden.2 Population migration and travel have distributed the disease into the United States, Europe, and Asia where it poses a growing risk to public health.3 Current levels of global infection are estimated to be between 7 and 8 million people.1
An acute illness results after initial infection with T. cruzi, from which the majority of people recover in the absence of treatment but remain infected. The long-term prognosis of Chagas disease sufferers is complicated by the migration of parasites from blood into tissues, causing protracted damage to the heart, colon, and esophagus in particular, as a consequence of lesions, inflammation, and fibrosis. The most severe cases lead to cardiomyopathy, megacolon, and megaesophagus syndromes.4 Exact morbidity and mortality figures vary with disease stage and are hard to determine accurately. Recent reports state Chagas disease to be the leading cause of death in the elderly in Brazil5 and a significant contributing factor to the global burden of cardiovascular disease.6
Treatment options are severely limited with only two drugs currently approved, benznidazole and nifurtimox. Both drugs are contraindicated for pregnant women and nursing mothers, an important limitation given that congenital transmission is a common method of infection.7 The adverse effects of both drugs are well documented8 and are exacerbated by the requirement for long treatment times. Benznidazole is currently the preferred choice, treating the acute stage of the infection most successfully with cure rates of up to 60%,9 although cessation of treatment occurs regularly with up to 30% of adult patients experiencing serious adverse reactions.10,11 Children below the age of 7 tolerate the drug much better than adults,12 and a pediatric formulation is now available. Treatment of the chronic form of the disease is controversial because of the lack of a demonstrated cure in patients in the late stages of the disease. Advocates supporting treatment suggest that even slowing progression to severe forms of the disease is a beneficial outcome, especially to untreated individuals infected during childhood.4 The data remain controversial however,13 and the ongoing BENEFIT clinical trial investigating the treatment of chronic Chagas patients with cardiomyopathy is aimed at providing further information on this issue.14
Innovative efforts to address drug inadequacy and plug the treatment gap has propelled antifungal CYP51 inhibitors into the Chagas therapeutic area, and clinical trials are in progress with third generation azole antifungals posaconazole15 and E1224, a prodrug of ravuconazole.16 The attention generated through the investigation of this drug class has initiated drug discovery efforts focused on compounds specifically optimized against T. cruzi CYP51 (Tc CYP51), the homologous enzyme in the parasite.17,18 In a breakthrough for this class of compounds, parasitological cure of T. cruzi infected mice with a twice daily treatment for 30 days of Tc CYP51 inhibitor VNI was recently demonstrated in acute (compound administration 24 h postinfection) and chronic (compound administration 90 days postinfection) immunosuppressive laboratory models of Chagas disease.19 VNI belongs to the azole class of T. cruzi inhibitors, utilizing an imidazole as the heme-binding motif. Potent inhibition of cytochrome P450 drug metabolizing enzyme CYP3A4 is a limitation of this class of compounds, an important consideration for treatment strategies involving combination therapy to avoid drug resistance issues and for drug–drug interactions in general.20
We have reported our initial efforts to develop an efficacious, low-cost, and safer alternative to benznidazole for the treatment of Chagas disease from fenarimol (1), a non-azole CYP51 antifungal identified from a targeted screening campaign.21 Fenarimol was optimized to give 4-(hydroxy(pyridin-3-yl)[4-(trifluoromethyl)phenyl]methyl)benzonitrile (2) (Figure 1), a compound able to reduce parasitemia to microscopically undetectable levels in a subchronic mouse model of T. cruzi infection, following once daily oral dosing at 20 mg/kg for 20 days. Parasites re-emerged in blood after three cycles of immunosuppression, indicating that the compound had not achieved a parasitological cure. While outperforming benznidazole at this dose, 2 was not as efficacious as posaconazole which cured ∼60% of mice when administered under the same protocol.
Figure 1.

Key scaffold hops taken during the investigation of the fenarimol series SAR. Tc IC50 is the concentration of compound required to cause a 50% inhibition in growth of T. cruzi (amastigote form), and values are the mean of at least two experiments. L6 cells (rat myoblasts from skeletal muscle) were used for assessment of cytotoxicity.
From realization that this optimized fenarimol series was at a structure–activity dead end, a series of scaffold hops were undertaken to open up new areas of chemical space, providing alternative opportunities to optimize medicinal chemical parameters and hence in vivo efficacy. Evaluation of the network of compounds that subsequently evolved supported the development of a robust and efficient project flow scheme and defined the target efficacy characteristics of a potential clinical candidate based on early pharmacokinetic/pharmacodynamic (PK/PD) observations. As a culmination of those efforts, we report herein the lead optimization of two fenarimol-related scaffolds leading to the discovery of two potent inhibitors of T. cruzi, compounds 6 and (S)-7, which have curative activity in a subchronic mouse model of T. cruzi infection. These non-azole, pyridine-containing compounds have recently been confirmed to inhibit T. cruzi CYP51.22 They do not inhibit CYP3A4 to any significant pharmacological extent (IC50 >10 μM), and they have suitable druglike properties for preclinical evaluation.
Results
Compound Design
A summary of the key scaffold hops undertaken during the course of the SAR studies is illustrated in Figure 1. It was determined that the (heteroaryl)(aryl)methine moiety was a required pharmacophore for activity against T. cruzi, with a degree of flexibility allowed in the chemical space occupied by the third aromatic ring (Figure 1, inset). This was demonstrated in the first instance with the evaluation of a series of potent piperazine analogues, exemplified by 3.23 We went on to determine that a number of amino-substituted cyclic amines could be inserted between the central carbon atom and the distal aromatic ring, resulting in structurally related scaffolds as illustrated by compounds 4–7, characterized by various changes in activity and pharmacokinetic properties.
Compound Synthesis
The synthesis of 6 is very concise, using readily available starting materials and standard synthetic transformations (Scheme 1). The order of the steps is important such that reductive amination of 3-aminopyridine with 1-tert-butoxycarboxylpiperidin-4-one to give intermediate 8 must occur before palladium-catalyzed arylation to give 9. Both palladium(II) acetate and CombiPhos-Pd6 were required for successful transformation of 8 to 9. Compound 6 has been synthesized on a multigram scale using this route, with the potential for additional optimization of reaction conditions still remaining to further improve the overall yield. Analogues were prepared by varying the substituted aromatic motif introduced in step 2 and the electrophilic coupling partner in step 4.
Scheme 1. Synthesis of 6 and Intermediate 10 for Analogue Preparation.

Reagents and conditions: (1) NaBH(OAc)3, AcOH, DCM, reflux (64%); (2) 4-bromotrifluoromethylphenyl, Pd(OAc)2, CombiPhos-Pd6, xantphos, NaOtBu, toluene, reflux (42%); (3) TFA–H2O–DCM (8:1:5) (quant); (4) 2-Cl-5-CF3-pyridine, DiPEA, DMF, 95 °C (45%).
The synthesis of the parent racemate of (S)-7 ((rac)-7) is also very concise and high yielding, employing readily available starting materials and standard synthetic transformations (Scheme 2). Simple SNAr transformation of 3-fluoropyridine by (4-chlorophenyl)acetonitrile gave nitrile 11, which was hydrolyzed and coupled to 1-[4-trifluoromethyl)phenyl]piperazine (13) (prepared through palladium catalyzed amination of 4-trifluoromethylphenyl bromide by tert-butyl piperazine-1-carboxylate, followed by deprotection) to give the target compound (rac)-7. Preparation of (rac)-7 has been successfully carried out on a multigram scale with chromatographic chiral resolution performed by supercritical fluid chromatography (SFC). The absolute configuration of the most active enantiomer was determined by X-ray diffraction (see Supporting Information). SAR investigations within this series were focused on derivatizing key piperazine intermediate 14 prepared as shown in Scheme 3.
Scheme 2. Synthesis of (rac)-7 and Chiral Resolution by SFC to give (R)-7 and (S)-7.

Reagents: (1) KOtBu, NMP (71%); (2) 2 M NaOH(aq) (81%); (3) 1-[4-(trifluoromethyl)phenyl]piperazine (13), HATU, DMF, DiPEA (78%); (4) SFC.
Scheme 3. Preparation of Intermediate 14 for the Synthesis of Analogues of (rac)-7.

Reagents: (1) HATU, DMF, DiPEA, tert-butyl piperazine-1-carboxylate; (2) TFA, DCM, MeOH (69%, two steps).
In Vitro Screening Cascade
Inhibition of T. cruzi (Tulahuen strain (Tc VI) transfected with β-galactosidase) in a whole parasite assay (amastigote form) was used as the primary in vitro activity screen as previously reported.21,23 A gate of IC50 ≤ 30 nM was applied for progression into physicochemical and in vitro ADME assays. Cytotoxicty was measured in L6 cells as a counterscreen, but no overt cellular toxicity was observed, with selectivity indices >1000-fold in most cases. Primary physicochemical and ADME evaluation included estimation of log D (pH 7.4), kinetic solubility in phosphate buffer (pH 6.5), predicted hepatic clearance and extraction ratios (EH) following incubation with human liver microsomes and inhibition of cytochrome P450 CYP3A4.
Compound 6 SAR
The achiral scaffold represented by compound 6 arose from the direct replacement of the central, chiral carbon atom of the (heteroaryl)(aryl)methine pharmacophore with the primary nitrogen atom from 4-aminopiperidine to give an achiral (heteroaryl)(aryl)amine template. The SAR investigations of the N-centered achiral series from which 6 was identified were very focused, guided by SAR information obtained from the many fernarimol-related scaffolds and subseries generated over the lifetime of the project. On the basis of the results of our early SAR studies,21,23 the heteroaromatic ring was fixed as 3-pyridyl for the majority of the analogues synthesized, and a two-point SAR matrix was elaborated with various piperidine N1 motifs (R1 SAR, Figure 2, panel A) paired with changes to substituent X1 of the N-phenyl ring (X1 SAR, Figure 2, panel B) to optimize potency, metabolic stability, and oral exposure. Figure 2 shows a representative set of the compounds prepared in this SAR study. Several compounds in this series were equipotent with posaconazole (Tc IC50 = 0.7 nM; L6 IC50 > 100 μM), and all compounds were more active than benznidazole (Tc IC50 = 1.5 μM; L6 IC50 > 100 μM).
Figure 2.

T. cruzi growth inhibition data for 6 and analogues: (A) variation of the R1 substituent; (B) variation of substituents X1 and X2 for triaryl analogues; (C) pyrimidinyl analogue of 6. (†) Compound 16 was prepared on an N-(4-chlorophenyl)-N-(piperidin-4-yl)pyridin-3-amine scaffold.
Lipophilicity and a degree of space filling were required of the R1 substituent, and simple alkyl analogues such as 15 and other hydroxylated alkyl motifs (not shown) were unable to reach high potencies. Amides 16 and 19 achieved subnanomolar activities, exposing what could be an additional binding pocket not accessible to any of the 2-substituted benzyl analogues on this scaffold, e.g., 20–22, or from earlier scaffolds (Figure 1). Substituted aromatics exemplified by 23–28 were generally 2- to 3-fold less active than the amide set, with the exception of nitrile analogues 25 and 26. R1 as either substituted 2- or 3-pyridyl was not significantly different in activity from the phenyl analogues. The activity of the R1 = phenyl-X set was also good following a change of the X1 substituent to either chloro or nitrile as shown in panel B. Additional substitution on the N1-phenyl, e.g., X2 = 2CN, 4CF3Ph (X1 = Cl), served only to increase the molecular weight with no gain in activity or metabolic stability (data not shown). The 5-pyrimidinyl analogue of 6 (compound 34) was surprisingly more active than the parent compound (panel C). We have observed this on only a few occasions throughout the expansion of the fenarimol SAR, and in general less basic heteroaromatics such as pyrimidine and pyrazine were lower in potency, which we speculate is due to a weaker interaction with the heme motif in the CYP51 enzyme.22
Microsomal stability was used to triage the cohort of similarly active compounds, and the choice of R1 was a major influence on this parameter (Table 1). Analogues with the best balance of activity and stability to oxidative metabolism in vitro were derived from the “substituted aromatics” set, with the trade-off being high lipophilicity (except 33) and low solubility at pH 6.5.
Table 1. In Vitro ADME Properties of 6 and Analogues.
| compd | CYP3A4 inhibitiona (μM) | pKa b | glogDpH 7.4 f | aqueous solubility,g pH 2/6.5 (μg/mL) | predicted human EH h |
|---|---|---|---|---|---|
| 16 | N/A | 5.4 | 4.2 | >100/6.3 | 0.91 |
| 22 | 18 | 5.1/8.7c | >5.3 | >100/3.1 | 0.87 |
| 24 | N/A | 5.4/4.6c | >5.3 | 12.5/<1.6 | 0.44 |
| 25 | 5.2 | 5.4/2.2c | 4.9 | >100/<1.6 | 0.88 |
| 27 | >20 | 5.4/4.5c | >5.3 | >100/<1.6 | 0.65 |
| 31 | >20 | 5.4/4.6c | 4.7 | 25/<1.6 | 0.69 |
| 33 | 12 | 5.4/2.7c | 3.0 | 25/<1.6 | 0.47 |
| 6 | >20 | 5.6/5.0d | >5.3 | >100/<1.6 | 0.68 |
| 28 | 16 | 5.4/1.9d | 4.8 | 25/<1.6 | 0.74 |
| 34 | 6.3 | 5.3d/1.8e | 4.9 | 12.5/<1.6 | 0.63 |
Human CYP3A4 inhibition (IC50) determined in human liver microsomes using 6β-hydroxylation of testosterone as the probe reaction pathway; N/A = not available.
pKa values calculated using JChem for Excel; pKa refers to 3-pyridyl nitrogen unless otherwise indicated.
pKa for piperidine nitrogen.
pKa for 2-pyridyl nitrogen (R1).
pKa for pyrimidinyl nitrogen.
Partition coefficient values of the test compounds were estimated by correlation of their chromatographic retention properties against the characteristics of a series of standard compounds with known partition coefficient values, at pH 7.4.
Kinetic solubility at pH 2 and pH 6.5 determined by nephelometry.
Microsome predicted hepatic extraction ratio (EH) determined in human liver microsomes.
Compound 7 SAR
In an attempt to enhance the stability of the important pyridyl(phenylmethine) pharmacophore and improve in vivo efficacy, the central methanamino motif of 5 (Figure 1) was replaced with acetamide, a structural change leading to investigation of scaffold 14 (Scheme 3), which was readily amenable to analogue preparation. The optimization of this new series proceeded rapidly, driven by SAR insights gathered from earlier scaffolds.
Two small libraries of analogues were prepared and profiled. The first library (Figure 3) covered variation of the R1 group and included the synthesis of (rac)-7. The pyrimidine analogue of (rac)-7 (not shown) was prepared as part of this set but was less active (IC50 = 118 nM) than 7. The second library (Figure 4) expanded the scope of the “R1 = substituted aromatic” set in conjunction with a change in substituent X (X = 4-Cl and 4-CN). For compounds in this library the activity of the most active enantiomer is shown (absolute configuration was not determined). Compound (S)-7 and less active enantiomer (R)-7 are included for comparison. The potency range was similar for the equivalent SAR study of the N1-substituted phenyl motif of compound 6 (Figure 2). The chiral resolution of analogues of 7 was performed using SFC in a similar manner to that for the parent compound. Table 2 shows in vitro ADME data for these analogues.
Figure 3.

T. cruzi growth inhibition data for analogues prepared on acetamide-scaffold 14 leading to identification of (rac)-7.
Figure 4.

T. cruzi growth inhibition data for the most active enantiomer of analogues of (S)-7. ∗∗ denotes chiral center.
Table 2. In Vitro ADME Properties of 7 and Analogues.
| compd | CYP3A4 inhibitiona (μM) | pKa b | glogDpH 7.4 e | aqueous solubility,f pH 2/6.5 (μg/mL) | predicted human EH g |
|---|---|---|---|---|---|
| (rac)-7 | 16 | 4.8/2.9c | 4.5 | >100/1.6 | 0.50 |
| (R)-7 | 17 | 4.8/2.9c | 4.5 | >100/1.6 | 0.61 |
| (S)-7 | 15 | 4.8/2.9c | 4.5 | >100/1.6 | 0.53 |
| 40 | >20 | 4.8/2.9c | 3.8 | >100/3.1 | 0.70 |
| 41 | >20 | 4.8/1.9d | 3.7 | 50/6.3 | 0.77 |
| 42 | >20 | 4.8/1.9d | 3.0 | >100/25 | 0.56 |
| 43 | 7 | 4.7/5.4d | 4.2 | 25/3.1 | 0.70 |
| 44 | >20 | 4.7/5.4d | 3.5 | 50/6.3 | 0.56 |
| 45 | 4 | 4.8/0.8c | 3.0 | >100/6.3 | 0.82 |
| 46 | >20 | 4.8/0.8c | 2.4 | 25/3.1 | 0.46 |
Human CYP3A4 inhibition (IC50) determined in human liver microsomes using 6β-hydroxylation of testosterone as the probe reaction pathway.
pKa values calculated using JChem for Excel; pKa refers to 3-pyridyl nitrogen unless otherwise indicated.
pKa for piperazine nitrogen.
pKa for 2-pyridyl nitrogen (R1).
Partition coefficient values of the test compounds were estimated by correlation of their chromatographic retention properties against the characteristics of a series of standard compounds with known partition coefficient values, at pH 7.4.
Kinetic solubility at pH 2 and pH 6.5 determined by nephelometry.
Microsome predicted hepatic extraction ratio (EH) determined in human liver microsomes.
Stability toward in vitro oxidative metabolism by human liver microsomes ranged from low to moderate across the series as indicated by the predicted EH values in Table 2 (low stability, EH > 0.7; moderate stability, EH = 0.3–0.7). The nitrile analogues 40, 42, 44, 46 were more metabolically stable (except for the (S)-7, 40 comparative pair), with modestly improved solubility (except for the 45, 46 comparative pair), and had lower log D values than their chloro counterparts (S)-7, 41, 43, and 45.
Initial PK/PD Assessment
The decision to progress compounds into in vivo efficacy and exposure profiling was driven by the EH value with a gate set at ≤0.7. While quite generous, this was a pragmatic choice to provide the best chance of capturing compounds suitable for testing in the mouse efficacy assay. Infected mice were treated once daily at 20 mg/kg for up to 10 days, and parasitemia was monitored throughout the dosing period. Single dose exposure profiles (three time points) were assessed in a parallel group of noninfected mice at the same dose. While not providing rigorous PK parameters, this three-point exposure profile proved useful as an initial indication of exposure times in relation to the in vitro IC50 while still providing a moderate level of throughput. As shown in Figure 5, all compounds tested achieved relatively high initial concentrations but varied in the rate of compound clearance. All compounds that effectively reduced blood parasitemia to nondetectable levels within the dosing period (Table 3 and Figure 5, panel A) exhibited sustained exposure profiles over the 24 h dosing interval, with final concentrations significantly exceeding the in vitro IC50 range for the compounds in this group. In contrast, the compounds that were less effective at reducing blood parasitemia (Table 3 and Figure 5, panel B), displayed a more rapid decrease in plasma concentrations and an overall reduced exposure profile, with the 24 h plasma concentrations being at or well below the IC50 range.
Figure 5.

Three-point mouse exposure profiles for compounds that effectively reduced blood parasitemia following once daily dosing of 20 mg/kg for 10 days (A) and compounds that did not reduce parasitemia using the same dosing protocol (B). Compounds shown in panel A are 5 (filled circle), 2 (open circle), 52 (filled triangle), 6 (open triangle), (rac)-7 (filled square), and posaconazole (open square) and in panel B are 47 (filled circle), 48 (open circle), 49 (filled triangle), 4 (open triangle), 50 (filled square), and 51 (open square).
Table 3. Preliminary PK/PD Assessment for Compounds Dosed Orally at 20 mg/kg for up to 10 Days to Mice Infected with T. cruzia.
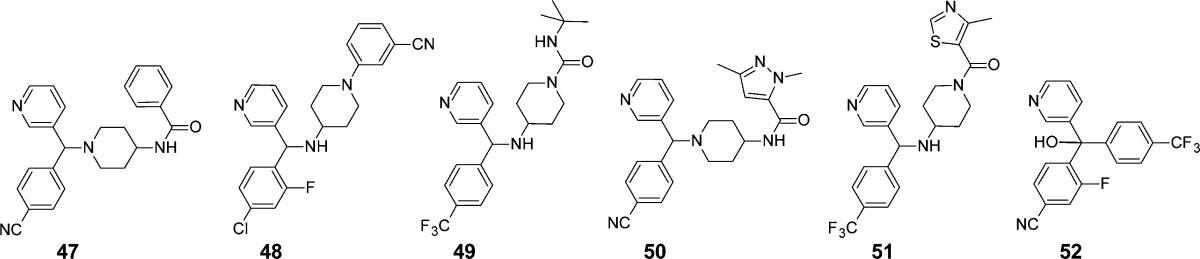
| compd | IC50 (μM) | plasma concn, 0.5 h (μM) | plasma concn, 24 h (μM) | parasites cleared from blood during 10-day dosing? |
|---|---|---|---|---|
| 47 | 0.024 | 3.31 | 0.016 | no |
| 48 | 0.013 | 1.29 | 0.008 | no |
| 49 | 0.061 | 3.26 | 0.001 | no |
| 4 | 0.019 | 2.42 | 0.002 | no |
| 50 | 0.047 | 5.29 | 0.002 | no |
| 51 | 0.058 | 3.28 | 0.001 | no |
| 5 | 0.011 | 0.85 | 0.490 | yes |
| 2 | 0.012 | 2.49 | 0.030 | yes |
| 52 | 0.002 | 1.37 | 0.047 | yes |
| 6 | 0.017 | 5.54 | 0.790 | yes |
| (rac)-7 | 0.007 | 1.55 | 0.370 | yes |
| posaconazole | 0.001 | 3.04 | 2.89 | yes |
Plasma exposure data were obtained in a parallel group of noninfected mice following a single oral dose of 20 mg/kg.
Representative exposure profiles of compounds from the 6 and 7 series are shown in the Supporting Information, and the plasma concentrations at 0.5 and 24 h following a single 20 mg/kg oral dose to noninfected mice are shown in Table 4. The desired pharmacokinetic profile of high plasma levels, extended exposure over the dosing interval, and high exposure relative to the in vitro IC50 was achieved within both compound sets, reflective of the highly optimized nature of the scaffolds. While this abbreviated exposure analysis does not allow the determination of pharmacokinetic parameters and does not take into account potential accumulation following repeat administration, it was used as a practical mechanism for prioritizing compounds for in vivo efficacy testing. Of this group, the three sulfones (33, 45, and 46) exhibited the most rapid clearance and had the lowest plasma concentrations at 24 h.
Table 4. Plasma Concentrations in Noninfected Mice at 0.5 and 24 h after a Single 20 mg/kg Oral Dosea.
| compd | IC50 (μM) | plasma concn, 0.5 h (μM) | plasma concn, 24 h (μM) |
|---|---|---|---|
| 6 | 0.017 | 5.54 | 0.79 |
| 24 | 0.017 | 8.30 | 0.94 |
| 31 | 0.003 | 0.51 | 1.11 |
| 33 | 0.002 | 3.15 | 0.01 |
| 34 | 0.005 | 7.77 | 0.69 |
| (S)-7 | 0.012 | 4.42 | 0.65 |
| 40 | 0.012 | 4.74 | 0.51 |
| 41 | 0.008 | 6.6 | 0.10 |
| 43 | 0.012 | 5.4 | 0.20 |
| 44 | 0.012 | 11.1 | 0.10 |
| 45 | 0.002 | 18.8 | 0.01 |
| 46 | 0.024 | 15.7 | 0.001 |
| posaconazole | 0.001 | 3.83 | 2.89 |
See also graphical presentation in Supporting Information.
For 6 and (S)-7, a more complete assessment of the pharmacokinetic properties in mice was also obtained following intravenous and oral administration. As shown in Table 5 and Figure 6, both compounds exhibited relatively low clearance, high volume of distribution, long half-life, and high oral bioavailability.
Table 5. Pharmacokinetic Parameters in Mice after Intravenous and Oral Administration of 6 and (S)-7.
| parameter | 6 | (S)-7 |
|---|---|---|
| iv dose (mg/kg) | 2 | 2 |
| po dose (mg/kg) | 20 | 20 |
| plasma CL (mL min–1 kg–1) | 8.1 | 7.7 |
| plasma Vss (L/kg) | 4.4 | 4.2 |
| half-life, iv/po (h) | 8.6/12.5 | 5.9/8.2 |
| oral bioavailability (%) | 95 | 64 |
Figure 6.

Plasma concentration versus time profiles following intravenous administration of 2 mg/kg (A) or oral administration of 20 mg/kg (B) to noninfected mice. Data represent the mean of three measurements (±SD) at each time point for 6 (filled circles) and (S)-7 (open triangles).
Efficacy of Lead Compounds in Vivo
Compounds with the desired oral exposure profile were progressed into a rigorous efficacy study to assess curative activity in mice infected with T. cruzi. Swiss mice inoculated with 40 000 trypomastigotes were administered test compounds 8 days postinfection on a once daily basis for 20 days. If parasites were not detected in blood following this treatment duration, cyclophosphamide-induced immunosuppression was commenced 10 days after cessation of dosing and in three cycles, with each cycle comprising 4 days of treatment followed by a 3-day rest. The absence of parasites in organs was evaluated by PCR analysis on tissue samples (spleen, heart, skeletal muscle, and colon) and blood to determine whether or not the mice were cured. A curative outcome reflected a negative result for all samples. The efficacy of lead compounds 6, (rac)-7, and (S)-7 is shown in Table 6 and in Figure 7, along with posaconazole (used as a positive control) and benznidazole.
Table 6. Survival and Cures after Once Daily Oral Dosing for 20 Days to T. cruzi Infected Mice.
| mouse survival after infectiona |
|||||||
|---|---|---|---|---|---|---|---|
| compd | dose (mg/kg) | day 8 | day 28 | day 37 | day 58 | –ve PCRb | cure ratec (%) |
| 6 | 20 | 5 of 5 | 4 of 5 | 4 of 5 | 3 of 5 | 3 of 3 | 60 |
| benznidazole | 100 | 5 of 5 | 4 of 5 | 4 of 5 | 0 of 5 | ||
| posaconazoled | 20 | 5 of 5 | 4 of 5 | 4 of 5 | 1 of 5 | 1 of 1 | 20 |
| (rac)-7 | 20 | 5 of 5 | 5 of 5 | 5 of 5 | 3 of 5 | 0 of 3 | |
| (S)-7 | 5 | 4 of 4 | 4 of 4 | 4 of 4 | 3 of 4 | 0 of 3 | 0 |
| (S)-7 | 10 | 4 of 4 | 4 of 4 | 4 of 4 | 3 of 4 | 2 of 3 | 50 |
| posaconazoled | 20 | 3 of 3 | 3 of 3 | 3 of 3 | 2 of 3 | 2 of 2 | 66 |
| vehiclee | 5 of 5 | 0 of 5 | |||||
Number of mice surviving out of the number of mice starting the experiment. Mice observed to have reoccurrence of parasites in blood were euthanized; day 8 = first compound dose; day 28 = first day of 10-day nontreatment period; day 37 = last day of nontreatment period prior to immunosuppression; day 58 = experiment end after three cycles of immunosuppression.
PCR analysis of blood and tissue samples taken from spleen, heart, skeletal muscle, and colon. Cures were defined as mice being PCR negative (−ve) for parasite DNA in all samples.
Percentage of mice that were cured of infection.
Posaconazole was used as a positive control. Cure rates for this compound vary approximately 20–60% in this assay.
Vehicle-treated mice were euthanized by day 13–14.
Figure 7.

Efficacy data in T. cruzi infected mice following once daily oral treatment for 20 days beginning on day 8 postinfection. The three panels represent separate experiments, and data points are the mean ± SEM. Symbols represent the following: filled circle (vehicle control, n = 5 in panel A, n = 3 in panels B and C); open square (100 mg/kg benznidazole, n = 5); filled diamond (20 mg/kg posaconazole, n = 5 in panel A, n = 3 in panels B and C); open triangle (20 mg/kg 6, n = 5); open diamond (20 mg/kg (rac)-7, n = 5); filled triangle (5 mg/kg (S)-7, n = 4); open circle (10 mg/kg (S)-7, n = 4). Crosses represent animal deaths (panel A, day 12 (posaconazole), day 16 (compound 6)), and the treatment phase is shown as the gray shaded area. The limit of detection (LOD) of the microscopic method is 20 000 parasites/mL.
Compound 6 dosed at 20 mg/kg significantly improved mouse survival compared to vehicle-treated mice, and 4 out of 5 mice were parasite free at the end of dosing. Following three cycles of immunosuppression, 3 out of 4 mice remained microscopically parasite free. PCR analyses of organs and blood from these mice were negative for parasite DNA, indicating an overall cure rate of 60%. Posaconazole, as the positive control, achieved a cure rate of 20% at the same dose level in this experiment. Mice dosed orally with benznidazole at 100 mg/kg experienced a re-emergence of parasites in blood during the immunosuppression phase of the experiment. All of the vehicle-treated mice exhibited increasing parasitemia and were euthanized by day 14. Administration of lower doses of 6 (5 and 10 mg/kg) improved mouse survival relative to vehicle-treated mice, but parasitological cure was not observed (data not shown). No other analogues in this series were as efficacious as compound 6.
The efficacy and cure rate of (rac)-7 was assessed following daily oral dosing at 20 mg/kg. Mouse survival relative to vehicle-treated mice was significantly improved with 5 out of 5 (100%) surviving and being microscopically parasite free in blood until day 37 (the end of the rest period). At the end of the experiment following three rounds of immunosuppression, 3 out of 5 mice (60%) did not show signs of parasite rebound, but PCR analyses of organs and blood from these mice were positive for parasite DNA, indicating no cures were obtained.
Further investigations were conducted with active enantiomer (S)-7 dosed at 5 and 10 mg/kg. Mouse survival and general health were significantly improved at the 5 mg/kg dose relative to vehicle-treated mice, with 3 out of 4 mice (75%) surviving until the end of the experiment. Results from PCR analyses of organs and blood from these mice were positive for parasite DNA in colon and skeletal muscle in all three mice, indicating cure had not been achieved. Compound (S)-7 dosed at 10 mg/kg attained the same level of mouse survival as the 5 mg/kg dose and in addition, achieved a parasitological cure in 2 out of the 3 mice remaining at the end of the experiment as determined by PCR analyses of organs and blood, representative of a 50% cure rate for (S)-7 in this experiment. The mouse in which cure was not obtained was only positive for parasite DNA in spleen and negative in all other samples.
Discussion
The CYP51 enzyme is of critical importance to organisms requiring sterol biosynthesis for membrane function.17 Inhibition of T. cruzi CYP51 affects sterol composition, damages parasite ultrastructure, and leads to parasite kill.17 A growing number of T. cruzi CYP51 inhibitors have been reported in the literature encompassing early hits,23−27 thoroughly investigated leads,28,29 and potential clinical candidates.19,30 The widely different experimental protocols used to evaluate these inhibitors (and other T. cruzi inhibitor compounds) make a comparison of compound effectiveness challenging,31 particularly in in vivo efficacy experiments. Important variables include the type of parasite strain used (or discrete typing unit (DTU)32), use of inbred vs wild type mice (influencing assay variability), dosage and frequency (e.g., once or twice daily), treatment duration, time of compound administration after infection, use of immunosuppression to “flush out” parasites from tissues, and the number of tissues sampled by PCR to evaluate cure. The existence of acute and chronic disease states in the human condition adds further complexity to the area, and feedback from ongoing clinical trials will help define the relationship of laboratory T. cruzi mouse infection models to the human disease.
During the course of our discovery efforts, we developed an efficient testing flow scheme to evaluate T. cruzi CYP51 inhibitors based on the non-azole antifungal compound fenarimol. Extensive in vivo evaluation of a broad range of molecular scaffolds prepared during lead optimization facilitated the development of a PK/PD hypothesis to aide compound triage into an in vivo mouse model of subchronic T. cruzi infection in wild type mice. The ability of compounds to clear parasites from the blood of mice with an established infection following once daily oral dosing for a 10-day period provided an initial and relatively rapid readout of compound efficacy. By relating the outcome of these studies to compound exposure in a parallel group of noninfected mice following oral dosing at the same dose level, it became evident that efficacy was associated with high plasma concentrations relative to the in vitro IC50 and an extended exposure profile over the full dosing interval (24 h). This profile became a point of differentiation for late stage compounds and selected for inhibitors with the highest probability of reducing parasite burden and improving mouse survival into the late-stage immunosuppressive phase of the in vivo efficacy model.
On the basis of the iv pharmacokinetic data generated for 6 and (S)-7, these extended profiles were also associated with high volumes of distribution, a feature previously suggested to be necessary for efficacy of other CYP51 inhibitors.13,33 Further definition of PK/PD relationships for T. cruzi is ongoing.
In summary, two highly optimized T. cruzi CYP51 inhibitors, 6 and (S)-7 developed following a series of scaffold hops from screen hit fenarimol, show curative effects in an immunosuppressive model of subchronic T. cruzi infection in mice as determined by PCR analyses of tissues and blood. Cure was obtained following once daily oral dosing of 20 mg/kg 6 and 10 mg/kg (S)-7 for 20 days. The efficacy of 6 and (S)-7 was comparable to the clinical candidate posaconazole (20 mg/kg) and better than benznidazole (100 mg/kg), the current standard of care, when evaluated in the same model. Both compounds are potent inhibitors of T. cruzi in vitro (Tulahuen TcVI), are noncytotoxic, and did not cause any adverse events on extended dosing in vivo in the mouse model. These non-azole, pyridine-based compounds do not inhibit cytochrome P450 3A4 in microsomes, a general liability of the azole class of CYP51 inhibitors and an important contributing factor to adverse drug–drug interactions and drug resistance. Compounds 6 and (S)-7 are prepared by short and efficient synthetic procedures suggestive of a low cost of goods. The extremely promising efficacy and druglike characteristics of both compounds have initiated further profiling with regard to their potential development as new treatments for Chagas disease.
Experimental Section
Posaconazole was purchased as an oral suspension (Noxafil Schering Corporation, 40 mg/mL). For in vivo efficacy and pharmacokinetic studies the oral suspension was diluted to the appropriate concentration with water. For in vitro studies, posaconazole was first isolated from the suspension by dilution with water and centrifugation, followed by extraction and recrystallization from hot isopropanol. Benznidazole was synthesized by Epichem Pty Ltd.
In Vitro Activity and ADME Properties
Inhibition of T. cruzi (Tulahuen strain TcVI transfected with β-galactosidase) was assessed using a whole parasite assay (amastigote form) in accordance with the procedure described by Buckner et al.34 and as previously described.21 Compounds were added to infected cells in seven-point serial dilutions performed in triplicate and incubated for 96 h. A gate of IC50 ≤ 30 nM was applied for progression into physicochemical and in vitro ADME assays. Cytotoxicity was assessed in L6 cells as previously described.21
In silico physicochemical parameters were calculated using JChem for Excel, version 5.1 (ChemAxon, Budapest). The log D (pH 7.4) was determined using a chromatographic method. Data were collected using a Waters 2795 HPLC instrument with a Waters 2487 dual channel UV detector (operated at 220 and 254 nm) with a Phenomenex Synergi Hydro-RP 4 μm (30 mm × 2 mm) column. The mobile phase comprised aqueous buffer (50 mM ammonium acetate, pH 7.4) and acetonitrile with an acetonitrile gradient of 0% to 100% over 10 min. Compound retention properties were compared to a set of nine standard compounds with known partition coefficients determined using shake flask methods.
Kinetic solubility in phosphate buffer (pH 6.5) was determined by serial dilution of a concentrated stock solution prepared in DMSO. The solubility range was determined by nephelometry, and reported values represent the minimum of this range.
Hepatic clearance and extraction ratio (EH) values were determined by incubating compounds with human liver microsomes (BD Gentest, Discovery Labware Inc., Woburn, MA) suspended in 0.1 M phosphate buffer (pH 7.4) at 37 °C with a final protein concentration of 0.4 mg/mL and compound concentration of 1 μM. An NADPH-regenerating system (1 mg/mL NADP, 1 mg/mL glucose 6-phosphate, 1 U/mL glucose 6-phosphate dehydrogenase) and MgCl2 (0.67 mg/mL) were added to initiate the metabolic reactions, which were subsequently quenched with ice-cold acetonitrile at time points ranging from 0 to 60 min. Control samples were incubated in the absence of NADPH to monitor for non-cytochrome P450-mediated metabolism. Following quenching, samples were centrifuged and the concentration of parent compound remaining in the supernatant was determined by LCMS. The first-order rate constant for substrate depletion was determined by fitting the data to an exponential decay function, and these values were used to calculate the in vitro intrinsic clearance which was scaled to predict the in vivo intrinsic clearance as described previously.35 The blood clearance and the predicted hepatic extraction ratio (EH) were calculated using the well-stirred model of hepatic clearance. Scaling factors and hepatic blood flow were taken from Ring et al.36
In Vivo Mouse Exposure and Pharmacokinetic Studies
All animal studies were conducted using established procedures in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and the study protocols were reviewed and approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee. The systemic exposure of lead compounds was assessed in nonfasted, noninfected male Swiss Outbred mice following administration of compounds by oral gavage. Compounds were formulated in either PEG400 (51, 47, 49, 4, 50) or as suspensions in an aqueous vehicle containing 0.5% (w/v) hydroxypropylmethylcellulose (Methocel E4M premium grade, Dow Chemical Co., Midland, MI), 0.4% (v/v) Tween 80 (Sigma Chemical Co., St Louis, MO), 0.5% (v/v) benzyl alcohol, and 5% v/v DMSO (all remaining compounds) and administered at a dose of 20 mg/kg. Blood samples were taken at 0.5, 7.5, and 24 h after dosing and plasma concentrations determined by LCMS following comparison to calibration standards prepared in blank mouse plasma. No adverse reactions or compound-related side effects were observed after administration of any of the test compounds at a dose of 20 mg/kg.
The in vivo mouse pharmacokinetic properties of 6 and (S)-7 were assessed following iv and oral administration at doses of 2 and 20 mg/kg, respectively. The iv formulations were prepared as aqueous solutions containing 40% (v/v) propylene glycol, 10% (v/v) ethanol, and 50% 0.01 M HCl (6 iv formulation also contained 0.4% v/v Tween 80), and the oral formulation was an aqueous suspension containing 0.5% (w/v) hydroxypropylmethylcellulose, 0.4% (v/v) Tween 80, and 0.5% (v/v) benzyl alcohol (6 oral formulation also contained 5% v/v DMSO). The iv dosing (50 μL) was conducted via the tail vein, and oral dosing (0.2 mL) was via gavage. A maximum of two blood samples were obtained from each mouse, via either submandibular bleed (approximately 120 μL, conscious sampling) or terminal cardiac puncture (0.6 mL, under isofluorane anesthesia), and transferred to a tube containing heparin and a stabilization cocktail (cOmplete protease inhibitor cocktail (Roche), potassium fluoride, and EDTA) to minimize the potential for ex vivo degradation. Samples were centrifuged, and plasma was separated and stored at −20 °C prior to analysis.
Plasma samples were assayed by LCMS using a Waters Micromass Xevo TQ triple quadrupole instrument coupled to a Waters Acquity UPLC. The column was a Supelco Ascentis Express RP Amide column (2.7 μm particle size, 50 mm × 2.1 mm i.d.) equipped with a Phenomenex Security Guard column with Synergy Polar packing material, and both columns were maintained at 40 °C. The mobile phase consisted of methanol and water, both containing 0.05% formic acid, and was delivered using a linear gradient over 3.3 min followed by re-equilibration to the starting conditions. The flow rate was 0.4 mL/min, and the injection volume was 5 μL. LCMS analysis was conducted in positive mode electrospray ionization and elution of the analytes monitored in MRM mode. Quantitation was accomplished by comparing the sample response (peak area) to a calibration curve prepared in blank mouse plasma and processed in the same way as the samples. The assays were validated for linearity over the sample concentration range, accuracy, and precision.
Pharmacokinetic parameters were estimated by noncompartmental methods using WinNonlin (version 5.2, Pharsight, Mountain View, CA). Oral bioavailability was estimated by comparing the dose normalized area under the curve following oral administration to that following iv dosing.
In Vivo Efficacy
Outbred female Swiss mice approximately 8 weeks old were obtained from the Animal Resources Centre (Perth, Western Australia). All animal experimentation was carried out with approval of the Animal Ethics Committee of Murdoch University. T. cruzi parasites (Tulahuen TcVI) were passaged through adult female Swiss mice to maintain virility. Trypomastigote-infected blood was collected and frozen in liquid nitrogen for subsequent infections in mice with an inoculum of 50 000 parasites.
Formulation of Test Compounds
Compounds 6 and (rac)-7 were made up to a concentration of 3 mg/mL in HPMC-SV (0.5% w/v hydroxypropylmethylcellulose + 0.4% v/v Tween 80 + 0.5% v/v benzyl alcohol in deionized H20 with 5% v/v DMSO), and (S)-7 was made up to concentrations of 0.75 and 1.5 mg/mL in HPMC-SV (0.5% w/v hydroxypropylmethylcellulose + 0.4% v/v Tween 80 + 0.5% v/v benzyl alcohol in deionized H20 with 5% v/v DMSO). Benznidazole was weighed into a clean glass vial and made up to 15 mg/mL in PBS containing 0.4% v/v Tween 80 and 10% v/v PEG400, shaking with glass beads to achieve an even suspension. Posaconazole (Noxafil, 40 mg/mL) was diluted with WFI for a final concentration of 3 mg/mL. All compounds were evenly suspended and made up fresh every 4–5 days.
In Vivo Acute Toxicity Testing
Compounds 6, (rac)-7, and (S)-7 were tested for acute in vivo toxicity through stepwise cumulative dosing using two mice per compound. Mice were given 25 and/or 50 mg/kg oral doses twice a day for a total of 2.5 days, eventually reaching cumulative doses of 100 and 200 mg/kg. Mice were observed for 1 week following the last dose, and no adverse effects or weight loss was noted for any of the compounds tested in this experiment.
In Vivo Efficacy Studies
Compounds were administered once daily at a maximum dose of 20 mg/kg (test compounds and posaconazole) or 100 mg/kg benznidazole commencing on day 8 postinfection (pi). For the initial PK/PD assessment, compounds were administered for 10 days and parasitemia was monitored throughout the dosing period by microscopy. More promising compounds and the positive controls posaconazole and benznidazole were progressed to a more rigorous efficacy study with dosing once daily for 20 days, with the last dose being administered on day 27 pi. Blood parasitemia was monitored over the course of the 20-day treatment. If parasites were not detected in blood following this treatment duration, cyclophosphamide-induced immunosuppression was commenced 10 days after cessation of dosing. Cyclophosphamide (50 mg kg–1 day–1) was administered ip in three cycles, with each cycle comprising 4 days of treatment followed by 3 days of rest. The absence of parasites from organs following this treatment was confirmed by PCR analysis targeting the nuclear satellite DNA of T. cruzi on tissue samples (spleen, heart, skeletal muscle, and colon) and blood.37,38 Euthanasia was carried out if parasitemia exceeded 9 × 106 parasites/mL, parasites rebounded in blood after dosing was completed, there were three minor abnormal clinical observations or one major clinical observation, or more than 20% weight loss occurred.
Chemistry
Reagents were purchased from commercial suppliers and used without further purification. Commercially available anhydrous solvents were used and stored under nitrogen unless indicated otherwise. Reactions involving moisture sensitive reagents were conducted under an atmosphere of dry nitrogen in glassware dried with a heat gun. Thin layer chromatography (TLC) using silica gel Merck 60 F254 plates and detection with UV light was used to monitor reactions. 1H and 13C NMR spectra were recorded in CDCl3 or DMSO-d6 solutions on a Varian 200, Bruker 300, or Varian 400 MHz NMR spectrometer. Chemical shifts are reported in parts per million (δ) downfield of tetramethylsilane (TMS). GCMS data were acquired on an Agilent 5973 network instrument. LCMS data were acquired on an Applied Biosystems/MDS Sciex API-2000 system. Flash chromatography was carried out with silica gel (0.04–0.06 μm, 230–400 mesh), with reverse phase silica gel (C18 35–70 μm), or on a Flashmaster II system using cartridges prepared in-house. All compounds tested in the in vitro and in vivo biological screens were purified to >95%. Purity was determined by HPLC analysis on an Agilent 1100 series instrument, fitted with a C8 reverse phase Agilent zorbax eclipse DB-LB 4.6 mm × 150 mm, 5 μm column (flow rate of 1.2 mL/min), 1H NMR spectra (accounting for noncompound peaks and residual solvent), and LCMS. Microwave reactions were carried out using a Biotage Initiator EXP machine.
Preparation of N-[4-(Trifluoromethyl)phenyl]-N-(1-[5-(trifluoromethyl)pyridin-2-yl]piperidin-4-yl)pyridin-3-amine (6)
tert-Butyl 4-(Pyridin-3-ylamino)piperidine-1-carboxylate (8). Sodium triacetoxyborohydride (63.82 g, 301.2 mmol) was added to a mixture of 1-tert-butoxycarboxylpiperidin-4-one (40.0 g, 200.8 mmol), 3-aminopyridine (20.78 g, 220.8 mmol), acetic acid (13.26 g, 220.8 mmol), and dichloromethane (60 mL). The mixture was heated under reflux overnight (20 h). After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (120 mL), washed with 2 M sodium hydroxide (6 × 70 mL) so that the pH was >7, washed with brine (2 × 70 mL), dried (magnesium sulfate), and concentrated to afford a light orange solid. The solid was washed with diethyl ether (100 mL), then filtered under vacuum to afford 8 (35.5 g, 64%) as a white solid. 1H NMR (CDCl3) 8.00–8.06 (m, 1H), 7.91–7.99 (m, 1H), 7.04–7.13 (m, 1H), 6.83–7.92 (m, 1H), 3.98–4.20 (m, 2H), 3.62 (br s, 1H), 3.38–3.50 (m, 1H), 2.82–3.00 (m, 2H), 1.96–2.12 (m, 2H), 1.45 (s, 9H), 1.22–1.42 (m, 2H).
tert-Butyl 4-[4-(Trifluoromethyl)phenyl](pyridin-3-yl)aminopiperidine-1-carboxylate (9). Sodium tert-butoxide (40.2 g, 418 mmol) was added slowly, portionwise to a mixture of 8 (29.0 g, 104.6 mmol), CombiPhos-Pd6 (1.57 g, 3.14 mmol), palladium(II) acetate (704 mg, 3.14 mmol), xantphos (2.42 g, 4.18 mmol), and 4-bromobenzotrifluoride (23.5 g, 104.6 mmol) in toluene (200 mL) under (and flushed with) an atmosphere of nitrogen. The reaction mixture was heated at 110 °C for 6 h under nitrogen. Upon cooling to room temperature, the reaction mixture was diluted with ethyl acetate (150 mL) and filtered through a short plug of Celite. The resulting filtrate was washed with water (3 × 100 mL), washed with brine (100 mL), dried (magnesium sulfate), filtered, and concentrated to give crude 9 (30 g) as a dark orange oil. This material was dissolved in ethyl acetate (150 mL) and adsorbed onto silica gel (approximately 30 g). Purification by column chromatography (eluent 100% dichloromethane increasing to 20% ethyl acetate in dichloromethane) afforded 9 (18.40 g, 42%) as a light, pale brown solid. 1H NMR (CDCl3) 8.55 (t, J = 3.2 Hz, 1H), 8.29–8.39 (m, 1H), 7.40–7.48 (m, 2H), 7.34–7.40 (m, 2H), 6.65 (d, J = 4.8 Hz, 2H), 4.10–4.38 (m, 2H), 3.98–4.10 (m, 1H), 2.72–2.93 (m, 2H), 1.91–2.03 (m, 2H), 1.42 (s, 9H), 1.22–1.38 (m, 2H).
N-(Piperidin-4-yl)-N-[4-(trifluoromethyl)phenyl]pyridin-3-amine (10). Compound 9 (18.32 g, 43.5 mmol) was dissolved in dichloromethane (50 mL) and water (10 mL). The resulting mixture was cooled in an ice bath and trifluoroacetic acid (80 mL) added slowly with stirring. The mixture was allowed to warm to room temperature and stirred overnight (16 h). TLC analysis (miniworkup) showed only baseline material (all starting material consumed). The reaction mixture was evaporated to dryness, dissolved in ethyl acetate (150 mL), and washed with 2 M sodium hydroxide until the pH of the aqueous layer was 10. The organic layer was separated, dried (magnesium sulfate), filtered, and concentrated to afford 10 (13.90 g, quantitative yield) as a sticky, dark orange resin. 1H NMR (CDCl3) 8.50–8.55 (m, 1H), 8.32–8.38 (m, 1H), 7.39–7.45 (m, 4H), 6.65 (d, J = 0.8 Hz, 2H), 3.96–4.09 (m, 1H), 3.07–3.25 (m, 2H), 2.69–2.85 (m, 2H), 1.91–2.06 (m, 2H), 1.29–1.45 (m, 2H).
N-[4-(Trifluoromethyl)phenyl]-N-(1-[5-(trifluoromethyl)pyridin-2-yl]piperidin-4-yl)pyridin-3-amine (6). N,N-Diisopropylethylamine (62.74g, 485 mmol) was added to a mixture of 10 (39.0 g, 121 mmol) and 2-chloro-5-(trifluoromethyl)pyridine (33.05 g, 182 mmol) in dimethylformamide (120 mL). The resulting mixture was heated at 100 °C overnight (18 h) under an atmosphere of nitrogen. Upon cooling to room temperature, the reaction mixture was diluted with ethyl acetate (200 mL) and filtered through a plug of Celite. The resultant solution was washed with water (3 × 100 mL), washed with brine (200 mL), dried (magnesium sulfate), absorbed onto silica gel (approximately 70 g), and purified by column chromatography (eluent 5% acetone in hexanes increasing to 33% acetone) to afford crude 6 (40.6 g) as a dark orange gel. This material was dissolved in ethyl acetate (250 mL), adsorbed onto silica gel (approximately 50 g), and repurified by column chromatography (eluent hexanes, then dichloromethane in hexanes increasing to 100% dichloromethane) to afford 39.3 g of semicrude material which was dissolved in the minimum volume of boiling hexanes (1250 mL). Upon seeding with a spatula tip of purified 6 (from a previous experiment) and standing overnight at room temperature, a solid formed. The solid material was filtered on a sinter funnel under vacuum to afford 6 (25.5 g, 45%) as a light, pale yellow solid. 1H NMR (CDCl3) 8.53 (d, J = 2.0 Hz, 1H), 8.31–8.39 (m, 1H), 7.59 (d, J = 2.8 Hz 1H), 7.41–7.49 (m, 2H), 6.65–6.73 (m, 2H), 6.68 (d, J = 8.4 Hz 2H), 6.61 (d, J = 9.2 Hz, 2H), 4.48–4.63 (m, 2H), 4.15–4.28 (m, 1H), 2.93–3.12 (m, 2H), 2.00–2.16 (m, 2H), 1.32–1.50 (m, 2H); 13C NMR (101 MHz, CDCl3) 159.7, 150.5, 150.0, 147.5, 145.7, 139.1, 136.6, 134.6, 126.7, 125.0, 125.0, 124.4, 120.8, 115.7, 115.0, 105.5, 55.7, 44.4, 30.5; LCMS [M + H]+ = 467.5; mp 95–97 °C. Anal. Calcd for C23H20F6N4: C, 59.23; H, 4.32; N, 12.01. Found: C, 59.49; H, 4.44; N, 12.06.
The mother liquor was concentrated to give a further 12.5 g of material as a dark orange gel, which was subjected to cycles of the solidification process with boiling hexanes to afford a further 7.3 g, 1.3 g, and 400 mg of 6. These batches were combined, dissolved in diethyl ether (50 mL), and washed with 4 M hydrochloric acid (180 mL). The aqueous layer was then extracted with more diethyl ether (50 mL). The aqueous layer was then basified (to pH 13) using 2 M sodium hydroxide (approximately 350 mL). The resulting cloudy white aqueous layer was extracted with ethyl acetate (2 × 100 mL) and the organic layer separated. Activated carbon was added to the ethyl acetate layer and the resulting suspension heated to 70 °C for 2 h. Magnesium sulfate was added and the suspension filtered. The solution was then concentrated to afford 6 (8.0 g) as a light, pale yellow resin. Upon standing in the fridge for 12 days, the resin had formed a white solid. This was further dried to give 6 (7.93g) as a white solid.
2-(4-Chlorophenyl)-1-{4-[4-(trifluoromethyl)phenyl]piperazin-1-yl}-2-(pyridin-3-yl)ethanone (7) and (R)- and (S)-7
(4-Chlorophenyl)(pyridin-3-yl)acetonitrile (11). 4-Chlorophenylacetonitrile (27.6 g, 182 mmol) and 3-fluoropyridine (9.00 g, 91.8 mmol) were dissolved in N-methyl-2-pyrrolidone (80 mL), and the solution was to 0 °C (ice/water bath). Potassium tert-butoxide (28.0 g, 250 mmol) was added in portions (the mixture became red/purple/orange in color), and then the reaction vessel was placed in an oil bath and heated to 85 °C and stirred overnight. The flask was removed from the heat and cooled to 0 °C, quenched with 1 M HCl (aq) to approximately pH 6–7, and diluted with ethyl acetate. A white solid material was present and removed by filtration through a sinter funnel, and the phases were separated. The organic layer was washed again with water (×2), dried (MgSO4), filtered, and concentrated under vacuum and the oily residue was purified by column chromatography (10% ethyl acetate in hexanes increasing to 30% ethyl acetate in hexanes) to obtain the desired product still contaminated with unreacted 4-chlorophenylacetonitrile and an unknown impurity. This material was partitioned into 2 M HCl (aq) and diethyl ether, and the phases were separated. The aqueous layer was washed again with diethyl ether and then treated with 2 M NaOH (aq) to pH ∼8 and extracted with ethyl acetate (×2). The ethyl acetate extracts were washed with brine, dried (MgSO4), filtered, and concentrated under vacuum and the residue was repurified by column chromatography (10% ethyl acetate in hexanes increasing to 30% ethyl acetate in hexanes) to provide intermediate nitrile 11 as an amber oil (15.4 g, 73%). 1H NMR (400 MHz, CDCl3) 8.60–8.62 (m, 2H), 7.65–7.69 (m, 1H), 7.37–7.41 (m, 2H), 7.32–7.36 (m, 1H), 7.27–7.31 (m, 2H), 5.16 (s, 1H).)
(4-Chlorophenyl)(pyridin-3-yl)acetic Acid (12). Nitrile 11 (7.5 g, 32.3 mmol) was suspended in 2 M NaOH (aq) (75 mL) and the suspension degassed with nitrogen for approximately 5 min. The flask was fitted with a balloon containing nitrogen and the mixture heated to 110 °C and stirred overnight. The mixture was cooled to room temperature and then washed with diethyl ether. The aqueous phase was separated and treated with 4 M HCl (aq) to pH ∼5 (precipitation observed) and extracted with ethyl acetate (×2). The combined organic washings were dried (MgSO4), filtered, and concentrated under vacuum to afford acid 12 as a pale yellow solid (4.4 g, 55%). 1H NMR (400 MHz, DMSO-d6) 13.04 (br s, 1H), 8.54 (d, J = 1.2 Hz, 1H), 8.46 (d, J = 3.9 Hz, 1H), 7.68–7.78 (m, 1H), 7.30–7.48 (m, 5H), 5.21 (s, 1H).
[NB: In other repeat preparations it has been observed that significant precipitation of some product can occur during the acidification process and will not dissolve in the ethyl acetate washings. This material can be isolated by filtration and is of comparable purity or higher purity than that isolated from the organic washings.]
2-(4-Chlorophenyl)-1-{4-[4-(trifluoromethyl)phenyl]piperazin-1-yl}-2-(pyridin-3-yl)ethanone (7). Acid 12 (3.0 g, 12.1 mmol), HATU (4.60 g, 12.1 mmol), and 1-[4-(trifluoromethyl)phenyl]piperazine (13) (2.78 g, 12.1 mmol) were dissolved in N,N-dimethylformamide (30 mL) and treated with diisopropylethylamine (3.18 mL, 18.2 mmol). The mixture was stirred at room temperature for 3 h and then partitioned between ethyl acetate and water. The organic layer was washed with water (×3) and then brine, dried (MgSO4), filtered, and concentrated under vacuum, and the crude residue was purified by column chromatography (Flashmaster II ethyl acetate/hexane/methanol gradient) to give 7 as an off-white foam (4.2 g, 75%). 1H NMR (400 MHz, CDCl3) 8.52 (dd, J = 1.2, 4.7 Hz, 1H), 8.48 (d, J = 1.6 Hz, 1H), 7.56–7.65 (m, 1H), 7.45–7.53 (m, 2H), 7.30–7.38 (m, 2H), 7.24–7.29 (m, 1H), 7.18–7.24 (m, 2H), 6.83–6.92 (m, 2H), 5.21 (s, 1H), 3.88–3.98 (m, 1H), 3.75–3.85 (m, 1H), 3.57–3.71 (m, 2H), 3.19–3.37 (m, 2H), 3.08–3.18 (m, 1H), 2.93–3.04 (m, 1H); 13C NMR (101 MHz, CDCl3) 169.0, 152.6, 149.8, 148.8, 136.8, 136.6, 134.8, 133.7, 129.9, 129.3, 126.5, 124.7, 123.5, 121.3, 115.0, 51.6, 48.1, 47.9, 45.6, 41.9; LCMS [M + H] = 460.3; HPLC (water/ACN + 0.1% TFA gradient) 99.5% at 254 nm. Anal. d for C24H21F3N3O: C, 62.68; H, 4.60; N, 9.14. Found: C, 62.70; H, 4.71; N, 9.13.
Chiral Resolution of 2-(4-Chlorophenyl)-1-{4-[4-(trifluoromethyl)phenyl]piperazin-1-yl}-2-(pyridin-3-yl)ethanone (7) to Give (R)-7 and (S)-7. Compound 7 was resolved by preparative chiral HPLC using the following conditions. First eluted enantiomer = (R)-7 (98.6% ee), second eluted enantiomer = (S)-7 (98.0% ee). Instrument: Thar 350 preparative SFC. Column: ChiralCel OJ, 10 μm, 300 mm × 50 mm i.d. Mobile phase: A, CO2; B, methanol. Gradient: B, 45%. Flow rate: 240 mL/min. Back pressure: 100 bar. Column temperature: 38 °C. Wavelength: 220 nm. Cycle time: 6 min. The NMR spectral data for the separated enantiomers were identical with the data for the racemate.
Preparation of 1-[4-(Trifluoromethyl)phenyl]piperazine (13)
To a stirred, degassed (with N2) solution of 1-(tert-butoxycarbonyl)piperazine (15 g, 80 mmol), 4-bromobenzotrifluoride (19 g, 84 mmol), palladium acetate (1.1 g, 2 mol %), and BINAP (2.5 g, 5 mol %) in a 1:1 mixture of ethyl acetate (80 mL) and 1,4-dioxane (80 mL) was added cesium carbonate (52 g, 160 mmol), and the mixture was heated to reflux under an N2 atmosphere. After 1 h, precipitation of palladium black had occurred and TLC analysis indicated consumption of the piperazine starting material. The mixture was cooled to room temperature and quenched with 10% NH4Cl (aq) (5 mL) and passed through a small plug of silica gel, which was washed with dichloromethane. The collected filtrate was concentrated under vacuum and the residue purified by column chromatography (eluent dichloromethane) to afford intermediate tert-butyl 4-[4-(trifluoromethyl)phenyl]piperazine-1-carboxylate as an off-white solid (24.7 g, 93%). 1H NMR (400 MHz, CDCl3) 7.45–7.53 (m, 2H), 6.89–6.96 (m, 2H), 3.54–3.64 (m, 4H), 3.19–3.29 (m, 4H), 1.49 (s, 9H).
tert-Butyl 4-[4-(trifluoromethyl)phenyl]piperazine-1-carboxylate (24.7 g, 74.5 mmol) was taken up in dichloromethane (100 mL) and methanol (5.5 mL) and then treated with trifluoroacetic acid (250 mL). The mixture was stirred overnight at room temperature. Then the volatiles were removed under vacuum and the was treated with 2 M NaOH(aq). The aqueous phase was extracted with ethyl acetate (×2), and the combined organic washings were dried (MgSO4), filtered, and concentrated under vacuum to afford the 1-[4-(trifluoromethyl)phenyl]piperazine 13 as a tan solid (15.6 g, 91%). 1H NMR (400 MHz, CDCl3) 7.40–7.57 (m, 2H), 6.85–7.02 (m, 2H), 3.17–3.30 (m, 4H), 2.98–3.10 (m, 4H).
Compound 52
52 was prepared as previously reported.21
Acknowledgments
This study was funded by DNDi and by a grant from the Australian Research Council. For the work described in this paper, DNDi received financial support from the following donors: Department for International Development (DFID) (U.K.), Reconstruction Credit Institution—Federal Ministry of Education and Research (KfW-BMBF) (Germany), Ministry of Foreign and European Affairs (MAEE) (France), Spanish Agency for International Development Cooperation (AECID) (Spain), Directorate-General for International Cooperation (DGIS) (The Netherlands), Médecins Sans Frontières (Doctors without Borders) (international), and a Swiss foundation. Partial funding support from the Australian Research Council is also gratefully acknowledged. The donors had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors thank Eric Voight and Rodger Henry from the Abbott crystallography group for studies relating to compound 7.
Glossary
Abbreviations Used
- Tc
Trypanosoma cruzi (T. cruzi)
- PK/PD
pharmacokinetic/pharmacodynamic
- Tc IC50
concentration of compound required to cause a 50% inhibition in growth of T. cruzi
Supporting Information Available
Synthetic methods for the preparation of compounds 4, 5, 14–51; abbreviated mouse exposure profiles of compounds 6, (S)-7, 24, 31, 33, 34, 40, 41, and 43–46; and X-ray data for (S)-7. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ A.K.: School of Medicine and Pharmacology, The University of Western Australia, Nedlands 6009, Western Australia, Australia.
Author Present Address
# S.C.: Faculty of Health Sciences, Curtin University, GPO Box U1987, Perth, Western Australia, 6845, Australia.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Chagas Disease (American Trypanosomiasis). WHO Fact Sheet No. 340. http://www.who.int/mediacentre/factsheets/fs340/en/ (accessed June 11, 2013).
- Navarro M.; Navaza B.; Guionnet A.; Lopez-Velez R. Chagas Disease in Spain: Need for Further Public Health Measures. PLoS Neglected Trop. Dis. 2012, 6, e1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascon J.; Bern C.; Pinazo M. J. Chagas Disease in Spain, the United States and Other Non-Endemic Countries. Acta Trop. 2010, 115, 22–27. [DOI] [PubMed] [Google Scholar]
- Coura J. R.; Borges-Pereira J. Chronic Phase of Chagas Disease: Why Should It Be Treated? A Comprehensive Review. Mem. Inst. Oswaldo Cruz 2011, 106, 641–645. [DOI] [PubMed] [Google Scholar]
- Martins-Melo F. R.; Alencar C. H.; Ramos A. N.; Heukelbach J. Epidemiology of Mortality Related to Chagas’ Disease in Brazil, 1999–2007. PLoS Neglected Trop. Dis. 2012, 6, e1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolani Y.; Bukhman G.; Hotez P. J. Neglected Tropical Diseases as Hidden Causes of Cardiovascular Disease. PLoS Neglected Trop. Dis. 2012, 6, e1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier Y.; Torrico F.; Sosa-Estani S.; Russomando G.; Luquetti A.; Freilij H.; Vinas P. A. Congenital Chagas Disease: Recommendations for Diagnosis, Treatment and Control of Newborns, Siblings and Pregnant Women. PLoS Neglected Trop. Dis. 2011, 5, e1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J. A.; de Mecca M. M.; Bartel L. C. Toxic Side Effects of Drugs Used To Treat Chagas’ Disease (American Trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [DOI] [PubMed] [Google Scholar]
- Haberland A.; Saravia S. G. M.; Wallukat G.; Ziebig R.; Schimke I. Chronic Chagas Disease: From Basics to Laboratory Medicine. Clin. Chem. Lab. Med. 2013, 51, 271–294. [DOI] [PubMed] [Google Scholar]
- Pinazo M. J.; Guerrero L.; Posada E.; Rodriguez E.; Soy D.; Gascon J. Benznidazole-Related Adverse Drug Reactions and Their Relationship to Serum Drug Concentrations in Patients with Chronic Chagas Disease. Antimicrob. Agents Chemother. 2013, 57, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasslocher-Moreno A. M.; do Brasil P.; de Sousa A. S.; Xavier S. S.; Chambela M. C.; da Silva G. M. S. Safety of Benznidazole Use in the Treatment of Chronic Chagas’ Disease. J. Antimicrob. Chemother. 2012, 67, 1261–1266. [DOI] [PubMed] [Google Scholar]
- Altcheh J.; Moscatelli G.; Moroni S.; Garcia-Bournissen F.; Freilij H. Adverse Events after the Use of Benznidazole in Infants and Children with Chagas Disease. Pediatrics 2011, 127, E212–E218. [DOI] [PubMed] [Google Scholar]
- Urbina J. A. Specific Chemotherapy of Chagas Disease: Relevance, Current Limitations and New Approaches. Acta Trop. 2010, 115, 55–68. [DOI] [PubMed] [Google Scholar]
- BENEFIT: Evaluation of the Use of Antiparasital Drug (Benznidazole) in the Treatment of Chronic Chagas’ Disease. http://clinicaltrials.gov/show/NCT00123916 (accessed August 28, 2013)
- STOPCHAGAS: A Study of the Use of Oral Posaconazole (POS) in the Treatment of Asymptomatic Chronic Chagas Disease. http://clinicaltrials.gov/showNCT0137780 (accessed September 27, 2013).
- Proof of Concept Study of E1224 To Treat Adults Patients with Chagas Disease. http://clinicaltrials.gov/showNCT01489228 (accessed September 27, 2013).
- Lepesheva G. I.; Waterman M. R. Sterol 14alpha-Demethylase (Cyp51) as a Therapeutic Target for Human Trypanosomiasis and Leishmaniasis. Curr. Top. Med. Chem. 2011, 11, 2060–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove T. Y.; Kim K.; Soeiro M. D. C.; da Silva C. F.; Batista D. D. J.; Batista M. M.; Yazlovitskaya E. M.; Waterman M. R.; Sulikowski G. A.; Lepesheva G. I. Cyp51 Structures and Structure-Based Development of Novel, Pathogen-Specific Inhibitory Scaffolds. Int. J. Parasitol.: Drugs Drug Resist. 2012, 2, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta F.; Dobish M. C.; Nde P. N.; Kleshchenko Y. Y.; Hargrove T. Y.; Johnson C. A.; Waterman M. R.; Johnston J. N.; Lepesheva G. I. VNI Cures Acute and Chronic Experimental Chagas Disease. J. Infect. Dis. 2013, 208, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin S.; Chandrasekar P. H. Safety of Triazole Antifungal Drugs in Patients with Cancer. J. Antimicrob. Chemother. 2010, 65, 410–416. [DOI] [PubMed] [Google Scholar]
- Keenan M.; Abbott M. J.; Alexander P. W.; Armstrong T.; Best W. M.; Berven B.; Botero A.; Chaplin J. H.; Charman S. A.; Chatelain E.; von Geldern T. W.; Kerfoot M.; Khong A.; Nguyen T.; McManus J. D.; Morizzi J.; Ryan E.; Scandale I.; Thompson R. A.; Wang S. Z.; White K. L. Analogues of Fenarimol Are Potent Inhibitors of Trypanosoma cruzi and Are Efficacious in a Murine Model of Chagas Disease. J. Med. Chem. 2012, 55, 4189–4204. [DOI] [PubMed] [Google Scholar]
- Hargrove T. Y.; Wawrzak Z.; Alexander P. W.; Chaplin J. H.; Keenan M.; Charman S. A.; Perez C. J.; Waterman M. R.; Chatelain E.; Lepesheva G. I. T. cruzi CYP51 Complexes with Two Pyridine-Based Drug Candidates for Chagas Disease: Structural Basis for Pathogen-Selectivity. J. Biol. Chem. 2013, 288, 31602–31615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan M.; Alexander P. W.; Diao H.; Best W. M.; Khong A.; Kerfoot M.; Thompson R. C. A.; White K. L.; Shackleford D. M.; Ryan E.; Gregg A. D.; Charman S. A.; von Geldern T. W.; Scandale I.; Chatelain E. Design, Structure–Activity Relationship and in Vivo Efficacy of Piperazine Analogues of Fenarimol as Inhibitors of Trypanosoma cruzi. Bioorg. Med. Chem. 2013, 21, 1756–1763. [DOI] [PubMed] [Google Scholar]
- Chen C. K.; Doyle P. S.; Yermalitskaya L. V.; Mackey Z. B.; Ang K. K. H.; McKerrow J. H.; Podust L. M. Trypanosoma cruzi Cyp51 Inhibitor Derived from a Mycobacterium Tuberculosis Screen Hit. PLoS Neglected Trop. Dis. 2009, 3, e372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunatilleke S. S.; Calvet C. M.; Johnston J. B.; Chen C. K.; Erenburg G.; Gut J.; Engel J. C.; Ang K. K. H.; Mulvaney J.; Chen S.; Arkin M. R.; McKerrow J. H.; Podust L. M. Diverse Inhibitor Chemotypes Targeting Trypanosoma cruzi Cyp51. PLoS Neglected Trop. Dis. 2012, 6, e1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friggeri L.; Scipione L.; Costi R.; Kaiser M.; Moraca F.; Zamperini C.; Botta B.; Di Santo R.; De Vita D.; Brun R.; Tortorella S. New Promising Compounds with in Vitro Nanomolar Activity against Trypanosoma cruzi. ACS Med. Chem. Lett. 2013, 4, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriani G.; Amata E.; Beatty J.; Clements Z.; Coffey B. J.; Courtemanche G.; Devine W.; Erath J.; Juda C. E.; Wawrzak Z.; Wood J. T.; Lepesheva G. I.; Rodriguez A.; Pollastri M. P. Antitrypanosomal Lead Discovery: Identification of a Ligand-Efficient Inhibitor of Trypanosoma cruzi Cyp51 and Parasite Growth. J. Med. Chem. 2013, 56, 2556–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle P. S.; Chen C. K.; Johnston J. B.; Hopkins S. D.; Leung S. S. F.; Jacobson M. P.; Engel J. C.; McKerrow J. H.; Podust L. M. A Nonazole Cyp51 Inhibitor Cures Chagas’ Disease in a Mouse Model of Acute Infection. Antimicrob. Agents Chemother. 2010, 54, 2480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner F. S.; Bahia M. T.; Suryadevara P. K.; White K. L.; Shackleford D. M.; Chennamaneni N. K.; Hulverson M. A.; Laydbak J. U.; Chatelain E.; Scandale I.; Verlinde C.; Charman S. A.; Lepesheva G. I.; Gelb M. H. Pharmacological Characterization, Structural Studies, and in Vivo Activities of Anti-Chagas Disease Lead Compounds Derived from Tipifarnib. Antimicrob. Agents Chemother. 2012, 56, 4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro M. d. N. C.; de Souza E. M.; da Silva C. F.; Batista D. d. G. J.; Batista M. M.; Pavao B. P.; Araujo J. S.; Aiub C. A. F.; da Silva P. B.; Lionel J.; Britto C.; Kim K.; Sulikowski G.; Hargrove T. Y.; Waterman M. R.; Lepesheva G. I. In Vitro and in Vivo Studies of the Antiparasitic Activity of Sterol 14alpha-Demethylase (Cyp51) Inhibitor VNI against Drug-Resistant Strains of Trypanosoma cruzi. Antimicrob. Agents Chemother. 2013, 57, 4151–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner F. S.Experimental Chemotherapy and Approaches to Drug Discovery for Trypanosoma cruzi Infection. In Advances in Parasitology, Chagas Disease, Part A; Weiss L. M., Tanowitz H. B., Kirchhoff L. V., Eds.; Academic Press: London, 2011; Vol. 75, pp 89–119. [DOI] [PubMed] [Google Scholar]
- Zingales B.; Miles M. A.; Campbell D. A.; Tibayrenc M.; Macedo A. M.; Teixeira M. M. G.; Schijman A. G.; Llewellyn M. S.; Lages-Silva E.; Machado C. R.; Andrade S. G.; Sturm N. R. The Revised Trypanosoma cruzi Subspecific Nomenclature: Rationale, Epidemiological Relevance and Research Applications. Infect., Genet. Evol. 2012, 12, 240–253. [DOI] [PubMed] [Google Scholar]
- Urbina J. A. Chemotherapy of Chagas’ Disease: The How and the Why. J. Mol. Med. 1999, 77, 332–338. [DOI] [PubMed] [Google Scholar]
- Buckner F. S.; Verlinde C.; LaFlamme A. C.; vanVoorhis W. C. Efficient Technique for Screening Drugs for Activity against Trypanosoma cruzi Using Parasites Expressing Beta-Galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach R. S. Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: An Examination of in Vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- Ring B. J.; Chien J. Y.; Adkison K. K.; Jones H. M.; Rowland M.; Do Jones R.; Yates J. W. T.; Ku M. S.; Gibson C. R.; He H. D.; Vuppugalla R.; Marathe P.; Fischer V.; Dutta S.; Sinha V. K.; Bjornsson T.; Lave T.; Poulin P. Phrma Cpcdc Initiative on Predictive Models of Human Pharmacokinetics, Part 3: Comparative Assessement of Prediction Methods of Human Clearance. J. Pharm. Sci. 2011, 100, 4090–4110. [DOI] [PubMed] [Google Scholar]
- Vallejo G. A.; Guhl F.; Chiari E.; Macedo A. M. Species Specific Detection of Trypanosoma cruzi and Trypanosoma rangeli in Vector and Mammalian Hosts by Polymerase Chain Reaction Amplification of Kinetoplast Minicircle DNA. Acta Trop. 1999, 72, 203–212. [DOI] [PubMed] [Google Scholar]
- Moser D. R.; Kirchhoff L. V.; Donelson J. E. Detection of Trypanosoma-cruzi by DNA Amplification Using the Polymerase Chain-Reaction. J. Clin. Microbiol. 1989, 27, 1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.