

Abstract
The complexes (Bu4N)(LMeM(II)-OH) (LMe = 2,6-dimethylphenyl-substituted pyridine(dicarboxamide); M = Cu or Ni) react with CH3CN to yield (Bu4N)(LMeM-CH2CN), novel cyanomethide complexes that were fully characterized, including by X-ray crystallography. These conversions contrast with the usual reactions of metal-hydroxide complexes with nitriles, which typically involve attack at the nitrile carbon and formation of amides or carboxylic acids. Kinetic studies (M = Cu) revealed a first-order dependence on the complex and a kinetic isotope effect (k(CH3CN)/k(CD3CN) of 4. Various mechanisms involving either intra- or intermolecular deprotonation steps are proposed.
In addition, (Bu4N)(LMeCu-OH) was oxidized by Fc+PF6− to a proposed Cu(III) complex LMeCuOH at low temperature, and comparisons of its stability and reactivity with dihydroanthracene were drawn to its previously described congener having isopropyl substituents on the phenyl rings of the supporting ligand. The cyanomethide complex (Bu4N)(LMeCu(CH2CN)) also was reversibly oxidized both electrochemically (E1/2 = −0.345 V vs. Fc/Fc+) and chemically (Fc+PF6−, −25 °C). The product was formulated as LMeCu(III)(CH2CN), a novel Cu(III)-alkyl complex relevant to such species proposed during copper-catalyzed organic reactions.
Keywords: metal-hydroxide, bioinorganic, nitrile, copper(III)
Introduction
Copper- and nickel-hydroxide complexes have been the subject of numerous investigations aimed at understanding their structural, magnetic, spectroscopic, and reactivity properties.[1] These studies are motivated by both fundamental issues as well as the proposed intermediacy of metal-hydroxide complexes in reactions of industrial and biological importance. For example, fixation of CO2 by M(II)-OH species (M = Cu, Ni) to yield carbonate or bicarbonates has been amply demonstrated;[1d–h,2] in the case of (Et4N)1 it proceeds at rates competitive with carbonic anhydrase and the rates for analogs vary as a function of the R groups on the ligand.[3] Analog (Bu4N)2 containing a more hindered supporting ligand was oxidized by one electron to yield a novel Cu(III)-OH species that rapidly attacks C-H bonds in a reaction of possible relevance to enzymatic and other catalytic oxidations.[4] Metal-hydroxides are also commonly proposed intermediates in the metal-promoted hydrolysis of nitriles, with a typical route involving nucleophilic attack of the bound hydroxide at the nitrile carbon.[5] These examples illustrate diverse facets of M(II)-OH chemistry, the understanding of which may impact the development of new reactants and catalysts.
With the original aim of comparing its redox chemistry with more sterically hindered (Bu4N)2, we targeted (Bu4N)3 for synthesis and study. Herein we report these results, as well as the serendipitous findings that (Bu4N)3 and (Bu4N)1 react with CH3CN to yield M(II)-CH2CN complexes. These products represent the first of their kind for M = Ni or Cu[6]; cyanomethide complexes of Pt/Pd,[7] Ir,[8] Rh,[9] Hg,[10] Au,[11] and Fe[12] have been prepared, albeit via different routes. The preparative conversions we discovered stand in distinct contrast to the usual mode of reactivity of metal-hydroxides with nitriles,[5] and represent new methods for the synthesis of rare cyanomethide complexes. The results also complement reports of scission of the C-C bond in CH3CN by Cu(II) complexes.[13] While this work was nearing completion, a report of the synthesis and X-ray structure of (Et4N)3 appeared.[14]
Results and Discussion
The convenient starting material LMeCu(MeOH) (5, Scheme 1) was isolated in ~70% yield by addition of NaOMe to a mixture of LMeH2 and CuCl2 in MeOH. Key characterization data in support of the formulation of 5 included an accurate CHN analysis, an axial EPR spectrum with distinguishable N-superhyperfine coupling typical for Cu(II) complexes of pyridine(dicarboxamide) ligands (Figure S1, Table S1), and the appropriate isotope pattern in the positive ion electrospray ionization mass spectrum (ESI-MS) for [NaLMeCu(MeOH)]+ (Figure S2).
Scheme 1.

Complexes characterized in this work.
Treatment of 5 with Ph4PCl in acetone yielded (Ph4P)(LCuCl) ((Ph4P)8). The UV-vis [λmax, nm (ε, cm−1M−1): 390 (2600), 620 (340) in CH3CN; Figure S3] and EPR (Figure S4, Table S1) spectroscopic properties of this complex are similar to those reported previously for the more hindered analog (Bu4N)(LiPrCuCl) [cf. λmax, nm (ε, cm−1M−1): 400 (3330), 625 (430) in acetone].[4] Further corroboration of the structure of (Ph4P)8 was provided by CHN analysis, a parent ion in its negative ion ESI-MS (Figure S5), and an X-ray crystal structure (Figure 1a). Overall, the structure of 8− is unremarkable, with bond parameters closely similar to those of (LiPrCuCl)−.[4]
Figure 1.
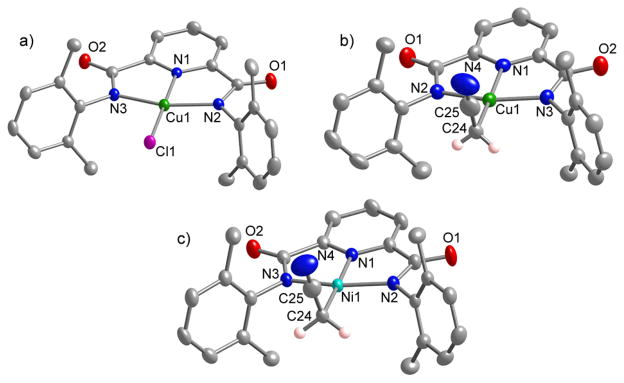
Representation of the X-ray structure of the anionic portions of (a) (Ph4P)8, (b) (Bu4N)6, and (c) (Bu4N)7, showing all non-hydrogen atoms as 50% thermal ellipsoids (except the two hydrogen atoms shown affixed to C24 in (b) and (c)). See Table 1 for selected interatomic distances and angles.
Reaction of 5 with Bu4NOH in MeOH/Et2O yielded (Bu4N)(LMeCuOH) ((Bu4N)3) in 85% yield. This compound is an alternative salt to that reported previously (Et4N+),[3] and was characterized by CHN analysis, UV-vis spectroscopy [λmax, nm (ε, cm−1M−1): 378 (1800), 550 (230) in acetone], and EPR spectroscopy (Figure S6). With the aim of drawing comparisons to the previously reported Cu(III)-OH complex LiPrCuOH derived from oxidation of 2,[4] we explored oxidation of (Bu4N)3. Treatment of a solution of (Bu4N)3 in 1,2-difluorobenzene (1,2-DFB) with Fc+PF6− at −25 °C resulted in a rapid color change from light blue to intense purple that then bleached over ~2 min.[15] The UV-vis spectrum of the initial product (blue spectrum in Figure 2) contains an intense feature at λmax = 560 nm that is closely analogous to that reported for LiPrCuOH (λmax 540 nm) in acetone[4] and which we have now measured in 1,2-DFB (560 nm; black spectrum in Figure 2). Generation of LMeCuOH is indicated by this diagnostic feature, identified previously as a ligand-based π → Cu(III) transition via TD-DFT calculations.[4] This product is significantly less stable than LiPrCuOH; both complexes decay in first-order fashion in 1,2-DFB,[16] but for LMeCuOH (k = 3.6 × 10−2 s−1) the rate is 105 times faster than that for LiPrCuOH (k = 3.3 × 10−7 s−1) at −25 °C. However, comparison of the second order rate constants for hydrogen atom abstraction from dihydroanthracene (DHA) by both complexes under identical conditions (−25 °C in 1,2-DFB, [DHA]0 = 1.2 mM, [LCuOH]0 = 0.07 mM) revealed them to be similar (k = 1.2 vs. 1.5 × 102 M−1s−1). Thus, while the decrease in steric hindrance resulting from the presence of Me rather than iPr groups on the flanking aryl groups of the supporting ligand results in higher rates for decomposition of the Cu(III)-OH unit, the reactivity with DHA is unperturbed.
Figure 2.

UV-vis spectra of the solutions resulting from the reactions with Fc+PF6+ of (Bu4N)2 (to yield LiPrCuOH, black, −25 °C, 1,2-DFB) and (Bu4N)3 (to yield LMeCuOH, blue, −25 °C, 1,2-DFB).
Interestingly, a color change from blue to red-purple was observed upon dissolution of (Bu4N)3 in CH3CN; the conversion is illustrated by the UV-vis spectra measured as a function of time shown in Figure 3. An apparent isosbestic point is seen at 604 nm. Complex (Bu4N)6 was isolated as a crystalline solid in 85% yield from the reaction solution and was identified on the basis of CHN analysis, a parent ion 6− in the negative ion ESI-MS (Figure S7), a signal in the EPR spectrum indicative of a Cu(II) assignment (Figure S8), and an X-ray crystal structure (Figure 1b). The clearly evident linear cyanomethide (CH2CN-) ligand features C-C and C-N distances of 1.412(5) and 1.165(4) Å that are consistent with single and triple bonds, respectively, and are similar to those seen in other M-CH2CN complexes. The Cu1-C24 bond (1.986(3) Å) represents a rare example of a Cu(II)-C(sp3) bond in a discrete molecule; the only other cases involve bonds between Cu(II) and a C atom within a macrocycle or a tripodal ligand,[17,18] whereas in (Bu4N)6 the metal is bound to a simple, ‘exogenous’ organic fragment. The complex thus provides important precedent for organocopper(II) species proposed as intermediates in various catalytic processes.[19]
Figure 3.
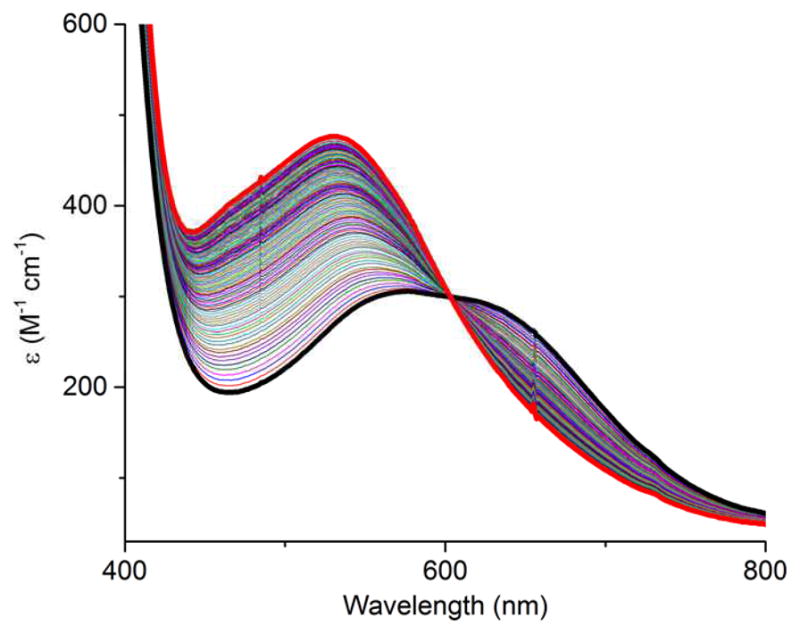
UV-vis spectra as a function of time of a solution of (Bu4N)3 (15 mM) dissolved in CH3CN (black: first spectrum taken within seconds of dissolution; red: final spectrum after 2 h; intermediate spectra taken at 1 min intervals).
In similar chemistry, dissolution of (Bu4N)(LMeNiOH) ((Bu4N)4)[3a] in CH3CN yielded (Bu4N)7. This product was characterized by NMR spectroscopy, negative ion ESI-MS (parent ion observed, Figure S9), and X-ray crystallography (Figure 1c). The structure of 7− is similar to 6−, albeit with different metal-ligand bond distances due to the different central metal ion.
The time course of the reaction of the hydroxide (Bu4N)3 with excess CH3CN (14.9–18.3 M, 5200–140,000 equiv) to yield the cyanomethide (Bu4N)6 was evaluated by multiple component analysis of UV-vis spectra (Figure 3). This analysis (see Supporting Information, Figures S12–S13) showed that the reaction follows pseudo-first order kinetics with kobs = 4(1) × 10−4 s−1 (20 °C, 1,2-DFB). The order in [CH3CN] was more difficult to assess; under conditions of large excesses of CH3CN varied between 14.9–18.3 M at [3−]0 values ranging between 0.13–2.89 mM the kobs values were essentially invariant (Figure S12). This zeroth-order dependence on [CH3CN] may be explained by a mechanism involving rapid pre-equilibrium binding of CH3CN followed by rate-determining attack by the hydroxide moiety, with [CH3CN]0 sufficiently high to cause the rate to be saturated. Under this working assumption, we attempted to see if the order in [CH3CN] changed at significantly lower [CH3CN]0 values (0.95 M). Unfortunately, under these conditions the rates were too slow to measure their dependence on [CH3CN] accurately, and we have not been able to discern the order in this reagent. In further kinetic studies, we compared the rates of reaction in CH3CN and CD3CN under identical conditions ([LMeCuOH]0 = 1.11 mM, 20 °C). A kinetic isotope effect (kH/kD) of 4.1(1) was observed and the identity of the product in the reaction with CD3CN was confirmed by ESI-MS and FT-IR spectroscopy. We note parenthetically that in the IR spectrum two peaks were observed for the nitrile stretch at 2162 and 2192 cm−1 for the deuterio complex versus one at 2173 cm−1 for the protio case. On the basis of DFT calculations, we attribute this disparity to coupling of the C ≡ N and C–D vibrations (see Supporting Information, Table S2).
Three possible mechanisms for the novel conversions of the hydroxide complexes (Bu4N)3 and (Bu4N)4 to the cyanomethide species (Bu4N)6 and (Bu4N)7, respectively, that are consistent with the combined experimental data are shown in Scheme 2. Paths A and B feature reversible binding of CH3CN to the starting M(II)-OH complex to yield a 5-coordinate intermediate. In path A, intramolecular deprotonation of the coordinated nitrile occurs (via a transition state with a 6-membered ring, albeit requiring distortion of the linear CH3CN ligand), which would have to be followed by a rapid isomerization and loss of H2O to yield the final C-bound cyanomethide product. In path B, the deprotonation is intermolecular with an exogenous CH3CN molecule, to yield a cyanomethide anion that would rapidly displace the bound H2O to afford the product. In path C, CH3CN displaces the hydroxide, which then deprotonates the coordinated nitrile to yield an N-bound cyanomethide. Isomerization to the C-bound form in the product would then occur. All three mechanisms are consistent with observation of a first order dependence on [(Bu4N)3], saturation of the rate at high [CH3CN], and the kinetic isotope effect. Distinction between paths A–C will therefore depend on further more in-depth mechanistic study.
Scheme 2.

Proposed mechanisms for the conversion of the hydroxide complexes (Bu4N)3 and (Bu4N)4 to (Bu4N)6 and (Bu4N)7, respectively, upon reaction with CH3CN. Ar = 2,6-dimethylphenyl.
Finally, we note that the cyanomethide complex (Bu4N)6 may be oxidized, as revealed by cyclic voltammetry. A pseudo-reversible wave was observed with E1/2 = −0.345 V vs. Fc/Fc+ and ΔEp = 105 mV (scan rate 200 mV/s, 0.1 M Bu4NPF6 in CH3CN; Figure S10). Consistent with this result, addition of Fc+PF6− to a solution of (Bu4N)6 in CH3CN at −30 °C resulted in an immediate color change from red-purple to bright orange and generation of an intense feature in the UV-vis spectrum with λmax ~ 465 nm, ε ~ 7700 M−1 cm−1 (red spectrum, Figure 4). This reaction could be reversed by addition of Cp*2Fe and the cycle repeated (Figure 4). The species giving rise to the intense electronic absorption feature is EPR silent. It is fairly stable at −30 °C (t1/2 ~ 90 minutes) and does not react at an appreciable rate with DHA (30 eq DHA at −30 °C). We tentatively hypothesize on the basis of the available data that this species is the Cu(III) complex LMeCu(CH2CN), a novel example of a Cu(III)-alkyl compound[20] that is notable because such Cu(III)-alkyl moieties are often postulated as intermediates in copper-catalyzed organic reactions.[19]
Figure 4.

UV-vis spectra showing the reversible oxidation of (Bu4N)6 (0.19 mM) in CH3CN at −30 °C. (solid black) (Bu4N)6; (solid red) after addition of 1 equiv. Fc+PF6−; (dashed black) subsequent addition of 1 equiv. Cp*2Fe; (dashed red) addition of second 1 equiv. Fc+PF6−. Note: the feature at ~790 nm corresponds to ferrocene.
In support of this assignment, DFT calculations revealed a singlet ground state structure with metal-ligand bond distances contracted by ~0.1 Å relative to 6− (Table S3) that is 22 kcal/mol more stable than a distorted triplet, similar to what was reported previously for LiPrCuOH.[4] In addition, TD-DFT calculated electronic transitions for the singlet structure (Table S4) showed an intense transition at 495 nm that matched well with the experimentally observed feature at λmax ~ 465 nm (Figure 5). This transition is dominated by a ligand(amide)-based → Cu(dx2-y2) excitation (a → c in Figure 5).
Figure 5.

(top) Comparison of experimental UV-vis spectrum of LCuCH2CN (black line) with calculated (B98) electronic transitions for restricted singlet (red) and triplet (blue) structures. (bottom) Orbitals involved in the TD-DFT calculated excitations at 495 and 622 nm. For the former, the major contribution is from (a) to (c), while for the latter, it is from (b) to (c).
Conclusions
In summary, we have discovered that the M(II)-OH (M = Cu, Ni) complexes (Bu4N)3 and (Bu4N)4 supported by a relatively unhindered pyridyl(dicarboxamide) ligand react with CH3CN in unprecedented fashion to yield M(II)-CH2CN complexes (Bu4N)6 and (Bu4N)7, respectively. Speculative pathways for this reaction are proposed on the basis of preliminary mechanistic data (Scheme 2). Oxidation of the copper(II)-hydroxide complex (Bu4N)3 results in the formation of LMeCuOH, identified as a Cu(III) species on the basis of comparison of its spectroscopic properties to the previously reported analog LiPrCuOH.[4] The less sterically hindered complex LMeCuOH decays significantly faster than LiPrCuOH, but reacts with DHA at a similar rate under identical conditions (−25 °C in 1,2-DFB). The copper(II)-cyanomethide complex also may be oxidized reversibly both electrochemically and chemically to yield a species that we formulate as a unique Cu(III)-alkyl complex on the basis of spectroscopy and theory.
Experimental Section
General Considerations
All solvents and reagents were obtained from commercial sources and used as received unless otherwise noted. LMeH2 was prepared according to the literature procedure.[3a] The solvents Et2O and pentane were passed through purification columns (Glass Contour, Laguna, California) before use. Acetonitrile was dried over CaH2, degassed, distilled under vacuum and stored over CaH2 in a glovebox. Acetone was dried over 3Å molecular sieves, degassed, vacuum transferred and stored over 3Å molecular sieves in a glovebox. 1,2-Difluorobenzene was dried over CaH2, degassed, distilled under vacuum and stored over 3Å molecular sieves in a glovebox. Anydrous DMF was stored over 3Å molecular sieves in a glovebox for 2 days and dried over a separate portion of 3Å molecular sieves for an additional 2 days before use. All reactions of LCu(CH3OH) (5) were performed in glovebox under a dry N2 atmosphere. Complex (Bu4N)(LNiOH) ((Bu4N)4) was prepared according to the previously published procedure and identified by the similarity of its UV-vis and 1H NMR spectroscopic features to the reported values.[3a] Complex LiPrCuOH was prepared as described in the literature.[4]
Physical Methods
NMR spectra were recorded on a 300 MHz Varian Inova spectrometer; for clarity, cation resonances are omitted from listings of data. UV-Vis spectra were recorded on an HP8453 (190–1100 nm) diode-array spectrophotometer equipped777 with a Unisoku low-temperature cryostat. Electrospray ionization mass spectra (ESI-MS) were recorded on a Bruker BioTOF II instrument in negative ion mode for all complexes except for LMeCuCH3OH, the spectrum of which was recorded in positive ion mode. Electron paramagnetic resonance (EPR) spectra were recorded a Bruker Continuous Wave EleXsys E500 spectrometer at 10 K. EPR simulations were performed using Bruker Simfonia software. Infrared spectra were collected on a Nicolet Avatar 370FT-IR. Elemental analyses were performed by Robertson Microlit Laboratory (Ledgewood, New Jersey) and Complete Analysis Laboratories Inc. (Parsippany, New Jersey). X-ray crystallography data collection and structure solution were conducted using a SMART Apex II instrument and the current SHELXTL suite of programs.[21]
LCu(MeOH) (5)
LH2 (1.015 g, 2.72 mmol), anhydrous CuCl2 (0.367 g, 2.73 mmol) and MeOH (100 mL) were added to a 250 mL round bottom flask to give a light green solution. Addition of a solution of NaOMe in MeOH (0.5 M, 9.2 mL, 4.6 mmol) yielded a deep forest green solution, which was stirred for 30 min. The solvent was removed in vacuo to give a green oil. This oil was dissolved in CH3CN (~ 20 mL) and toluene (100 mL) was added, after which a precipitate formed and the solution became mahogany in color. The mixture was filtered through a fine porosity frit and solvent was removed from the filtrate in vacuo to produce a bright green powder. Subsequent washings with acetonitrile (3 × 5 mL) and hexanes (3 × 5 mL) afforded the product as a bright green solid, which was dried under vacuum at 40 °C overnight (0.75 g, 69%). ESI-MS (CH3OH, m/z): calcd 489.11 [Na+M]+, found 489.23. UV-Vis [λmax, nm (ε, M−1 cm−1) in CH3CN]: 397 (1400), 545 (720). EPR (1:3-acetone:toluene, 100 K): gx = 2.028, gy = 2.055, gz = 2.189; A values (× 10−4 cm−1): A//(Cu) =193, A(N) =15.0. Anal. Calcd. for C24H25CuN3O3: C, 61.72; H, 5.40; N, 9.00. Found: C, 61.83; H, 5.29; N, 8.93.
(Bu4N)(LCuOH) ((Bu4N)3)
To 5 (147 mg, 0.314 mmol) was added Et2O (~10mL) to give a green reaction mixture. Upon the addition of Bu4NOH in MeOH (1.0 M, 0.31 mL, 0.31 mmol) a blue sticky precipitate formed. The reaction was stirred for ~5 min and the solvent was removed in vacuo to give a dark blue oil. The oil was washed with Et2O (3 × 5 mL) to yield the product as a bright blue-purple solid (129 mg, 85%). UV-Vis [λmax, nm (ε, M−1 cm−1) in acetone]: 378 (1800), 550 (230). EPR (1:3-acetone:toluene, 10 K): gx = 2.02, gy = 2.06, gz = 2.19; A values (x 10−4 cm−1): A//(Cu) =192, A(Npy) =18.2, A (Nam) =13.5. Anal. Calcd. for C39H58CuN4O3: C, 67.45 H, 8.42; N, 8.07. Found: C, 66.86; H, 8.15; N, 8.02.
(Bu4N)(LCu(CH2CN)) ((Bu4N)6)
Compound (Bu4N)3 (109 mg, 0.185 mmol) was added to CH3CN (~ 10 mL) to give a dark blue solution, which quickly became red-purple (<2 min). The reaction was stirred for 30 min and the solvent was removed in vacuo. The resulting solid was washed with Et2O (2 × 3 mL) to yield the product as a deep purple solid (129 mg, 85%). Negative ion ESI-MS (CH3OH, m/z): calcd 474.11 [M-Bu4N]1−, found 473.96. UV-Vis [λmax, nm (ε, M−1 cm−1) in CH3CN]: 315 (3800), 476 (380). FT-IR(Nujol mull): 2173 cm−1. EPR (1:3-CH3CN:toluene, 100 K): gx = 2.003, gy = 2.06, gz = 2.3; A values (x 10−4 cm−1): A//(Cu) =100, A(N) =15.0. Anal. Calcd. for C41H59CuN5O2: C,68.63; H,8.29; N,9.76. Found: C, 68.58; H, 8.22; N, 9.66. The deuterated version of this compound was prepared according to the same procedure used to prepare (Bu4N)6, except using CD3CN (99.8%) on a 52.5 mg (0.0756 mmol) scale, to give the product as a deep purple solid (54.4 mg, 73.2 %). ESI-MS (CH3OH, m/z): calcd 476.12 [M-Bu4N]1−, found 476.09. FT-IR (Nujol mull): 2162, 2192 cm−1.
(Bu4N)(LNi(CH2CN)) ((Bu4N)7)
The synthesis of this compound was performed similarly to that used to prepare (Bu4N)6. Briefly, (Bu4N)4 (115 mg, 0.167 mmol) was added to CH3CN (~ 10 mL) to give a yellow orange solution which became red-orange upon stirring overnight. The solvent was removed in vacuo to give a sticky red solid to which 5 mL of Et2O was added and stirred for 3 h to give the product (80.8 mg, 67.7%). ESI-MS (CH3OH, m/z): calcd 469.12 [M-Bu4N]1−, found 469.20. UV-Vis [λmax, nm (ε, M−1 cm−1) in CH3CN]: : 400, (4500), 478 (sh, 1500). FT-IR(Nujol mull): 2190 cm−1. 1H NMR (CD2Cl2): d −0.89 (s, 1), 2.29 (s, 12), 6.84 (m, 6), 7.58 (d, 2), 7.88 (t,1). Unfortunately, repeated attempts to obtain satisfactory CHN analysis were unsuccessful.
(Ph4P)(LCuCl) ((Ph4P)8)
A suspension of Ph4PCl (54.6 mg, 0.146 mmol) in acetone (4 mL) was added drop-wise to a mahogony solution of compound 5 (68.0 mg, 0.146 mmol) in acetone (10 mL). The resulting deep emerald green solution was stirred for ~10 minutes. The solvent was removed in vacuo to yield a dark green residue. The residue was triturated with Et2O (4 × 5 mL) until a fine green precipitate was observed. Residual solvent was removed in vacuo to give a bright green powder (87.6 mg, 75.8%). ESI-MS (CH3OH, m/z): calcd 469.06 [M-Ph4P]1-, found 469.09. UV-Vis [λmax, nm (ε, M−1 cm−1) in CH3CN]: 390 (2600), 620 (340). EPR (1:3-acetone:toluene, 10 K): gx = 2.026, gy = 2.062, gz = 2.182; A values (x 10−4 cm−1): A//(Cu) =195, A(N) =15.3. Anal. Cald. for C47H41ClCuN3O2P: C, 69.71; H, 5.10; N, 5.19; Cl, 4.38. Found: C, 68.88; H, 4.96; N, 5.11; Cl, 4.29.
Oxidation of (Bu4N)6
To a cuvette, 0.15 mL of a 3 mM solution of 6 in CH3CN was added to 2.50 mL of CH3CN. The cuvette was cooled to −30 °C and 0.30 mL of a 1.5 mM solution of Fc+PF6− was added to give rise to an intense chromophore (λmax ~ 465 nm, ε ~ 5500 M−1 cm−1).
General procedure for kinetics experiments
All reactions were prepared in a nitrogen filled glovebox and cuvettes were sealed under inert atmosphere with a septum. Reactions were monitored by UV-Vis spectroscopy and performed via the following illustrative procedure (see Supporting Information for more details). To a cuvette, 0.10 mL of a solution of the complex ((Bu4N)2 or -3) in 1,2-DFB (3 mM) was added to 2.70 mL of 1,2-DFB. In reactions with DHA, 0.07 mL of a 3mM solution of the complex in 1,2-DFB and 0.14 mL of 30 mM DHA in 1,2-DFB were added to 2.64 mL of 1,2-DFB. The cuvette was cooled to −25 °C and 1.5 mM FcPF6 (0.10 mL) was added to give rise to an intense chromophore (λmax ~560). The decay of the chromophore was monitored until no further spectral changes were observed. With one exception, the data were then analyzed using the Olis GlobalWorks software package using a single value decomposition (SVD) fitting protocol. The data was fit to several reaction order types using the SVD protocol until a suitable fit was achieved where the error was reduced and the spectral contributions calculated by the Olis software were in good agreement with experimentally observed spectra (Figure S12). The exception to this method was for the slow self-decay of LiPrCuOH (10% decay after 75 min), which was monitored at a single wavelength (560 nm) to yield estimated t1/2 and k values.
Supplementary Material
Table 1.
Selected interatomic distances (Å) and angles (deg) for the X-ray crystal structures of (Bu4N)6, (Bu4N)7, and (Ph4P)8.(a)
| (Bu4N)6 | (Bu4N)7 | ||
|---|---|---|---|
| Cu1-C24 | 1.986(3) | Ni1-C24 | 1.9414(18) |
| Cu1-N1 | 1.945(2) | Ni1-N1 | 1.8501(15) |
| Cu1-N2 | 2.015(2) | Ni1-N3 | 1.9129(14) |
| Cu1-N3 | 2.011(2) | Ni1-N2 | 1.9178(15) |
| C24-C25 | 1.412(5) | C24-C25 | 1.436(3) |
| C25-N4 | 1.165(4) | N1-Ni1-N3 | 82.08(6) |
| N1-Cu1-C24 | 175.37(11) | N1-Ni1-N2 | 82.63(6) |
| N2-Cu1-C24 | 101.07(11) | N3-Ni1-N2 | 164.68(6) |
| N2-Cu1-N1 | 79.91(9) | N1-Ni1-C24 | 177.90(7) |
| N3-Cu1-C24 | 99.96(11) | N3-Ni1-C24 | 97.64(7) |
| N3-Cu1-N1 | 79.21(9) | N2-Ni1-C24 | 97.68(7) |
| N3-Cu1-N2 | 158.95(9) | N4-C25-C24 | 178.0(2) |
| N4-C25-C24 | 176.8(3) | Ni1-N1 | 1.8501(15) |
|
|
|||
| (Ph4P)8 | |||
|
|
|||
| Cu1-N1 | 1.930(2) | ||
| Cu1-N2 | 2.013(2) | ||
| Cu1-N3 | 2.013(2) | ||
| Cu1-Cl1 | 2.2017(7) | ||
| N1-Cu1-N3 | 80.57(9) | ||
| N1-Cu1-N2 | 79.64(9) | ||
| N3-Cu1-N2 | 160.20(9) | ||
| N1-Cu1-Cl1 | 179.64(8) | ||
| N3-Cu1-Cl1 | 99.23(6) | ||
| N2-Cu1-Cl1 | 100.56(7) | ||
Standard deviations in parentheses.
Acknowledgments
This research was supported by the National Institutes of Health (grant R37GM47365 to W. B. T.). We thank Victor G. Young, Jr. for assistance with X-ray crystallography. Some experiments reported in this paper were performed at the Biophysical Spectroscopy Center in the University of Minnesota Department of Biochemistry, Molecular Biology, and Biophysics.
Footnotes
Supporting information for this article is available on the WWW under http://www.eurjic.org/ or from the author.
Supporting Information (see footnote on the first page of this article): Selected ESI-MS, spectroscopic, and kinetic data (pdf). X-ray crystallographic information has been deposited to the Cambridge Crystallographic Data Centre (CCDC 926419 – 926421).
References
- 1.Selected references: Hodgson DJ. Prog Inorg Chem. 1975;19:173–242.Kitajima N, Koda T, Hashimoto S, Kitagawa T, Moro-oka Y. J Am Chem Soc. 1991;113:5664–5671.Fujisawa K, Kobayashi T, Fujita K, Kitajima N, Moro-oka Y, Miyashita Y, Yamada Y, Okamoto K-i. Bull Chem Soc, Jpn. 2000;73:1797–1804.Kitajima N, Hikichi S, Tanaka M, Moro-oka Y. J Am Chem Soc. 1993;115:5496–5508.Murthy NN, Karlin KD. J Chem Soc, Chem Commun. 1993:1236–1238.Bazzicalupi C, Bencini A, Bencini A, Bianchi A, Corana F, Fusi V, Giorgi C, Paoli P, Paoletti P, Valtancoli B, Zanchini C. Inorg Chem. 1996;35:5540–5548. doi: 10.1021/ic9603262.Looney A, Han R, McNeill K, Parkin G. J Am Chem Soc. 1993;115:4690–4697.Company A, Jee JE, Ribas X, Lopez-Valbuena JM, Gómez L, Corbella M, Llobet A, Mahía J, Benet-Buchholz J, Costas M, van Eldik R. Inorg Chem. 2007;46:9098–9110. doi: 10.1021/ic700692t.
- 2.Acharya AN, Das A, Dash AC. Adv Inorg Chem. 2004;55:127–199. [Google Scholar]
- 3.a) Huang D, Holm RH. J Am Chem Soc. 2010;132:4693–4701. doi: 10.1021/ja1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang D, Makhlynets OV, Tan LL, Lee SC, Rybak-Akimova EV, Holm RH. Inorg Chem. 2011;50:10070–10081. doi: 10.1021/ic200942u. [DOI] [PubMed] [Google Scholar]; c) Huang D, Makhlynets OV, Tan LL, Lee SC, Rybak-Akimova EV, Holm RH. Proc Natl Acad Sci USA. 2011;108:1222–1227. doi: 10.1073/pnas.1017430108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donoghue PJ, Tehranchi J, Cramer CJ, Sarangi R, Solomon EI, Tolman WB. J Am Chem Soc. 2011;133:17602–17605. doi: 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Kukushkin VY, Pombeiro AJL. Inorg Chim Acta. 2005;358:1–21. [Google Scholar]; b) Murthy NN, Mahroof-Tahir M, Karlin KD. J Am Chem Soc. 1993;115:10404–10405. [Google Scholar]
- 6.Copper(I)-cyanomethide species useful in organic transformations have been described: Corey EJ, Kuwajima I. Tetrahedron Lett. 1972:487–489.Tsuda T, Nakatsuka T, Hirayama T, Saegusa T. J Chem Soc Chem Comm. 1974:557–558.
- 7.a) Suzuki K, Yamamota H, Kanie S. J Organomet Chem. 1974;73:131–136. [Google Scholar]; b) Ros R, Michelin RA, Bataillard R, Roulet R. J Organomet Chem. 1977;139:355–359. [Google Scholar]; c) del Pra A, Zanotti G, Bombieri G, Ros R. Inorg Chim Acta. 1979;36:121–125. [Google Scholar]; d) Henderson W, Oliver AG. Acta Crystallgr. 1999;C55:1406–1408. [Google Scholar]; e) McCrindle R, Ferguson G, McAlees AJ, Parvez M, Roberts PJ. J Chem Soc, Dalton Trans. 1982:1699–1708. [Google Scholar]
- 8.a) English AD, Herskovitz T. J Am Chem Soc. 1977;99:1648–1649. [Google Scholar]; b) Crestani MG, Steffen A, Kenwright AM, Batsanov AS, Howard JAK, Marder TB. Organometallics. 2009;28:2904–2914. [Google Scholar]
- 9.Tanabe T, Evans ME, Brennessel WW, Jones WD. Organometallics. 2011;30:834–843. [Google Scholar]
- 10.Liu Q-X, Li S-J, Zhao X-J, Zang Y, Song H-b, Guo J-H, Wang X-G. Eur J Inorg Chem. 2010;2010:983–988. [Google Scholar]
- 11.Emeljanenko D, Peters A, Vitske V, Kaifer E, Himmel HJ. Eur J Inorg Chem. 2010;2010:4783–4789. [Google Scholar]
- 12.Chambers MB, Groysman S, Villagrán D, Nocera DG. Inorg Chem. ASAP; 2013. [DOI] [PubMed] [Google Scholar]
- 13.a) Marlin DS, Olmstead MM, Mascharak PK. Angew Chem Int Ed. 2001;40:4752–4754. doi: 10.1002/1521-3773(20011217)40:24<4752::aid-anie4752>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]; b) Li L, Narducci Sarjeant AA, Vance MA, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2005;127:15360–15361. doi: 10.1021/ja054948a. [DOI] [PubMed] [Google Scholar]; c) Lu T, Zhuang X, Li Y, Chen S. J Am Chem Soc. 2004;126:4760–4761. doi: 10.1021/ja031874z. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Huang D, Chen YS, Holm RH. Inorg Chem. 2012;51:11017–11029. doi: 10.1021/ic301506x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attempts to perform the oxidation reactions in acetone, which was found to be useful for generating and studying LiPrCuOH, were less successful; the oxidized product was apparent by UV-vis spectroscopy, but it decayed more quickly and at rates that varied in irreproducible manner.
- 16.Despite repeated attempts, we have been unable to identify the product(s) of these decay reactions.
- 17.a) Kinoshita I, James Wright L, Kubo S, Kimura K, Sakata A, Yano T, Miyamoto R, Nishioka T, Isobe K. Dalton Trans. 2003:1993. [Google Scholar]; b) Miyamoto R, Santo R, Matsushita T, Nishioka T, Ichimura A, Teki Y, Kinoshita I. Dalton Trans. 2005:3179. doi: 10.1039/b507073b. [DOI] [PubMed] [Google Scholar]
- 18.Grzegorzek N, Pawlicki M, Szterenberg L, Latos-Grażyński L. J Am Chem Soc. 2009;131:7224–7225. doi: 10.1021/ja902310n. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 19.a) Wendlandt AE, Suess AM, Stahl SS. Angew Chem Int Ed. 2011;50:11062–11087. doi: 10.1002/anie.201103945. [DOI] [PubMed] [Google Scholar]; b) Schröder K, Konkolewicz D, Poli R, Matyjaszewski K. Organometallics. 2012;31:7994–7999. [Google Scholar]
- 20.a) Naumann D, Roy T, Tebbe KF, Crump W. Angew Chem Int Ed. 1993;32:1482–1483. [Google Scholar]; b) Bertz SH, Cope S, Murphy M, Ogle CA, Taylor BJ. J Am Chem Soc. 2007;129:7208–7209. doi: 10.1021/ja067533d. [DOI] [PubMed] [Google Scholar]; c) Bartholomew ER, Bertz SH, Cope S, Dorton DC, Murphy M, Ogle CA. Chem Commun. 2008:1176. doi: 10.1039/b717290g. [DOI] [PubMed] [Google Scholar]; d) Bartholomew ER, Bertz SH, Cope S, Murphy M, Ogle CA. J Am Chem Soc. 2008;130:11244–11245. doi: 10.1021/ja801186c. [DOI] [PubMed] [Google Scholar]; e) Bartholomew ER, Bertz SH, Cope SK, Murphy MD, Ogle CA, Thomas AA. Chem Commun. 2010;46:1253. doi: 10.1039/b924000d. [DOI] [PubMed] [Google Scholar]
- 21.SHELXTL V6.14. Bruker Analytical X-Ray Systems; Madison, WI: 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.