

Abstract
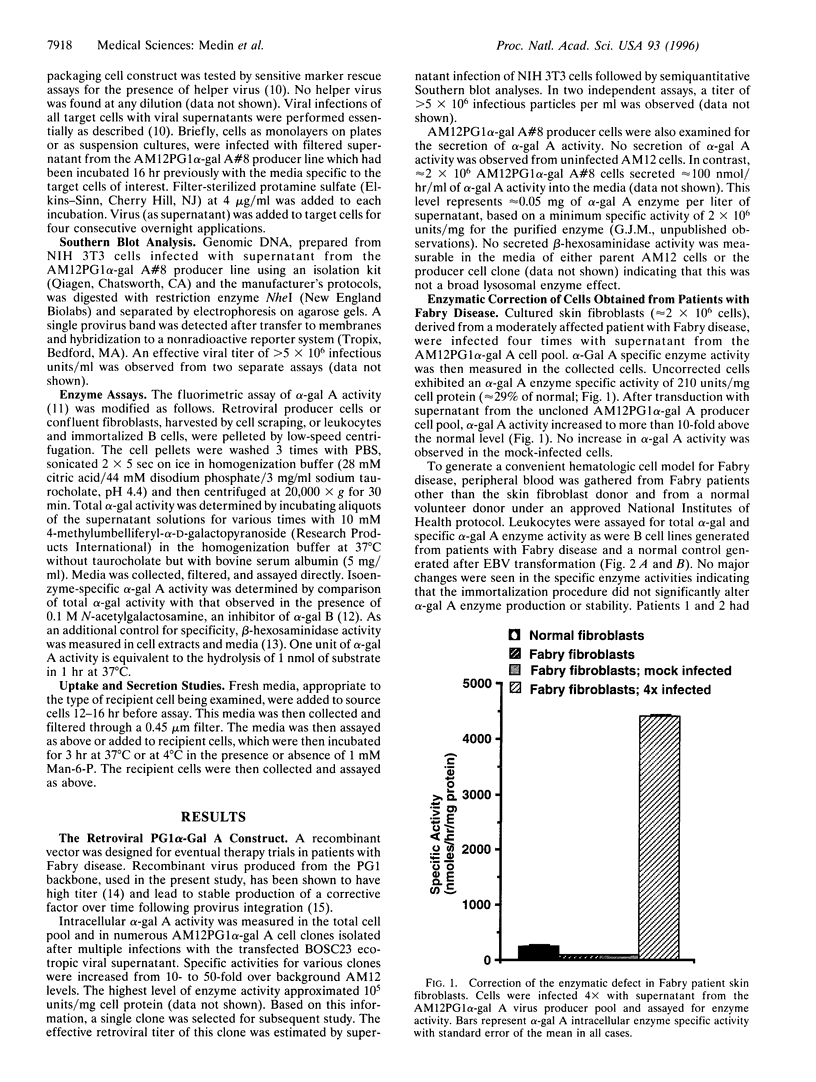
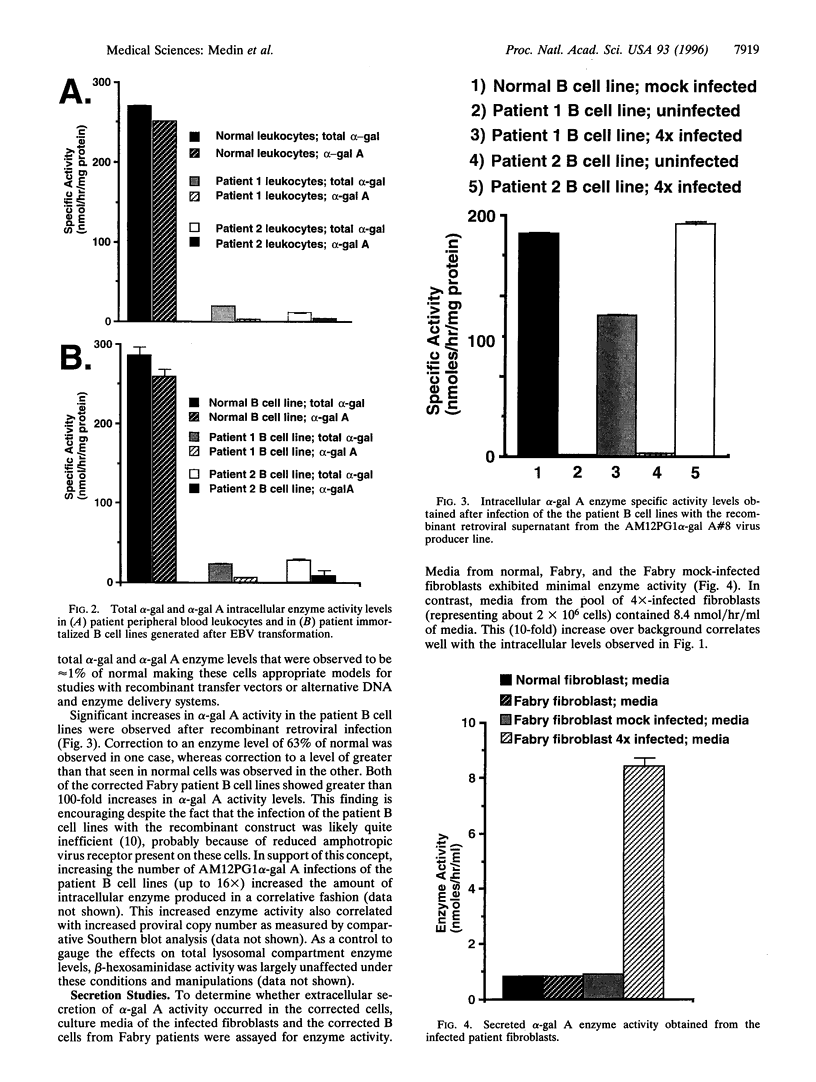
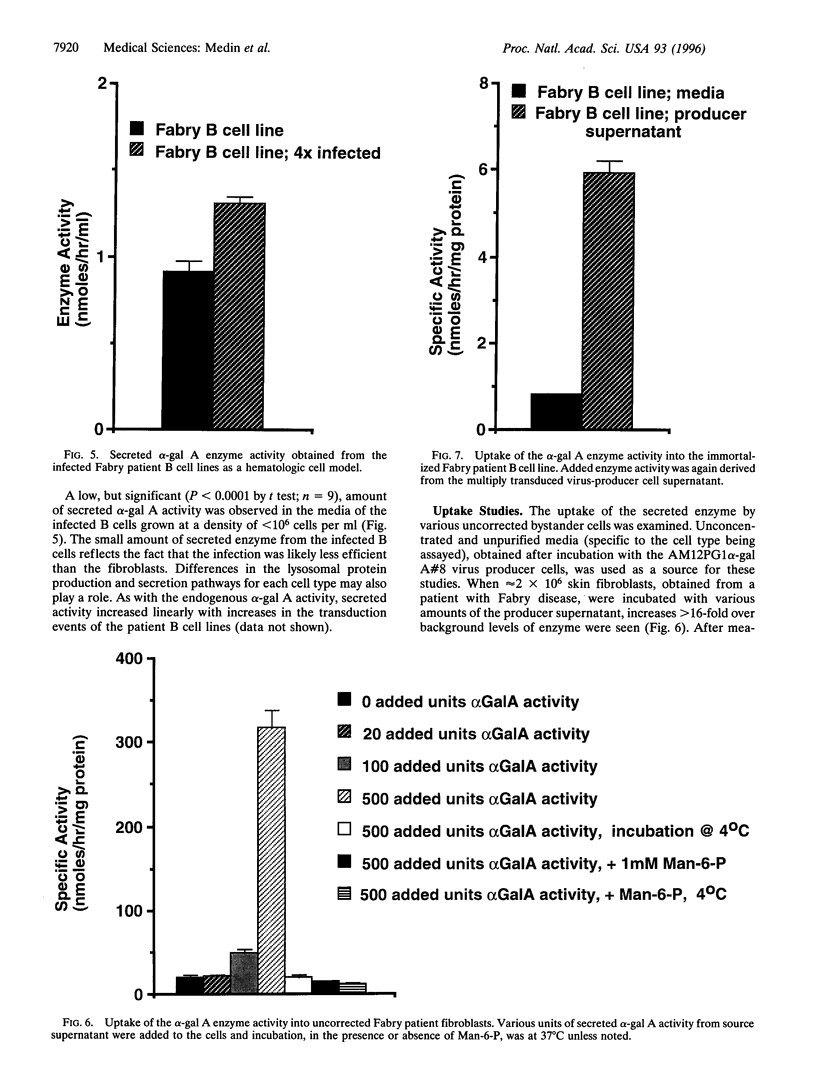
Fabry disease is an X-linked metabolic disorder due to a deficiency of alpha-galactosidase A (alpha-gal A; EC 3.2.1.22). Patients accumulate glycosphingolipids with terminal alpha-galactosyl residues that come from intracellular synthesis, circulating metabolites, or from the biodegradation Of senescent cells. Patients eventually succumb to renal, cardio-, or cerebrovascular disease. No specific therapy exists. One possible approach to ameliorating this disorder is to target corrective gene transfer therapy to circulating hematopoietic cells. Toward this end, an amphotropic virus-producer cell line has been developed that produces a high titer (>10(6) i.p. per ml) recombinant retrovirus constructed to transduce and correct target cells. Virus-producer cells also demonstrate expression of large amounts of both intracellular and secreted alpha-gal A. To examine the utility of this therapeutic vector, skin fibroblasts from Fabry patients were corrected for the metabolic defect by infection with this recombinant virus and secreted enzyme was observed. Furthermore, the secreted enzyme was found to be taken up by uncorrected cells in a mannose-6-phosphate receptor-dependent manner. In related experiments, immortalized B cell lines from Fabry patients, created as a hematologic delivery test system, were transduced. As with the fibroblasts, transduced patient B cell lines demonstrated both endogenous enzyme correction and a small amount of secretion together with uptake by uncorrected cells. These studies demonstrate that endogenous metabolic correction in transduced cells, combined with secretion, may provide a continuous source of corrective material in trans to unmodified patient bystander cells (metabolic cooperativity).
Full text
PDF



Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Brady R. O., Gal A. E., Bradley R. M., Martensson E., Warshaw A. L., Laster L. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967 May 25;276(21):1163–1167. doi: 10.1056/NEJM196705252762101. [DOI] [PubMed] [Google Scholar]
- Brady R. O., Tallman J. F., Johnson W. G., Gal A. E., Leahy W. R., Quirk J. M., Dekaban A. S. Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry's disease. N Engl J Med. 1973 Jul 5;289(1):9–14. doi: 10.1056/NEJM197307052890103. [DOI] [PubMed] [Google Scholar]
- Braun S. E., Pan D., Aronovich E. L., Jonsson J. J., McIvor R. S., Whitley C. B. Preclinical studies of lymphocyte gene therapy for mild Hunter syndrome (mucopolysaccharidosis type II). Hum Gene Ther. 1996 Feb 10;7(3):283–290. doi: 10.1089/hum.1996.7.3-283. [DOI] [PubMed] [Google Scholar]
- Correll P. H., Colilla S., Dave H. P., Karlsson S. High levels of human glucocerebrosidase activity in macrophages of long-term reconstituted mice after retroviral infection of hematopoietic stem cells. Blood. 1992 Jul 15;80(2):331–336. [PubMed] [Google Scholar]
- Correll P. H., Colilla S., Karlsson S. Retroviral vector design for long-term expression in murine hematopoietic cells in vivo. Blood. 1994 Sep 15;84(6):1812–1822. [PubMed] [Google Scholar]
- Desnick R. J., Dean K. J., Grabowski G., Bishop D. F., Sweeley C. C. Enzyme therapy in Fabry disease: differential in vivo plasma clearance and metabolic effectiveness of plasma and splenic alpha-galactosidase A isozymes. Proc Natl Acad Sci U S A. 1979 Oct;76(10):5326–5330. doi: 10.1073/pnas.76.10.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbairn L. J., Lashford L. S., Spooncer E., McDermott R. H., Lebens G., Arrand J. E., Arrand J. R., Bellantuono I., Holt R., Hatton C. E. Long-term in vitro correction of alpha-L-iduronidase deficiency (Hurler syndrome) in human bone marrow. Proc Natl Acad Sci U S A. 1996 Mar 5;93(5):2025–2030. doi: 10.1073/pnas.93.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou Y. A., Bishop D. F., Desnick R. J. Overexpression of human alpha-galactosidase A results in its intracellular aggregation, crystallization in lysosomes, and selective secretion. J Cell Biol. 1992 Dec;119(5):1137–1150. doi: 10.1083/jcb.119.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S. Treatment of genetic defects in hematopoietic cell function by gene transfer. Blood. 1991 Nov 15;78(10):2481–2492. [PubMed] [Google Scholar]
- Kusiak J. W., Quirk J. M., Brady R. O. Purification and properties of the two major isozymes of alpha-galactosidase from human placenta. J Biol Chem. 1978 Jan 10;253(1):184–190. [PubMed] [Google Scholar]
- Mapes C. A., Anderson R. L., Sweeley C. C., Desnick R. J., Krivit W. Enzyme replacement in Fabry's disease, an inborn error of metabolism. Science. 1970 Sep 4;169(3949):987–989. doi: 10.1126/science.169.3949.987. [DOI] [PubMed] [Google Scholar]
- Markowitz D., Goff S., Bank A. Construction and use of a safe and efficient amphotropic packaging cell line. Virology. 1988 Dec;167(2):400–406. [PubMed] [Google Scholar]
- Mayes J. S., Scheerer J. B., Sifers R. N., Donaldson M. L. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry's disease. Clin Chim Acta. 1981 May 5;112(2):247–251. doi: 10.1016/0009-8981(81)90384-3. [DOI] [PubMed] [Google Scholar]
- Medin J. A., Migita M., Pawliuk R., Jacobson S., Amiri M., Kluepfel-Stahl S., Brady R. O., Humphries R. K., Karlsson S. A bicistronic therapeutic retroviral vector enables sorting of transduced CD34+ cells and corrects the enzyme deficiency in cells from Gaucher patients. Blood. 1996 Mar 1;87(5):1754–1762. [PubMed] [Google Scholar]
- Pear W. S., Nolan G. P., Scott M. L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993 Sep 15;90(18):8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y., Aksentijevich I., Murray G. J., Brady R. O., Pastan I., Gottesman M. M. Retroviral coexpression of a multidrug resistance gene (MDR1) and human alpha-galactosidase A for gene therapy of Fabry disease. Hum Gene Ther. 1995 Jul;6(7):905–915. doi: 10.1089/hum.1995.6.7-905. [DOI] [PubMed] [Google Scholar]
- Tallman J. F., Brady R. O., Quirk J. M., Villalba M., Gal A. E. Isolation and relationship of human hexosaminidases. J Biol Chem. 1974 Jun 10;249(11):3489–3499. [PubMed] [Google Scholar]
- Wolfe J. H. Recent progress in gene therapy for inherited diseases. Curr Opin Pediatr. 1994 Apr;6(2):213–218. doi: 10.1097/00008480-199404000-00016. [DOI] [PubMed] [Google Scholar]
- Wolfe J. H., Sands M. S., Barker J. E., Gwynn B., Rowe L. B., Vogler C. A., Birkenmeier E. H. Reversal of pathology in murine mucopolysaccharidosis type VII by somatic cell gene transfer. Nature. 1992 Dec 24;360(6406):749–753. doi: 10.1038/360749a0. [DOI] [PubMed] [Google Scholar]
- Zhou X. Y., Morreau H., Rottier R., Davis D., Bonten E., Gillemans N., Wenger D., Grosveld F. G., Doherty P., Suzuki K. Mouse model for the lysosomal disorder galactosialidosis and correction of the phenotype with overexpressing erythroid precursor cells. Genes Dev. 1995 Nov 1;9(21):2623–2634. doi: 10.1101/gad.9.21.2623. [DOI] [PubMed] [Google Scholar]
- deVeber G. A., Schwarting G. A., Kolodny E. H., Kowall N. W. Fabry disease: immunocytochemical characterization of neuronal involvement. Ann Neurol. 1992 Apr;31(4):409–415. doi: 10.1002/ana.410310410. [DOI] [PubMed] [Google Scholar]