

Abstract
Antibody-mediated rejection of solid organ transplants is characterized by intragraft macrophages. It is incompletely understood how donor specific antibody binding to graft endothelium promotes monocyte adhesion, and what, if any, contribution is made by the Fc region of the antibody. We investigated the mechanisms underlying monocyte recruitment by HLA class I antibody-activated endothelium. We used a panel of murine monoclonal antibodies of different subclasses to crosslink HLA I on human aortic, venous and microvascular endothelial cells, and measured the binding of human monocytic cell lines and peripheral blood monocytes. Both anti-HLA I murine IgG1 and mIgG2a induced endothelial P-selectin, which was required for monocyte adhesion to endothelium irrespective of subclass. Mouse IgG2a but not mIgG1 could bind human FcγRs. Accordingly, HLA I mIgG2a but not mIgG1 treatment of endothelial cells significantly augmented recruitment, predominantly through FcγRI, and, to a lesser extent, FcγRIIa. Moreover, HLA I mIgG2a promoted firm adhesion of monocytes to ICAM-1 through Mac-1, which may explain the prominence of monocytes during antibody mediated rejection. We confirmed these observations using human HLA allele specific monoclonal antibodies and IgG purified from transplant patient sera. HLA I antibodies universally elicit endothelial exocytosis leading to monocyte adherence, implying that P-selectin is a putative therapeutic target to prevent macrophage infiltration during antibody-mediated rejection. Importantly, the subclass of donor specific antibody may influence its pathogenesis. These results imply that hIgG1 and hIgG3 should have a greater capacity to trigger monocyte infiltration into the graft than IgG2 or IgG4 due to enhancement by FcγR interactions.
Introduction
Organ transplantation is a life-saving therapy for end-stage organ failure. Advances in histocompatiblity testing, patient management and immunosuppression have improved short-term graft survival, estimated at 75-90% for the majority of solid organ transplants at one year after surgery (Organ Procurement and Transplantation Network data as of April 20, 2012). However, long-term graft survival has continued to be low; 50% or more of all solid organ grafts are lost at 10 years post-transplant. The major challenge to achieving long-term graft survival is chronic rejection, or transplant vasculopathy, in which the blood vessels of the graft develop concentric neointimal thickening with ultimate lumen occlusion, necessitating retransplantation. Rejection of organ transplants is caused by alloimmune responses mediated by T cells and/or antibodies, primarily targeting the donor's polymorphic HLA molecules. Many studies have correlated the presence of anti-donor HLA antibodies with antibody-mediated rejection, poor graft outcome (1, 2), and chronic rejection (3, 4). A histological hallmark of antibody-mediated rejection (AMR) is the presence of intragraft macrophages (5), and macrophages rather than T cells associate with decreased renal allograft function and poor survival (6-10). Macrophages can comprise up to 60% of the cellular infiltrate in acute rejection, including acute cellular rejection (11), and are also found in the vascular lesions of transplant vasculopathy (12, 13). Depletion of macrophages ameliorates chronic rejection in experimental models (14), and recently Bruneau et al. reiterated the significance of intragraft leukocytes, including monocytes, proposing that the process of “leukocyte-induced angiogenesis” drives chronic rejection (15).
Donor specific HLA antibodies binding to the endothelial and smooth muscle cells of the graft vasculature can trigger activation of the complement cascade. However, complement deposition is not always observed in acutely injured allografts, even when patients have histological evidence of AMR and donor specific antibodies (DSA) (16). Our group has proposed that the pathogenesis of HLA class I (HLA I) antibodies derives in part from their ability to directly activate the graft vascular cells via crosslinking of HLA I molecules by the F(ab′)2 portion. We and others have demonstrated in vitro, in experimental animals models and in patient biopsies that HLA or MHC I antibody binding to endothelial and smooth muscle cells triggers intracellular signaling cascades mediating proliferation and cellular resistance to death (17-20), primarily via Src, PI3K/Akt, ERK and mammalian target of rapamycin (mTOR). Additionally, HLA I molecule ligation mobilizes endothelial vesicles called Weibel-Palade bodies (WPb), the exocytosis of which results in a rapid increase in von Willebrand Factor (vWF) secretion, cell surface P-selectin and adherence of neutrophilic cells (21, 22).
Activated endothelial cells express E- or P-selectin, which capture leukocytes from the blood by binding to PSGL-1, and support low affinity adhesion known as rolling or tethering. This interaction exposes the leukocyte to endothelial-bound factors such as chemokines or complement split products, which activate the leukocyte through its G protein coupled receptors or complement receptors. The initial binding to P-selectin primes the leukocyte for a proadhesive state and is required for efficient recruitment (reviewed in (23)). Upon activation, leukocyte integrins are converted from a low affinity to high ligand binding state, permitting them to firmly adhere to their endothelial cell expressed ligands, the cellular adhesion molecules VCAM-1 and ICAM-1 (reviewed in (24, 25)); subsequently they transmigrate into the tissue. Endothelial intracellular adhesion molecule 1 (ICAM-1) is bound by two leukocyte integrins that contain the β2 subunit. The αL subunit pairs with β2 integrin to form LFA-1, expressed on most lymphoid and myeloid subsets, while the αM subunit complexed with β2 integrin forms Mac-1, which is restricted to monocytes and activated neutrophils.
In addition to adhesion molecules, monocytes express receptors for the Fc region of IgG. Humans have three families of Fc gamma receptors (FcγRs). FcγRI (CD64) is the highest affinity receptor for IgG by several orders of magnitude, and is expressed on monocytes as well as activated neutrophils. Activating FcγRIIa and FcγRIIIa bind multimeric or immune complexed IgG (reviewed in (26, 27)), and mediate adherence to immune complexes deposited on endothelial cells or plastic (28-30). Antibody subclass controls its effector functions, including affinity for FcγRs. In humans, IgG3 and IgG1 are the most effective activators of complement among gamma globulins and have the highest affinity for FcγRs, while IgG2 and IgG4 have limited ability to activate the complement cascade and generally low binding to FcγRs. Murine IgG2a also has high affinity for human FcγRs and can bind all three family members, while murine IgG1, in general, has the lowest affinity for human FcγRs (27, 31). These attributes are reflected in the different capacities of antibody subclasses to trigger FcγR-mediated functions in neutrophils, NK cells and monocytes, such as antibody-dependent cellular cytotoxicity (cell depletion) in vivo (32, 33), phagocytosis (34), and FcγR-dependent cytokine production (35).
Macrophages play a critical role in both acute and chronic allograft rejection. We therefore focused on the recruitment of monocytes in an in vitro model of HLA I antibody-mediated rejection to gain a better understanding of how HLA I antibodies promote accumulation of intragraft macrophages. We stimulated primary human aortic endothelium, human umbilical vein endothelial cells, and the human microvascular endothelial cell line HMEC-1 with a panel of HLA I-specific murine monoclonal antibodies with high or low affinity for human FcγRs. We investigated recruitment of two monocytic cell lines (Mono Mac 6 and THP-1) and of peripheral blood-derived human monocytes in response to HLA I antibody binding to endothelial cells. Results were confirmed using human allele specific monoclonal antibodies and IgG purified from transplant recipient sera. We hypothesized that HLA I antibodies have a unique capacity compared with other endothelial cell antibodies to promote monocyte recruitment into the allograft because of their dual actions on the endothelium and on the monocyte. We report herein that HLA I crosslinking by antibodies rapidly increases endothelial cell surface P-selectin, which is sufficient to initiate the recruitment of monocytes. Moreover, the engagement of monocyte FcγRs with endothelial-bound HLA I antibody can enhance adherence, and thus the subclass of the HLA I antibody significantly influences its ability to augment P-selectin-mediated recruitment through FcγRI and FcγRII.
Methods
Reagents and Antibodies
Mouse monoclonal anti-human HLA-I antibodies (clone W6/32, murine IgG2a, from BioXCell; clone 246-B8.E7, murine IgG2a, clones MEM-147 and MEM-81, murine IgG1, from Abcam) were chosen as model antibodies because they are well-characterized, recognize monomorphic epitopes on all HLA class I antigens (36), and are available in distinct IgG subclasses. Human allele specific monoclonal antibodies (37) were derived from heterohybridomas established at the Leiden University Medical Center and purified by Protein A chromatography. Specificity for HLA was confirmed using complement dependent cytotoxicity assay (CDC) and flow cytometric cross-match against single HLA transfected cells (38). Clones used were human IgG1; clone MUL2C6 (L) recognizes HLA-A3/A11, while clone SN230G6 (L) recognizes HLA-A2/B17. Irrelevant control human IgG1 was obtained from Sigma. These antibodies were used to ligate HLA I on endothelial cells. Isotype matched control antibodies, nonspecific mIgG, anti-endoglin (CD105: murine IgG2a, clone P3D1, Millipore; murine IgG1, Abcam), or anti-integrin β3 mIgG1 (CD61, Abcam) were used as negative controls. Isotype control murine IgG2a (MOPC-173) and murine IgG1 (MOPC-21), low endotoxin, azide free format, were purchased from BioLegend.
Neutralizing antibodies to integrin β2 (murine IgG1 clone TS1/18, BioLegend), αL (BioLegend), αM (BioLegend), ICAM-1 (sheep IgG, R&D; murine IgG1 clone HCD54, BioLegend), PSGL-1 (murine IgG1 clone KPL-1, BioLegend), P-selectin (clone AK4, murine IgG1, BioLegend; sheep or goat IgG, R&D), FcγRI (clone 10.1, BioLegend), FcγRII (clone AT10, Abcam), FcγRIII (clone 3G8, BioLegend), and Fcα/μR (BioLegend) were used and were of isotypes shown to have low affinity for human FcγRs (31) to minimize nonspecific blockade of FcγR. Recombinant soluble PSGL-1 Fc chimera YSPSL (rPSGL-1) was obtained from Y's Therapeutics (San Bruno, CA), which has an Fc region from human IgG1 containing point mutations to eliminate interactions with complement and FcγRs.
Cells and Culture Conditions
The human microvascular endothelial cell line HMEC-1 (kindly provided by Dr. Judith Berliner, Department of Pathology and Laboratory Medicine, UCLA) was grown on 0.1% gelatin in IMDM supplemented with 10% heat-inactivated fetal bovie serum (FBS) and antibiotics. Primary human aortic endothelial cells (HAEC) were isolated from aortic rings of explanted donor hearts as previously described (19). Primary human umbilical vein endothelial cells (HUVEC) were obtained from ATCC. HAEC and HUVEC were grown to confluence on 0.1% gelatin in PBS, and cultured as previously described (22). All experiments were repeated with endothelial cells from at least two different donors, with different HLA genotypes (CAR: A*02, A*68; B*60, B*65; CAS: A*02, A*30; B*35; B*35; 3F1153: A*02, A*11; B*44, B*56; D121: A*01, A*02, B*08, B*60; H126: A*03, A*29; B*35, B*44). Primary endothelial cells were used at passages 4 through 8.
The human monocytic cell line Mono Mac 6 (39, 40) (kind gift of Dr. Judith Berliner, Department of Pathology and Laboratory Medicine, UCLA) was cultured in RPMI-1640 supplemented with 10% FBS, 10μg/mL insulin, sodium-pyruvate, penicillin-streptomycin, and nonessential amino acids (Gibco). THP-1 monocytic cells from ATCC were maintained in RPMI-1640 supplemented with 5% FBS and antibiotics. Monocytic cell lines were subcultured every 2-3 days and maintained at a density of less than 106 cells per mL.
Isolation of Primary Human Monocytes
Whole blood was obtained from healthy volunteers in accordance with UCLA IRB#10-001689. Monocytes were enriched from peripheral blood using Ficoll-Paque and magnetic-activated cell sorting. Briefly, peripheral blood mononuclear cells (PBMC) were isolated using Ficoll-Paque density centrifugation (GE Healthcare). Monocytes were enriched from PBMC using negative selection Pan-Monocyte Enrichment Kit according to the manufacturer's protocol (Miltenyi). Purity of enriched monocytes was greater than 85% as determined by flow cytometric staining for CD14.
Purification of IgG from Transplant Recipient Sera
Patient sera from sensitized transplant recipients were obtained in accordance with UCLA IRB#11-000577. Sera were selected for reactivity against HLA antigens expressed by the panel of endothelial cells, pooled (PS6), and subsequently used in experiments. The presence of polyclonal anti-HLA antibody was confirmed using LABScreen Single Antigen Beads (One Lambda) on the Luminex platform. Antibody reactivity was to the following antigens expressed by aortic endothelial cells: A3, A29, B35, B44 (donor H126); A2, A11, B44, B56 (donor 3F1153); A1, A2, B8, B60 (donor D121).
The IgG fraction from the pooled highly sensitized transplant recipient sera (PS6) or from negative serum lacking HLA antibodies (NS) was purified using Protein G column chromatography. Briefly, sera were diluted to 1:12 in 20mM phosphate buffer pH 7.0 (binding buffer). Diluted sera were mixed with beads at a ratio of 4mL sera to 1mL beads, and incubated in a centrifuge column (Thermo Scientific) with end-over-end rotation overnight at 4°C. The column was washed with binding buffer until the A280 returned to baseline. IgG was eluted using 0.1M citrate buffer pH 2.16 and immediately neutralized to physiological pH with 1M Tris pH 9.0. Purified IgG was concentrated and the buffer was exchanged to PBS using Amicon Ultra-0.5mL Centrifugal Filters for Protein (MWCO 50kDa, Millipore) according to the manufacturer's recommended methods. IgG concentration was measured using NanoDrop 8000 Spectrophotometer, assuming an extinction coefficient of 1.4 for absorbance at A280.
Binding of the IgG fraction of transplant recipient sera to unfixed HAEC was determined by cell-based ELISA method from Florey et al. (28). To evaluate the individual subclasses in human IgG bound to HAEC, biotinylated anti-hIgG1, anti-hIgG2 or anti-hIgG3 (Sigma Aldrich) were used as detection antibodies. Preliminary experiments confirmed that IgG from HLA-reactive recipient sera bound to endothelial cells, and contained hIgG1, hIgG2 and hIgG3 (unpublished observations), as previously reported (41-43).
Antibody Binding to Endothelial Cells
Endothelial cells were detached by Accutase, and stained with 1μg of HLA I antibody, anti-CD61 or anti-CD105 antibody in 0.1mL of PFA flow buffer (PBS with 2.5% FBS and 0.1% sodium azide) for 30 min on ice. Cells were washed twice and a secondary anti-mouse Fcγ FITC-conjugated antibody at 1:100 (Jackson Immuno) was used to detect antibody binding. Alternatively, cells were stained with PE-conjugated ICAM-1 (BD Biosciences). Fluorescence was measured by flow cytometry on a FACSCalibur cytometer (Becton Dickinson).
Flow Cytometric Determination of Monocyte Receptor Expression
Expression of FcγRs on unfixed monocytes was measured by flow cytometry. Antibodies to integrin β2, αL, αM (Biolegend), FcγRI (anti-CD64, Biolegend), FcγRII (anti-CD32, StemCell), FcγRIII (anti-CD16, Biolegend), and Fcα/μR (anti-CD351, Biolegend)—all murine monoclonal IgG1—were used at 1μg per 0.1mL PFA flow buffer on ice for 30-45 min, followed by secondary antibody anti-mouse Fcγ-FITC (Jackson Immuno). Results are shown as representative histograms or average median channel fluorescence ± SEM from three experiments.
The FcγRIIa allotype of monocytes was determined by a flow cytometric method as previously described (44-46). Briefly, monocytic cells were stained with pan-FcγRIIa antibody (clone AT10) or with clone 3D3, which recognizes only the R131 allele of FcγRIIa (BD Pharmingen). Cells homozygous for FcγRIIa-H131 stain negatively for 3D3, while cells homozygous for R131 will have approximately equal amounts of staining for pan-FcγRIIa and R131-specific antibody. Cells are considered heterozygous if the ratio of R131 staining to total FcγRIIa is approximately 0.5. As a control, the monocytic cell line U937 was stained in parallel, which is reportedly homozygous for R131 (46).
Determination of Cell Surface P-selectin
Cell surface expression of P-selectin was measured on adherent endothelial cells by cell-based ELISA (adapted from (47)). Briefly, confluent endothelial cells in a 96-well plate were treated with control or HLA I antibodies in M199 with 5% FBS for 30 min. Cells were washed once with PBS and fixed in freshly prepared 2.5% paraformaldehyde in PBS for 5 min at room temperature. Unpermeabilized cells were washed and blocked with 5% BSA in PBS. Sheep anti-P-selectin (R&D) was added at 1μg/mL for 2hr. Wells were washed three times and donkey anti-sheep-HRP (Millipore) was added at 1:1000 for 2hr at room temperature. After washing, TMB substrate was added and optical density (A650) was read on a SpectraMax plate reader (Molecular Devices).
Blockade of FcγRs
Since monocytes, Mono Mac 6 and THP-1 express high levels of Fc gamma receptors (FcγR), preliminary studies were undertaken to determine the ability of Mono Mac 6 to take up soluble monomeric murine IgG, and to assess the effectiveness of blocking strategies. According to these findings, in specified adhesion experiments monocytes, Mono Mac 6 or THP-1 were incubated with 20μg/mL of purified human IgG in PBS (Fisher Scientific) or 10μg/mL of monoclonal murine IgG2a (MOPC-173) for 15 min to block the FcγR from binding HLA I IgG on the surface of the endothelial cells.
Static Adherence Assay
Adhesion of monocytes to endothelial monolayers was measured as previously described (22). Briefly, confluent endothelial cells were treated with mIgG, anti-CD105 mIgG2a, HLA I antibodies, or positive control thrombin in M199 with 2% FBS (assay medium) for the indicated times. Monocytes or monocytic cells were fluorescently labeled with CFDA-SE (Vybrant Cell Tracer, Invitrogen) at 2μM in HBSS with Ca2+ and Mg2+ for 10 min at 37°C, then washed. Monocytes were added at approximately 2×105 cells per well in a 24 well plate (roughly equivalent ratio of 2-3monocytes:1EC) for 20min at 37°C. The coculture was washed three times, and fixed. Images of adherent monocytes were acquired by fluorescence microscopy on a Nikon Eclipse Ti, in 8 to 10 4× objective fields for each condition. The number of adherent cells in each field was quantified using automated software from MIT called CellProfiler (19, 22, 48). Results are expressed for each condition as the mean number of adherent monocytes per field, or as fold change in mean adherent monocytes per field normalized to untreated, ± standard error of the mean (SEM).
To determine the effect of adhesion molecules in monocyte adherence to antibody-activated endothelium, receptors were blocked on either the monocytes or the endothelial cells prior to coculture. To block P-selectin and ICAM-1, endothelial cells were treated as above, and functional grade P-selectin or ICAM-1 blocking antibodies were added at 10μg/mL for at least 10 min before coculture with monocytes. To block monocytic receptors, cells were preincubated with neutralizing antibodies to β2 integrin, αL (LFA-1), αM (Mac-1), PSGL-1, FcγRI, FcγRII, FcγRIII, or Fcα/μR at 10μg/mL for 20 min prior to the adhesion assay. All blocking antibodies were murine IgG1 isotype, except anti-P-selectin, which was goat immunoglobulin. Alternatively, to block selectins, rPSGL-1 was added at 20μg/mL, a concentration which maximally inhibited Mono Mac 6 adherence to purified immobilized P-selectin in preliminary experiments. Percent inhibition of adherence by each inhibitor was calculated as follows: ((fold change without inhibitor - 1) - (fold change with inhibitor − 1))/(fold change without inhibitor − 1) × 100%.
Immobilized Immunoglobulin Adherence Assay
Purified polyclonal human IgG (Fisher), murine IgG2a (MOPC-173), or murine IgG1 (AK4) were diluted in carbonate/bicarbonate buffer and coated onto high protein binding (Nunc Maxisorp) 96 well plates at 0.1mL per well, then incubated overnight at 4°C. The next day, the protein solution was removed, and the wells were blocked with 5% BSA in PBS for 2hr at room temperature. Mono Mac 6 cells were labeled with 2μM CFSE in HBSS for 10 min, then serum rescued with 5% FBS in HBSS, spun down and resuspended at 106 cells per mL. Cells were added to the plate at 105 cells in 0.1mL per well and incubated at 37°C for 30 min. Wells were washed three times with HBSS using the flick method, then fixed. Adherent cells were visualized by fluorescence microscopy using a 4× objective, and 5 fields per well were counted using Image J or CellProfiler.
Dynamic Adherence Assay
Confluent HAEC in a 12 well plate were treated with mIgG, anti-CD105 mIgG2a, or HLA I antibodies in M199 with 2% FBS (assay medium). Mono Mac 6 were fluorescently labeled with CFDA-SE as above, then washed. Mono Mac 6 were added at approximately 5×105 cells per well and allowed to adhere for 20 min at 37°C on an orbital shaker (New Brunswick) at 40RPM. At this speed, the τmax was calculated at 1.5 dynes/cm2, similar to shear stress in capillaries (49). Dynes/cm2 were estimated using the following formula: τmax= a√(ηρ(2Πf)3) (49, 50), where a is the radius of rotation for the shaker, η is the viscosity of the medium, ρ is the density of the medium, and f is the frequency of rotation in revolutions per second. The coculture was washed three times, and fixed. Adherent monocytes were counted as above. The center of the well was avoided, as shear stress becomes negligible here on under orbital conditions (50). Results are expressed for each condition as the mean number of adherent monocytes per field ± standard error of the mean (SEM).
Statistical Analysis
One-way analysis of variance (ANOVA) with Tukey's multiple comparisons test, protected Fisher's LSD test, or Dunnett's multiple comparisons test was used for statistical comparison of means, with p<0.05 considered significant. Data are presented as mean ± SEM.
Results
The magnitude of HLA I antibody-induced monocyte adhesion to endothelial cells is dependent upon HLA I antibody subclass
As a surrogate for donor specific HLA I antibody, we tested a panel of monoclonal murine antibodies each recognizing a monomorphic epitope on human HLA I molecules, and assessed recruitment of human monocytes to human endothelial cells stimulated with these pan HLA I antibodies. The monocytic cell line Mono Mac 6 bound to untreated human aortic endothelial cells (HAEC from donor 3F1153) at 297.7±24 cells per 4× field. Treatment of HAEC with HLA I antibody led to a large increase in the number of Mono Mac 6 adherent on the endothelial monolayer (Figure 1a). HLA I mIgG1 (clone MEM-147) increased the number of bound Mono Mac 6, as did endothelial stimulation with HLA I murine IgG1 (clone MEM-81), to a mean 508±25 and 445±7 adherent monocytes per field respectively (P≤0.001). Both clones of mIgG2a pan-HLA I antibodies (clones W6/32 and 246.B8-E7) dramatically increased adherence of Mono Mac 6 to endothelial cells, to an average of 777±21 and 999±60 per field, respectively. Similar observations were made when the panel was used to stimulate endothelial cells from a donor with a different HLA genotype (H126). Representative fluorescence microscopy fields demonstrating Mono Mac 6 adherent to the endothelial monolayer (3F1153) are given in Figure 1a.
Figure 1.

HLA I antibodies trigger adherence of monocytes to endothelium differentially depending on subclass.
a. Confluent HAEC (donor 3F1153, black bars; or donor H126, white bars) were stimulated with murine monoclonal HLA-I antibodies of isotype IgG1 (clones MEM-147 or MEM-81) or IgG2a (clones W6/32 or 246.B8-E7) at 1μg/mL for 30min. CFSE-labeled Mono Mac 6 (MM6) were overlaid and allowed to bind for 20min. Nonadherent cells were washed off and adherent cells were counted in 10 fields per condition. Results are displayed as the mean number of adherent MM6 ± SEM Groups were compared using Two-way ANOVA and Holm-Sidak's multiple comparisons test.
* P≤0.05, ** P≤0.01, *** P≤0.001, **** P≤0.0001 versus untreated. Representative 4× fluorescence microscopy fields for MM6 binding to 3F1153 HAEC are given in the bottom panel.
b. HAEC were treated as in (a), and adhesion of MM6 (black bars) or THP-1 (white bars) was measured. Results are expressed as the fold increase in the number of adherent monocytes from three or more independent experiments. Groups were compared using One-way ANOVA followed by Tukey's multiple comparisons test. ** P≤0.01, *** P≤0.001 comparing HLA I mIgG1 to mIgG2a.
c. HAEC were treated as in (a), and adhesion of CFSE-labeled human CD14+ monocytes enriched from peripheral blood was measured. Results are displayed as the mean number of adherent monocytes ± SEM. One-way ANOVA followed by Tukey's multiple comparisons test. **** P≤0.0001.
d. HAEC were stimulated with control anti-CD61 mIgG1, anti-CD105 mIgG2a, HLA I mIgG1 or mIgG2a at 1μg/mL for 30min, and adherence of MM6 (black bars) or THP-1 (white bars) was determined. Results are expressed as the fold increase in the number of adherent monocytes from three or more independent experiments. Groups were compared to untreated using One-way ANOVA followed by uncorrected Fisher's LSD. ns P>0.05, * P≤0.05, ** P≤0.01, **** P≤0.0001 versus untreated.
e. HUVEC (dark grey bars) or HMEC-1 (light grey bars) were stimulated with control anti-CD105 mIgG2a or HLA I antibody at 1μg/mL for 30min, and adherent MM6 were counted. Results are representative of two experiments. Two-way ANOVA, Sidak's multiple comparisons test. ns P>0.05, **** P<0.0001 versus untreated.
We observed a notable difference in the magnitude of Mono Mac 6 adherence stimulated by mIgG1 compared with mIgG2a HLA I antibodies, and confirmed these findings with a second monocytic cell line, THP-1 (Fig. 1b). Adhesion of both Mono Mac 6 and THP-1 monocytes was significantly increased to endothelial cells stimulated with HLA I mIgG1 (MEM-147), by 2.16±0.21 fold and 2.17±0.16 fold over untreated, respectively. Stimulation of endothelial cells with HLA I mIgG2a (W6/32) markedly increased adherence of Mono Mac 6 by 4.50±0.49 fold over control, and THP-1 by 4.64±0.7 fold over control. When the two isotypes were compared in parallel, the degree of monocytic cell adherence to HLA I mIgG2a (W6/32)-treated endothelial cells was 2-fold higher than to HLA I mIgG1 (MEM-147)-treated endothelial cells in more than four experiments (p<0.01) (Figure 1b). We further validated these observations with human CD14+ monocytes enriched from peripheral blood (PBMC-monocytes). Stimulation of HAEC with HLA I mIgG2a (W6/32) significantly increased PBMC-monocyte adherence, from 73.4±8.4 to 898.2±38.5 per field. HLA I mIgG1 (MEM-147) treatment also significantly increased monocyte binding, although to a lesser extent than mIgG2a, at 501.9±62.8 monocytes per field (Figure 1c). This observed difference in recruitment capacity between isotypes could not be attributed to unequal amounts of HLA I antibody bound to the endothelial cell surface, as the binding of each HLA I monoclonal was comparable on endothelia from several different donors (Supplemental Figure 1a).
Monocyte Recruitment by Endothelial Cells is a Specific Response to HLA I antibodies
To determine whether endothelial cell antibodies could promote monocyte adherence as observed with HLA I antibodies, human aortic endothelial cells were treated with non-HLA or HLA I antibodies at the same concentration (1μg/mL). All antibodies bound to endothelial cells (Supplemental Figure 1b). Coating of aortic endothelial cells with murine antibody to integrin β3 (anti-CD61 mIgG1) was not sufficient to cause adherence of Mono Mac 6 (1.03±0.025-fold over untreated) to HAEC. In contrast, treatment of endothelium with HLA I-specific mIgG1 (MEM-147) significantly increased adherence of Mono Mac 6 by 2.71±0.46-fold (Figure 1d). Similarly, while antibody recognizing endoglin (anti-CD105 mIgG2a) did not stimulate monocytic cell binding (1.17±0.11-fold over untreated), HLA I mIgG2a (W6/32) provoked extensive Mono Mac 6 recruitment (4.39±0.32-fold). Similar results were obtained when THP-1 monocytes were used (Figure 1d).
In addition, control anti-CD105 mIgG2a had no significant effect on Mono Mac 6 adherence to human umbilical vein endothelial cells (HUVEC), or the human dermal microvascular cell line HMEC-1. In contrast, treatment with HLA I mIgG1 (MEM-147) or HLA I mIgG2a (W6/32) at 1μg/mL significantly increased Mono Mac 6 binding to both HUVEC and HMEC-1 (Figure 1e). Therefore, monocyte recruitment is specific to antibodies recognizing HLA I molecules, and is not induced by isotype matched non-HLA I antibody bound to endothelia.
HLA I antibodies differentially recruit monocytes depending on their capapcity to engage FcγRs
Mono Mac 6 and THP-1 express FcγRI (CD64) and FcγRII (CD32), with little detectable FcγRIII (CD16) (Supplemental Figure 2a), which is similar to the predominant monocyte subset in peripheral blood (CD64+CD14+CD16−) (reviewed in (51)). The ability of human FcγRs to interact with murine immunoglobulin, particularly with mIgG2a, has been previously characterized (52, 53). Therefore, we postulated that the difference in the capacity of HLA I antibody IgG1 and IgG2a subclasses to stimulate monocyte adherence was due to unequal affinities of the Fc portion for human FcγRs on monocytes. To test the ability of each murine IgG isotype to engage FcγRs on the monocyte, we used a solid phase assay in which irrelevant immunoglobulin was immobilized and Mono Mac 6 adherence was measured (Supplemental Figure 2b). Mono Mac 6 did not adhere to wells coated with murine IgG1 or BSA alone. In contrast, Mono Mac 6 adhered to immobilized positive control polyclonal human IgG, or to murine IgG2a, to a significantly greater extent than to BSA alone. Thus, Mono Mac 6 monocytes have minimal ability to bind mIgG1, while binding to murine IgG2a through FcγRs was high, consistent with previous studies (54, 55).
In light of these findings, we determined whether HLA I mIgG2a-triggered monocyte recruitment had an Fc-mediated component. Mono Mac 6 or THP-1 were pretreated with a blocking antibody to FcγRI, FcγRII, FcγRIII, or with soluble IgG to inhibit all FcγRs, and adherence to HLA I mIgG2a (W6/32)-treated human aortic endothelial cells was measured. HLA I mIgG2a-stimulated endothelial cells demonstrated a more than 5-fold increase in THP-1 monocyte binding compared to untreated endothelium (Figure 2a). Inhibition of FcγRI significantly reduced the number of THP-1 adherent to HLA I mIgG2a (W6/32)-treated endothelial cells by 89.3±4.4%, while a blocking antibody against FcγRII had a minor inhibitory effect at 24.4±4.9%. In contrast, blocking antibodies to FcγRIII had no significant effect (Figure 2a and 2b), consistent with the absence of this receptor on the monocytic cell lines (Supplemental Figure 2a). Similar results were obtained when Mono Mac 6 binding was assessed. Inhibition of FcγRI reduced Mono Mac 6 binding to HLA I mIgG2a (W6/32)-stimulated endothelium by 82.6±8.1%, while a neutralizing antibody to FcγRII resulted in lesser inhibition of adherence (59.2±8.8%). FcγRIII antagonism did not significantly affect the ability of Mono Mac 6 monocytes to adhere to HLA I mIgG2a (W6/32)-treated endothelial cells, nor did a control antibody against Fcα/μR (unpublished observations). Pretreatment of monocytes with a total FcγR blockade inhibited THP-1 adherence by 72.0±8.8% and Mono Mac 6 adherence by 85.6±4.8% (Figure 2b). Mono Mac 6 recruitment by HAEC treated with the second clone of HLA I mIgG2a (246-B8.E7) was also FcγR-dependent (unpublished observations). These results demonstrate that FcγRI is predominantly involved in monocytic cell adherence to endothelial cells activated with HLA I mIgG2a.
Figure 2.
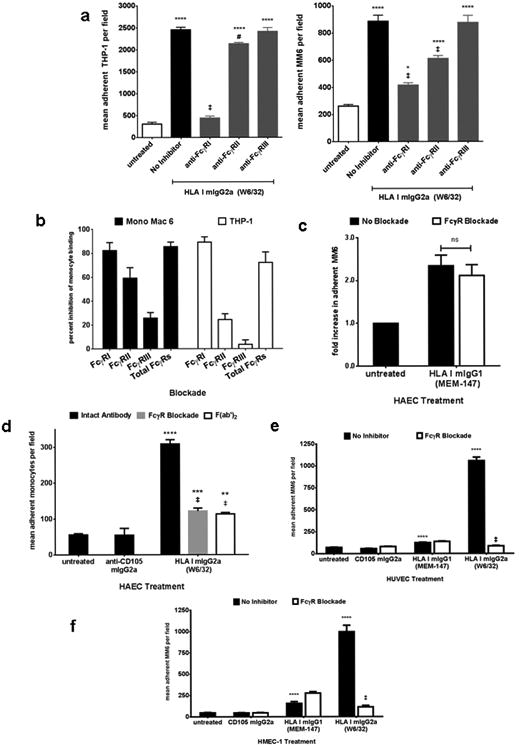
HLA I mIgG2a-mediated monocyte recruitment is dependent upon FcγRs.
a. HAEC were stimulated with HLA I mIgG2a at 1μg/mL for 30min. Mono Mac 6 (left panel) or THP-1 (right panel were pretreated with neutralizing antibodies to FcγRI, FcγRII or FcγRIII at 10μg/mL for 15min, and adherence to endothelium was measured. Results are from one representative experiment out of three.
b. HAEC were stimulated and MM6 (black bars) or THP-1 (white bars) adherence was measured with or without FcγR inhibition as in (a). Total FcγR were blocked by incubating monocytic cells with soluble human IgG at 10μg/mL. Results are expressed as the average ± SEM percent inhibition of monocyte binding from three or more independent experiments.
c. HAEC were stimulated with HLA I mIgG1 at 1μg/mL for 30min. Adherent MM6 with or without an FcγR blockade were counted. Results are shown as fold increase in the number of adherent MM6 normalized to untreated from threeindependent measurements (top panel). ns P>0.05 comparing no inhibitor to FcγR blockade. Average percentinhibition of adherence in 8 independent experiments is given in the bottom panel.
d. HAEC were stimulated with intact HLA I antibody at 1μg/mL, or with F(ab′)2 fragment of HLA I mIgG2a at 5μg/mL for 30min. Adherence of human CD14+ monocytes enriched from peripheral blood was measured with and without an FcγR blockade. Results are shown from one representative experiment of two.
e. HUVEC were stimulated with 1μg/mL of anti-CD105 mIgG2a, HLA I mIgG1 or HLA I mIgG2a for 30min, and adherence of MM6 was measured with and without an FcγR blockade as above. Results from one representative experiment o two are shown.
f. MM6 adherence to HMEC-1 was performed as in (e).
Groups were compared using One-way ANOVA followed by Tukey's multiple comparisons test.
* P<0.05, ** P≤0.01, *** P<0.001, **** P≤0.0001 versus untreated. ns P>0.05, # P<0.05, ‡ P≤0.01 versus no inhibitor.
We observed a difference in FcγR utilization between Mono Mac 6 and THP-1; blockade of FcγRIIa largely impaired Mono Mac 6 but not THP-1 recruitment (Figure 2a and 2b). We posited that this discrepancy was due to allelic differences in FcγRIIa, which is dimorphic in humans. The R131 and H131 alleles have divergent affinities for the various subclasses of murine and human IgG, and H131 has higher overall affinity for IgG than R131. We determined the allotype of FcγRIIa on each monocytic cell line using flow cytometry method as previously described (45, 56), where the binding of an R131 allele-specific antibody (clone 3D3) can be compared with staining for total FcγRIIa to determine homozygosity or heterozygosity. This method revealed that Mono Mac 6 are homozygous for FcγRIIa-H131, while THP-1 are heterozygous (H/R). As a control, U937 monocytic cell line carried only the R131 allele as previously documented (Supplemental Figure 2c) (45, 46, 56, 57).
Although our data indicated that FcγRs participated in monocyte recruitment in response to HLA I antibodies with a high affinity for FcγRs, previous studies (57, 58) and our preliminary data implied that mIgG1 would have a weaker capacity to engage human FcγRs, which may explain the lower capacity of HLA I mIgG1 to trigger adherence of monocytes. Therefore, to determine whether HLA I mIgG1-stimulated adhesion to endothelial cells was also dependent on FcγRs, we treated HAEC with HLA I mIgG1 (MEM-147), and Mono Mac 6 were left untreated or incubated with soluble IgG to block FcγRs prior to the adherence assay. HLA I mIgG1 (MEM-147) significantly increased the adherence of Mono Mac 6 to endothelium, by 2.34±0.24 fold over untreated. In contrast to HLA I mIgG2a, HLA I mIgG1-triggered Mono Mac 6 binding was not significantly different when FcγRs were blocked, at 2.11±0.25 fold over untreated (mean 18.16±5.19% inhibition over 8 experiments) (Figure 2c). Similar results were obtained when endothelial cells were stimulated with HLA I mIgG1 (MEM-81) (data not shown). Therefore, inhibition of FcγRs by preincubation with soluble IgG did not impair the ability of monocytes to bind to endothelial cells stimulated with HLA I mIgG1, in agreement with our observation that mIgG1 did not bind human FcγRs on Mono Mac 6.
Peripheral blood CD14+ monocytes highly express FcγRI and FcγRIIa, but only a small subset (<10%) express FcγRIII (unpublished observations, (51)). While treatment with anti-CD105 mIgG2a did not increase recruitment of monocytes, HLA I mIgG2a (W6/32) stimulated a 5.27-fold increase in peripheral blood monocytes bound to HAEC. Adherence of monocytes was partially dependent on interaction between FcγRs and antibody, as FcγR blockade significantly reduced adherence to HAEC to 2.08-fold over untreated (74.7% inhibition, Figure 2d). In contrast, FcγR blockade had no inhibitory effect on peripheral blood-monocyte recruitment to HLA I mIgG1 (MEM-147)-activated endothelial cells (data not shown). To validate the contribution of the Fc fragment of HLA I antibody to monocyte recruitment, we stimulated HAEC with an F(ab′)2 fragment of W6/32, which lacks the Fc portion of the antibody but retains antigen recognition sites. HLA I crosslinking with W6/32 F(ab′)2 resulted in significantly lower numbers of adherent monocytes compared with intact antibody, identical to results with an FcγR blockade (Figure 2d). Notably, however, monocyte binding in response to HLA I F(ab′)2 was 1.94-fold higher than to unstimulated endothelial cells (p<0.001), consistent with our previous observation (22). In addition, while FcγR antagonism significantly impaired Mono Mac 6 adherence to HUVEC (Figure 2e) and HMEC-1 (Figure 2f) stimulated with HLA I mIgG2a (W6/32), FcγR blockade had no effect on Mono Mac 6 recruitment in response to endothelial activation with HLA I mIgG1 (MEM-147).
Since leukocyte recruitment occurs in the blood vessels under shear stress, we recapitulated more physiological endothelial-monocyte interactions by performing the adhesion assay under dynamic conditions. HAEC were treated with anti-CD105 mIgG2a, HLA I mIgG1 (MEM-147) or HLA I mIgG2a (W6/32). Mono Mac 6 with or without an FcγR blockade were overlaid and incubated with stimulated endothelial cells on an orbital shaker. While inhibition of FcγRs had no effect on HLA mIgG1-induced recruitment, HLA I mIgG2a-induced recruitment was significantly inhibited when FcγR were blocked (Supplemental Figure 2d).
HLA I antibody-induced endothelial P-selectin is required for monocyte adherence irrespective of antibody isotype
We observed that HLA I mIgG1-stimulated monocyte adherence to endothelial cells was not affected by an FcγR blockade. Moreover, inhibition of FcγRs did not completely eliminate monocyte binding in response to HLA I mIgG2a, and that F(ab′)2 HLA I antibody stimulated a significant increase in monocyte binding over untreated (22). Taken together, these results pointed to an additional mechanism of recruitment unrelated to the Fc fragment and dependent upon HLA I recognition and dimerization. Therefore, we proposed that HLA I antibodies activate endothelia leading to increased adhesion molecule expression, which could independently support monocyte adhesion.
Given the rapidity of the recruitment response (<1hr), we first determined the effect of stimulation with monoclonal HLA I antibodies on P-selectin, which is stored in Weibel-Palade bodies and rapidly translocated to the cell surface upon endothelial activation. Confluent endothelial cells were left untreated or exposed to isotype control mIgG2a, non-HLA endoglin antibody (anti-CD105 mIgG2a), antibody recognizing integrin β3 (anti-CD61 mIgG1) or HLA I antibody (clones W6/32, MEM-147 and MEM-81) for various times. Cell surface P-selectin was measured by cell-based ELISA. Crosslinking of HLA molecules with HLA I mIgG2a (W6/32), HLA I mIgG1 (MEM-81) or HLA I mIgG1 (MEM-147) significantly elevated P-selectin on the endothelial cell surface by more than 2-fold over baseline (Figure 3a and 3b), with expression detected as early as 5min, peaking at 30min and sustained up to 45min after stimulation (Supplemental Figure 3a). P-selectin induction by HLA I mIgG2a was dose dependent with a maximal effect at 5μg/mL (unpublished observations), confirming previous findings (21, 22). Activation with classical Weibel-Palade body agonists histamine, PMA and thrombin significantly increased endothelial P-selectin (Figure 3a and 3b), as reported (59). In contrast, non-HLA anti-CD105 nor anti-CD61 antibodies did not stimulate cell surface P-selectin on HAEC (Figure 3b). Similarly, P-selectin was not detected on unstimulated HUVEC or HMEC-1, and was unchanged by isotype control mIgG or anti-CD105 mIgG2a. HLA I crosslinking by antibodies significantly increased P-selectin detected on the surface of HUVEC and HMEC-1 by 1.6-fold and 3-fold, respectively (Figure 3c). Thus, several clones of HLA I antibodies elicited exocytosis and increase P-selectin on aortic, venous and microvascular human endothelial cells at a similar magnitude as traditional Weibel-Palade body agonists.
Figure 3.

HLA I crosslinking by antibodies stimulates Weibel-Palade body exocytosis and increased cell surface P-selectin on endothelial cells.
a. HAEC were treated for 30min with HLA I antibody at 1μg/mL, 10min with 5U/mL thrombin, or 20min with 200nM PMA. Cell surface P-selectin was stained on unpermeabilized cells with sheep anti-P-selectin followed by anti-sheep-HRP, and detected by adding TMB substrate. Results are expressed as the mean optical density of duplicate wells for each condition. Results from one representative experiment out of three total is shown.
b. HAEC were stimulated with anti-CD105, anti-CD61 antibody or HLA I antibody at 1μg/mL for 30min, histamine for 5min, or PMA at 200nM for 15min. P-selectin was measured as in (a).
c. HUVEC (dark grey bars) or HMEC-1 (light grey bars) were stimulated with control or HLA I antibody as in (b) and cell surface P-selectin was measured.
d/e. HAEC (d) or HMEC-1 (e) were stimulated with HLA I antibody at 1μg/mL for 30min. Adherence of MM6 was measured with or without rPSGL-1 at 20μg/mL to block P-selectin or anti-PSGL-1 at 10μg/mL. Results from on representative experiment of three are shown as the mean number of adherent MM6 ± SEM in the top panel. Bottom panel (d) shows representative fluorescence microscopy fields.
Groups were compared by One-way ANOVA followed by Dunnett's multiple comparisons test.
ns P>0.05, * P≤0.05, ** P≤0.01, *** P≤0.001, **** P≤0.0001 versus untreated. ‡ P<0.01 versus no inhibitor.
We hypothesized that endothelial P-selectin elicited by HLA I ligation would engage its ligand PSGL-1, expressed on monocytic cell lines and monocytes (Supplemental Figure 2e). To test this hypothesis, endothelial cells were treated with HLA I mIgG2a (W6/32) or HLA I mIgG1 (MEM-147) and exposed to the soluble antagonist of P-selectin, rPSGL-1. Adherence of Mono Mac 6 was measured with and without the inhibitor. The presence of rPSGL-1 significantly inhibited Mono Mac 6 binding to endothelial cells stimulated with either HLA I mIgG2a (W6/32) or HLA I mIgG1 (MEM-147) by 69.4±4.3% and 70.4±8.4%, respectively (Figure 3d and 3e), demonstrating the effectiveness of a P-selectin blockade against recruitment induced by both isotypes of HLA I antibody. As a control, rPSGL-1 antagonist also reduced monocyte adherence to thrombin-stimulated endothelial cells by 88.6±6.9% (unpublished observations). Representative fields illustrating Mono Mac 6 adherence to HLA I antibody-activated endothelial cells with and without the antagonist are shown in Figure 3d.
As an alternative strategy, neutralizing antibody to P-selectin or to its receptor PSGL-1 reduced Mono Mac 6 adherence to HLA I antibody-activated endothelial cells by 40% to 65% (Supplemental Figure 3b). rPSGL-1 or neutralizing antibody to PSGL-1 also significantly attenuated HLA I antibody-mediated Mono Mac 6 adherence to HMEC-1 (Figure 3e). Similar results were observed with THP-1 (unpublished observations). Since selectin-mediated leukocyte capture is most prominent under shear stress (25), we confirmed our observations under dynamic conditions. P-selectin antagonism with rPSGL1- significantly impaired Mono Mac 6 adherence to HLA I antibody-treated endothelium under flow conditions (Supplemental Figure 3c). These results demonstrate that P-selectin is a critical mediator of HLA I antibody-triggered monocyte binding to endothelial cells irrespective of HLA I antibody subclass and under both static and flow conditions.
HLA I antibodies promote firm adherence to endothelial cells preferentially through Mac-1/ICAM-1 interactions
While P-selectin initiates capture of leukocytes from the blood and mediates rolling on endothelial cells, integrin binding to ICAM-1 or VCAM-1 is required for firm adherence and ultimately transmigration into the tissue. HAEC constitutively express ICAM-1 as previously documented (60, 61); however, we did not observe an alteration in ICAM-1 surface expression 1hr, 2hr or 4hr after exposure to HLA I antibodies (Figure 4a) consistent with previous reports (62). Since Mono Mac 6 and THP-1 express the ICAM-1 binding integrins LFA-1 (αLβ2) and Mac-1 (αMβ2) (Supplemental Figure 2e), we reasoned that ICAM-1/β2 integrins may be involved in stable adhesion of monocytes to HLA I antibody-stimulated endothelial cells after initial capture and activation by P-selectin.
Figure 4.
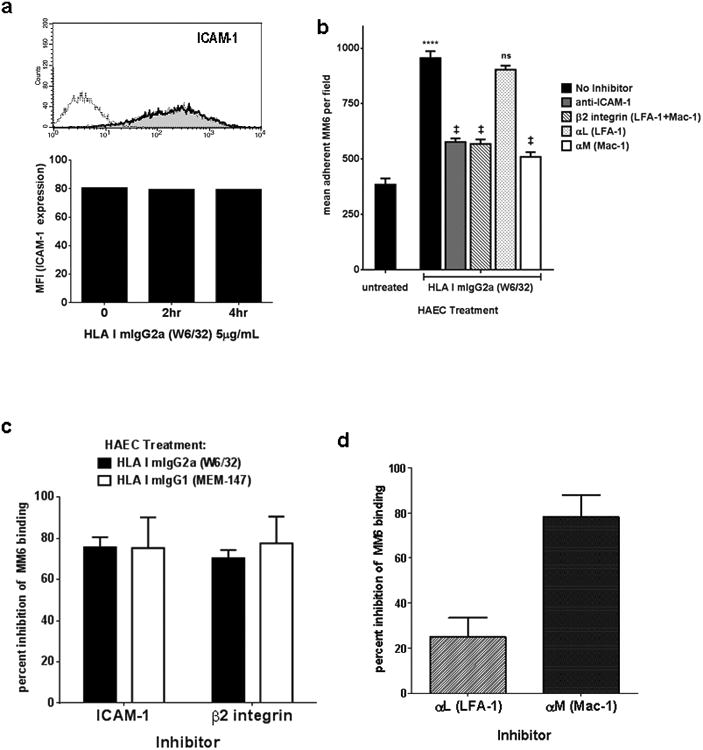
Endothelium activated with HLA I antibodies support firm adhesion of monocytes through ICAM-1 and Mac-1 interactions.
a. HAEC were treated with HLA I mIgG2a (W6/32) for 1hr, 2hr, or 4hr, then detached with Accutase. ICAM-1 was stained with a PE-conjugated antibody and expression was measured by flow cytometry. Histogram shows ICAM-1 basal expression on unstimulated endothelial cells (grey fill) and after exposure to HLA I antibodies for 1hr (dark line). Median fluorescence intensity of ICAM-1 at 2hr and 4hr after exposure to HLA I antibody is graphed. Results are representative of two independent measurements.
b. HAEC were treated for 30min with HLA I antibody at 1μg/mL. Adhesion of Mono Mac 6 was measured as above, with or without neutralizing antibody at 10μg/mL to the indicated adhesion molecules. Results are from one representative experiment out of three. *** P≤0.001, **** P≤0.0001 versus untreated. ns P>0.05, ‡ P≤0.001 versus no inhibitor.
c. Mono Mac 6 adhesion to HAEC stimulated with HLA I mIgG2a (black bars) or mIgG1 (white bars) was measured as in (b). Results are expressed as the average percent inhibition ± SEM of adherence by each inhibitor for each condition.
d. Mono Mac 6 adhesion to HAEC stimulated with HLA I mIgG2a was measured as in (b). Results are expressed as the average percent inhibition ± SEM of adherence by neutralizing antibody to LFA-1 or Mac-1 for each condition.
To test this hypothesis, HAEC were treated with HLA I antibody and a neutralizing antibody to ICAM-1 was added. HLA I mIgG2a (W6/32) increased adherence of Mono Mac 6 to endothelial cells to an average of 995±31 per field, compared with adherence to untreated endothelial cells at 384±27 per field. Mono Mac 6 adherence was significantly inhibited when ICAM-1 was blocked, by 75.7±5.3% for HLA I mIgG2a and 75.3±14.9% for HLA I mIgG1 (Figure 4b and 4c). We blocked ICAM-1 binding integrins by pretreating the Mono Mac 6 with a neutralizing antibody against β2 integrin prior to the adhesion assay. β2 integrin inhibition reduced Mono Mac 6 binding to HLA I mIgG2a (W6/32)-stimulated endothelium by 70.3±4.4% compared with no inhibitor. Blockade of β2 integrin also abolished Mono Mac 6 ability to bind to HLA I mIgG1 (MEM-147)-treated endothelium, reducing the number of adherent cells by 84.1±15.9% (Figure 4c).
Because monocytes express two integrins which bind to ICAM-1, we explored the relative contribution of each to HLA I mIgG2a-triggered adherence. The β2 subunit pairs with the αL subunit to form LFA-1 (αLβ2), while αM complexes with β2 to form the Mac-1 integrin. Mono Mac 6 were incubated with a neutralizing antibody to the αL subunit of LFA-1, or to the αM subunit of Mac-1 (Figure 4b and 4d). Pretreatment of Mono Mac 6 with a blocking antibody targeting αL (LFA-1) did not significantly alter recruitment in response to HLA I mIgG2a, at 903±17 per field. In contrast, inhibition of αM (Mac-1) on Mono Mac 6 significantly impaired the monocytes' ability to adhere to HLA I mIgG2a-activated endothelial cells, reducing the number of adherent monocytes to a mean 509±21 per field (Figure 4b). Neutralizing antibody to αL (LFA-1) had limited inhibitory effect, reducing adherence by 25.0±8.4% (p>0.01), while blockade of αM (Mac-1) abolished the adhesive capacity of Mono Mac 6, by 78.1±19.7% compared to monocytes without inhibitor (Figure 4b and 4d). This suggests that Mac-1 integrin is the dominant molecule mediating firm adhesion of monocytes to endothelium activated with HLA I antibodies, despite much higher expression of LFA-1. Similar results were obtained when THP-1 monocyte adherence was assessed (unpublished observations).
Human HLA I allele specific IgG1 triggers recruitment of peripheral blood monocytes through P-selectin, ICAM-1 and FcγRs
We next tested the effect of human HLA I antibodies on recruitment of peripheral blood-monocytes. We predicted that human monoclonal HLA I allele specific antibodies derived from sensitized volunteers would trigger endothelial-monocyte interactions by similar mechanisms as above. Only human IgG1 HLA I antibodies were available (37). We tested the reactivity of monoclonal human IgG1 recognizing HLA-A2 (clone SN230G6) and anti-HLA-A3 (clone MUL2C6) against HAEC with known HLA I genotypes (3F1153: A*02 A*68; H126: A*03 A*29), and confirmed that each HLA I antibody bound to endothelial cells expressing its antigen (data not shown).
HLA-typed HAEC were stimulated with relevant allele-specific human HLA-A antibodies. Binding of human HLA-A3 specific IgG1 on HLA-A*03 positive endothelial cells significantly increased adherence of PBMC-monocytes in a dose-dependent manner (0.01-1μg/mL), with a maximal effect at 1μg/mL (Figure 5a). Similar results were observed when HLA-A*02-positive HAEC were treated with anti-HLA-A2 hIgG1. In contrast, stimulation of HLA-A*03-positive HAEC with irrelevant anti-HLA-A2 hIgG1 did not significantly increase adhesion of peripheral blood monocytes.
Figure 5.

Activation of endothelial cells with human anti-HLA I IgG1 triggers adherence of peripheral blood monocytes via FcγRI, P-selectin and ICAM-1.
a. HAEC were stimulated with human mAb HLA I IgG1 at varying doses for 30min. Top panel, HLA-A*03 expressing endothelial cells (H126) were treated with anti-HLA-A3 hIgG1 (black bars) or irrelevant anti-HLA-A2 hIgG1 (grey bar). Bottom panel, HLA-A*02 expressing HAEC (3F1153) were treated with anti-HLA-A2 hIgG1. Measurement of peripheral blood CD14+ monocyte adherence was performed as above.
b. HLA-A*03+ HAEC were treated with anti-HLA-A3 hIgG1 at 1μg/mL as in (a). Peripheral blood monocytes were left untreated or preincubated with neutralizing antibody to FcγRs. Results are shown as mean number of adherent monocytes per field ± SEM from one representative experiment out of three.
c. HLA-A*03+ HAEC were treated as in (a), with or without indicated neutralizing antibodies at 10μg/mL. Results are from one representative experiment out of three.
d. Peripheral blood monocyte adherence was measured as above with or without neutralizing antibody to integrin β2 and rPSGL-1 antagonist.
* P≤0.01, **** P≤0.0001 versus untreated. ns P>0.05, # P≤0.05, ‡ P≤0.001 versus no inhibitor.
Given that the Fc region of human IgG1 isotype has a high affinity for human FcγRI, FcγRIIa and FcγRIII, we postulated that human IgG1 may engage monocyte FcγRs during the adhesion cascade. To determine whether stimulation of endothelial cells with HLA I specific hIgG1 resulted in FcγR-dependent monocyte recruitment, individual FcγRs were blocked with neutralizing antibodies. HLA-A3 hIgG1-mediated adhesion of monocytes to HLA-A*03-positive HAEC (H126) was significantly inhibited by antagonism of FcγRI, decreasing adherence from 3.1-fold over untreated to 1.8-fold (a 52% inhibition). Total FcγR blockade reduced monocyte adherence to 2.1-fold, a 50% inhibition. A minor effect was observed with neutralization of FcγRII (2.6-fold, 23% inhibition) and FcγRIII (2.9-fold, 10% inhibition) (Figure 5b). Similar results were obtained with HLA-A2 hIgG1 (Supplemental Figure 4a and 4b). Taken together, these results suggest that donor specific IgG1 predominantly engage FcγRI to promote monocyte-endothelial interactions, and that this interaction contributes to maximal HLA I antibody-triggered adhesion. Thus, antagonism of FcγR interactions with the Fc fragment of donor specific antibody can significantly attenuate the recruitment of monocytes to endothelium by human HLA I IgG1.
To define the adhesion molecules participating in human HLA I antibody-induced recruitment of PBMC-monocytes, we blocked ICAM-1/integrin β2 and PSGL-1/P-selectin during monocyte adherence. HLA-A3 hIgG1 significantly increased the number of monocytes bound to HLA-A*03-positive endothelial cells by more than 3-fold. Antagonism of PSGL-1 reduced monocyte binding by 50.1% (Figure 5c). Firm adhesion molecules were also required, as blockade of monocyte β2 integrins significantly prevented maximal recruitment 57.9% (Figure 5c). Similar results were obtained using HLA-A2 hIgG1 on A*02-expressing HAEC (Supplemental Figure 4c). Combinatorial blockade of P-selectin, FcγRs and integrin β2 fully abolished adhesion of monocytes to HLA-A2 hIgG1-treated HAEC, demonstrating the cooperative effect of these signals (Figure 5d). In conclusion, these experiments demonstrate that human HLA I antibodies can trigger FcγR, P-selectin and ICAM-1 dependent recruitment of peripheral blood monocytes by endothelial cells.
HLA-I Antibodies from Sensitized Transplant Recipients Stimulate binding of Monocytes
Finally, we purified the IgG fraction of sera from highly sensitized transplant recipients and tested the effect of polyclonal HLA-specific human IgG on endothelial cell recruitment of monocytes. Binding of IgG from HLA I antibody positive sera to HAEC was confirmed using cell-based ELISA (data not shown). For subsequent experiments, we selected a concentration of HLA-reactive IgG with similar binding capacity as the unfractionated serum diluted 1:4 on endothelial cells from two different donors.
Adhesion of peripheral blood monocytes to HAEC exposed to polyclonal HLA I reactive IgG or unfractionated sera at varying concentrations was measured. Stimulation of HAEC with unfractionated serum diluted 1:4 significantly increased adhesion of monocytes, as did HLA-specific IgG (0.2mg/mL, 0.4mg/mL or 1.7mg/mL) in a dose dependent manner (Figure 6a) compared with IgG from a negative control serum. Antagonism of FcγRs or FcγRI alone significantly attenuated monocyte binding by 52.3% and 46.6% (Figure 6b). Moreover, inhibition of P-selectin, ICAM-1, or β2 integrin or reduced monocyte binding by 46.6%, 69.5%, and 82.6%, respectively (Figure 6b). Finally, combined blockade of both β2 integrin and P-selectin fully abolished monocyte binding to background levels (Figure 6c).
Figure 6.

Anti-HLA antibodies from sensitized transplant recipients stimulates endothelial recruitment of peripheral blood monocytes.
a. HAEC (H126) were stimulated with unfractionated anti-HLA I-containing serum (diluted 1:4), the IgG fraction from anti-HLA I serum, or with the IgG fraction of a negative serum at the indicated concentrations for 30min. Adherence of peripheral blood monocytes was measured. Results are representative of two independent experiments with monocytes from twodifferent donors.
b/c HAEC (CAR) were stimulated with the IgG fraction of anti-HLA I positive serum at 1.7mg/mL. Inhibitors were added individually (b) or in combination (c) as above and adherence of peripheral blood monocytes was measured.
**** P≤0.0001 versus untreated. # P≤0.05, † P≤0.01 ‡ P≤0.001 versus no inhibitor.
Discussion
Taken together, our findings point to a complex model of monocyte-endothelial cell interactions during antibody-mediated rejection. We propose that crosslinking of HLA I is a universal function of HLA I antibodies irrespective of subclass (including human IgG2 and IgG4), and is sufficient to increase P-selectin and support an intermediate level of monocyte tethering through PSGL-1 (Figure 7a). Complement fixing subclasses of donor specific antibody such as human IgG1 or IgG3 can concurrently engage FcγRI (and to a lesser extent FcγRIIa) to amplify P-selectin-mediated adhesion of monocytes, intensifying infiltration during AMR (Figure 7b). We speculate that both P-selectin-PSGL-1 and antibody-FcγR interactions promote firm adhesion to ICAM-1 by activating monocyte Mac-1 integrin.
Figure 7.

Proposed model of monocyte recruitment during HLA I antibody-mediated rejection.
a. Human HLA I antibodies of subclasses with low affinity for human FcγRs include IgG2 and hIgG4. Crosslinking of HLA I on endothelial cells by IgG2 and IgG4 triggers endothelial cell exocytosis leading to increased P-selectin at the cell surface. Monocytes are recruited through tethering via P-selectin/PSGL-1, which promotes firm adherence throughICAM-1/Mac-1.
b. Human HLA I antibodies with high affinity for human FcγRs include IgG1 and IgG3. Antibodies that interact with FcγRs promote monocyte recruitment both through P-selectin as in (a), and through FcγRI- and FcγRIIa-dependent pathways. Engagement of PSGL-1 and FcγRs cooperatively augments firm adherence to ICAM-1.
Although macrophage infiltration frequently accompanies antibody-mediated rejection and intragraft macrophages are associated with poor outcome, the mechanisms by which HLA I antibodies promote mononuclear cell recruitment to the graft are not fully understood. We report that HLA I specific antibodies can trigger monocyte adherence through two parallel mechanisms. The antigen recognition function is mediated by the variable region, and, given its bivalent structure, IgG can dimerize (crosslink) its target. Our laboratory has demonstrated that intracellular signaling induced by HLA I antibodies is dependent upon this crosslinking ability (60). HLA I crosslinking by antibodies of different subclasses increased P-selectin on the cell surface of endothelium, a response that was not observed with non-HLA antibodies or third party human HLA I antibodies. Secondly, the degree to which HLA I antibody stimulated monocyte adherence varied depending on the immunoglobulin subclass, due to the capacity to interact with FcγRs on the monocyte. Antibodies against HLA I appear to have a unique capacity to trigger endothelial cell activation in addition to the canonical Fc fragment-mediated effector functions, derived from the signaling capacity of the target HLA I, which we have previously reported to transduce signals controlling proliferation, survival and cytoskeletal remodeling (19, 20, 63, 64).
We demonstrated that P-selectin induction in response to HLA I crosslinking was a common feature of endothelial cells derived from aorta, umbilical vein and dermal microvasculature, although responsiveness varied among cell types. P-selectin is stored in Weibel-Palade bodies as a rapidly releasable pool, which appears at the membrane intralumenally. Since the first description of von Willebrand Factor (vWF) localization to Weibel-Palade bodies (WPb) (65), endothelial cell biologists have determined that the density and content of WPb varies among the vascular beds and tissues (66, 67), suggesting that endothelia have unequal propensity to recruit leukocytes. The various vascular beds are also differentially susceptible to antibody-mediated rejection and transplant vasculopathy. Alloantibody deposition occurs most prominently the cardiac microvasculature, where intravascular macrophages are also commonly observed (68). In renal AMR, complement deposition affects the arteries or the peritubular capillaries. Veins are rarely affected in chronic allograft rejection, while the arterial vasculature and microvasculature undergo proliferative and fibrotic changes in renal and cardiac allografts. Our results point to a central role for HLA I-induced P-selectin across vascular beds, and suggest that a therapeutic P-selectin antagonist, such as rPSGL-1, may be clinically useful in ameliorating macrophage accumulation in the allograft during AMR.
The fusion protein rPSGL-1 effectively attenuated adhesion of monocytes to aortic, venous and microvascular endothelial cells activated by murine or human HLA I antibodies. In vivo P-selectin is bound by and expressed by platelets, which bridge the monocyte and endothelium to support shear-resistant adherence (69, 70). HLA I antibody directly activates platelet exocytosis, promoting platelet-monocyte aggregates (71). Under physiological conditions, activated platelets are likely to play an important role in antibody-mediated leukocyte recruitment to the graft. von Willebrand Factor (vWF), which is present in Weibel-Palade bodies and released during HLA I antibody-triggered exocytosis (21), has been identified as an adhesive substrate for neutrophils, through both PSGL-1 and the β2 integrin Mac-1 (72, 73). These considerations highlight the potential strength of using rPSGL-1-Fc chimera as a therapeutic agent, since P-selectin is a critical initiator of the leukocyte recruitment cascade and PSGL-1 responds to a variety of ligands.
HLA I antibodies were capable of both promoting P-selectin-induced recruitment and enhancing this recruitment via FcγR engagement, pointing to a unique model of HLA I antibody pathogenesis. In other models of inflammation, anti-endothelial cell antibody or immune complexes can cooperate with chemokines or selectins to augment recruitment of neutrophils by endothelial cells (74-76). Notably, antibodies alone cannot support leukocyte capture on endothelial cells without concurrent expression of selectins in vitro (28, 29) or in vivo (75, 77), consistent with our finding that non-HLA antibodies did not increase adherence of monocytes. Florey et al. proposed that the amplification of recruitment by endothelial-bound antibodies through FcγR-dependent mechanisms may cause inappropriately extensive leukocyte influx relative to the actual level of endothelial activation (28). In transplantation, the intragraft milieu may be altered by ischemia/reperfusion, infection or alloresponses, and endothelial cells may have a high background level of inflammation. Our data suggest that FcγR engagement by HLA class I antibodies can augment signaling events that increase firm adhesion of monocytes to ICAM-1, as has been shown for immune complexes (30, 78-80). Due to FcγR-mediated enhancement, anti-donor HLA I antibodies or MICA antibodies, are likely to magnify monocyte recruitment above what would otherwise occur in response to cytokine activation of graft endothelium. Therefore, selectin tethering to PSGL-1 may synergize with FcγR engagement to produce a highly adhesive conformation of monocyte integrins.
We found that FcγRI prominently participated in recruitment by certain subclasses of HLA I antibodies. While FcγRIIa is extensively distributed on lymphoid and myeloid cells, FcγRI expression is generally restricted to monocytes and activated neutrophils. Moreover, despite several fold higher expression of LFA-1 compared with Mac-1 on monocytes, Mono Mac 6 and THP-1 almost exclusively utilized Mac-1 to adhere to HLA I antibody-stimulated endothelial cells. Finally, there is a well-studied signaling cross-talk between Mac-1 and FcγRs, particularly with FcγRI (72, 81, 82). For these reasons, monocytes may be more responsive to HLA antibody bound on the endothelium compared with other myeloid or lymphoid cells.
Recipient polymorphisms in FcγR may influence the extent to which FcγR-bearing cells are recruited. While there are no known functional mutations in FcγRI, FcγRIIa (activating) exists in two major allotypes in the human population. FcγRIIa-H131 has very low affinity for the Fc portion of murine IgG1 but is the only FcγR which can bind human IgG2. In contrast, FcγRIIa-R131 has increased affinity for murine IgG1 but a lower overall affinity for human IgG (57, 83), and is associated with increased susceptibility to autoimmune diseases and bacterial infection (84, 85). THP-1 carry both H131 and R131 alleles (86), while Mono Mac 6 are homozygous for H131. We noted a difference between THP-1 and Mono Mac 6 in FcγR-dependent recruitment, as well as some variation among healthy volunteer donors of the peripheral blood monocytes, possibly attributed to FcγRIIa polymorphism, suggesting that FcγRIIa polymorphism may influence cellular infiltration during antibody-mediated rejection.
Our data indicate that the subclass of a donor specific HLA I antibody and the FcγR(s) it engages shape the degree of infiltration of FcγR-bearing immune cells by endothelial cells. Donor specific HLA antibodies found in transplant patients can be of any of the four IgG subclasses, although IgG1 dominates (87). Recent advances in antibody detection technologies can reveal the subclass or complement fixing capacity of donor specific antibodies (88, 89), but raise the question of whether subclass is clinically significant to rejection risk and graft outcome. For example, complement fixing donor specific antibodies predicted acute rejection episodes but not long-term outcome (42, 43, 90). In a murine model of antibody-mediated rejection, both complement fixing and non-complement fixing antibodies could cause chronic rejection lesions in transplanted allografts, indicating that MHC I antibodies can promote proliferative changes in a complement-independent manner (91, 92), as observed in vitro (19). Our group and others reported that administration of an F(ab′)2 fragment of donor specific MHC I antibody increased neutrophil and macrophage infiltration in vivo (21, 64) and in vitro (22). Thus donor specific HLA antibodies may have a dual capacity to cause graft injury, through both Fc-dependent and Fc-independent mechanisms.
We used monoclonal human IgG1 against two common HLA-A alleles and demonstrated increased P-selectin- and FcγR-dependent monocyte recruitment. However, we were unable to compare human subclasses as HLA I monoclonal IgG2 and IgG4 isotypes, which have low-to-no interaction with FcγRs, are not available. Hence there is a need to generate and study human or humanized HLA I monoclonals of multiple subclasses to assess their influence on adherence in a system that more closely mimics physiological conditions.
Our data differ from the findings of Yamakuchi et al., who reported that HLA I antibody-mediated binding of HL60 cells required P-selectin but not ICAM-1 (21). HL60 are less differentiated and more closely resemble neutrophils (93, 94). Mono Mac 6, THP-1 and primary blood monocytes express higher levels of FcγRs, CD14 and β2 integrins, including LFA-1 and Mac-1, than HL60 ((28) and unpublished observations). Thus the effects of FcγRs and β2 integrins may be less evident with neutrophilic cells. Our results thus add to this work, demonstrating that basal recruitment can be enhanced by concurrent engagement of PSGL-1 and FcγRI on monocytes.
In conclusion, we have shown that endothelial cell exocytosis of P-selectin downstream of HLA I crosslinking independently promotes recruitment of monocytes. The findings reiterate the fact that donor specific antibodies have pathogenic potential due to their direct effects on the graft vasculature, which are in part unrelated to their complement fixing or FcγR engaging capacity. Therefore selectin inhibition may alleviate monocyte trafficking into the allograft in response to donor specific HLA I antibodies, irrespective of subclass. Moreover, concurrent FcγR engagement by complement fixing donor specific antibodies potentiates selectin-dependent monocyte adherence. This evidence points to an increased pathogenic potential of donor specific antibodies of subclasses with high affinity for human FcγRs.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Frans Claas (Leiden University Medical Center, Leiden, The Netherlands) for his contributions to generating the human monoclonal antibodies. We thank Judith Berliner (Department of Pathology and Laboratory Medicine, University of California, Los Angeles) for providing monocytic cell lines, as well as for sharing her expertise and technical recommendations.
Footnotes
This work was supported by the NIH grant U01AI077821 and 5R01HL090995 (E.F.R.), NIH/NIAID AI042819-10A2 (E.F.R.); UCLA Dissertatation Year Fellowship (N.M.V.), and NRSA Vascular Biology Training Grant 5 T32 HL69766-10 (N.M.V.).
References
- 1.Amico P, Honger G, Mayr M, Steiger J, Hopfer H, Schaub S. Clinical relevance of pretransplant donor-specific HLA antibodies detected by single-antigen flow-beads. Transplantation. 2009;87:1681–1688. doi: 10.1097/TP.0b013e3181a5e034. [DOI] [PubMed] [Google Scholar]
- 2.Willicombe M, Brookes P, Santos-Nunez E, Galliford J, Ballow A, Mclean A, Roufosse C, Cook HT, Dorling A, Warrens AN, Cairns T, Taube D. Outcome of Patients with Preformed Donor-Specific Antibodies Following Alemtuzumab Induction and Tacrolimus Monotherapy. Am J Transplant. 2011;11:470–477. doi: 10.1111/j.1600-6143.2010.03421.x. [DOI] [PubMed] [Google Scholar]
- 3.Loupy A, Cazes A, Guillemain R, Amrein C, Hedjoudje A, Tible M, Pezzella V, Fabiani JN, Suberbielle C, Nochy D, Hill GS, Empana JP, Jouven X, Bruneval P, Duong Van Huyen JP. Very late heart transplant rejection is associated with microvascular injury, complement deposition and progression to cardiac allograft vasculopathy. Am J Transplant. 2011;11:1478–1487. doi: 10.1111/j.1600-6143.2011.03563.x. [DOI] [PubMed] [Google Scholar]
- 4.Michaels PJ, Espejo ML, Kobashigawa J, Alejos JC, Burch C, Takemoto S, Reed EF, Fishbein MC. Humoral rejection in cardiac transplantation: risk factors, hemodynamic consequences and relationship to transplant coronary artery disease. J Heart Lung Transplant. 2003;22:58–69. doi: 10.1016/s1053-2498(02)00472-2. [DOI] [PubMed] [Google Scholar]
- 5.Fishbein MC, Kobashigawa J. Biopsy-negative cardiac transplant rejection: etiology, diagnosis, and therapy. Curr Opin Cardiol. 2004;19:166–169. doi: 10.1097/00001573-200403000-00018. [DOI] [PubMed] [Google Scholar]
- 6.Girlanda R, Kleiner DE, Duan Z, Ford EA, Wright EC, Mannon RB, Kirk AD. Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am J Transplant. 2008;8:600–607. doi: 10.1111/j.1600-6143.2007.02109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pilmore HL, Painter DM, Bishop GA, McCaughan GW, Eris JM. Early up-regulation of macrophages and myofibroblasts: a new marker for development of chronic renal allograft rejection. Transplantation. 2000;69:2658–2662. doi: 10.1097/00007890-200006270-00028. [DOI] [PubMed] [Google Scholar]
- 8.Tinckam KJ, Djurdjev O, Magil AB. Glomerular monocytes predict worse outcomes after acute renal allograft rejection independent of C4d status. Kidney Int. 2005;68:1866–1874. doi: 10.1111/j.1523-1755.2005.00606.x. [DOI] [PubMed] [Google Scholar]
- 9.Magil AB, Tinckam K. Monocytes and peritubular capillary C4d deposition in acute renal allograft rejection. Kidney Int. 2003;63:1888–1893. doi: 10.1046/j.1523-1755.2003.00921.x. [DOI] [PubMed] [Google Scholar]
- 10.Magil AB. Monocytes/macrophages in renal allograft rejection. Transplant Rev (Orlando) 2009;23:199–208. doi: 10.1016/j.trre.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Wyburn KR, Jose MD, Wu H, Atkins RC, Chadban SJ. The role of macrophages in allograft rejection. Transplantation. 2005;80:1641–1647. doi: 10.1097/01.tp.0000173903.26886.20. [DOI] [PubMed] [Google Scholar]
- 12.Chomette G, Auriol M, Cabrol C. Chronic rejection in human heart transplantation. J Heart Transplant. 1988;7:292–297. [PubMed] [Google Scholar]
- 13.Cuda JD, Baldwin WM, 3rd, Steenbergen C, Judge DP, Dropulic LK, Halushka MK. Extensive cardiac allograft vasculitis and concurrent fat necrosis 6 years after orthotopic heart transplantation. J Heart Lung Transplant. 2007;26:1212–1216. doi: 10.1016/j.healun.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 14.Kitchens WH, Chase CM, Uehara S, Cornell LD, Colvin RB, Russell PS, Madsen JC. Macrophage depletion suppresses cardiac allograft vasculopathy in mice. Am J Transplant. 2007;7:2675–2682. doi: 10.1111/j.1600-6143.2007.01997.x. [DOI] [PubMed] [Google Scholar]
- 15.Bruneau S, Woda CB, Daly KP, Boneschansker L, Jain NG, Kochupurakkal N, Contreras AG, Seto T, Briscoe DM. Key Features of the Intragraft Microenvironment that Determine Long-Term Survival Following Transplantation. Front Immunol. 2012;3:54. doi: 10.3389/fimmu.2012.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sis B, Grynoch R, Murray AG, Campbell P, Solez K. Antibody-mediated rejection with a striking interstitial monocyte/macrophage infiltration in a renal allograft under FTY720 treatment. Am J Kidney Dis. 2008;51:127–130. doi: 10.1053/j.ajkd.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 17.Li F, Zhang X, Jin YP, Mulder A, Reed EF. Antibody ligation of human leukocyte antigen class I molecules stimulates migration and proliferation of smooth muscle cells in a focal adhesion kinase-dependent manner. Hum Immunol. 2011;72:1150–1159. doi: 10.1016/j.humimm.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jindra PT, Zhang X, Mulder A, Claas F, Veale J, Jin YP, Reed EF. Anti-HLA antibodies can induce endothelial cell survival or proliferation depending on their concentration. Transplantation. 2006;82:S33–35. doi: 10.1097/01.tp.0000231447.34240.3c. [DOI] [PubMed] [Google Scholar]
- 19.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, Reed EF. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin, and focal adhesion kinase in an actin-dependent manner. J Immunol. 2002;168:5415–5423. doi: 10.4049/jimmunol.168.11.5415. [DOI] [PubMed] [Google Scholar]
- 20.Jin YP, Korin Y, Zhang X, Jindra PT, Rozengurt E, Reed EF. RNA interference elucidates the role of focal adhesion kinase in HLA class I-mediated focal adhesion complex formation and proliferation in human endothelial cells. J Immunol. 2007;178:7911–7922. doi: 10.4049/jimmunol.178.12.7911. [DOI] [PubMed] [Google Scholar]
- 21.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, Wasowska BA, Baldwin WM, 3rd, Pober JS, Lowenstein CJ. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A. 2007;104:1301–1306. doi: 10.1073/pnas.0602035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valenzuela NM, Hong L, Shen XD, Gao F, Young SH, Rozengurt E, Weglinski JK, Fishbein MC, Reed EF. Blockade of P-Selectin Is Sufficient to Reduce MHC I Antibody-Elicited Monocyte Recruitment In Vitro and In Vivo. Am J Transplant. 2013;13:299–311. doi: 10.1111/ajt.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zarbock A, Muller H, Kuwano Y, Ley K. PSGL-1-dependent myeloid leukocyte activation. J Leukoc Biol. 2009;86:1119–1124. doi: 10.1189/jlb.0209117. [DOI] [PubMed] [Google Scholar]
- 24.Margadant C, Monsuur HN, Norman JC, Sonnenberg A. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol. 2011;23:607–614. doi: 10.1016/j.ceb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 26.Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Florey OJ, Johns M, Esho OO, Mason JC, Haskard DO. Antiendothelial cell antibodies mediate enhanced leukocyte adhesion to cytokine-activated endothelial cells through a novel mechanism requiring cooperation between Fc{gamma}RIIa and CXCR1/2. Blood. 2007;109:3881–3889. doi: 10.1182/blood-2006-08-044669. [DOI] [PubMed] [Google Scholar]
- 29.Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G, Luscinskas FW, Mayadas TN. Fc gamma RIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 2001;14:693–704. doi: 10.1016/s1074-7613(01)00150-9. [DOI] [PubMed] [Google Scholar]
- 30.Kocher M, Siegel ME, Edberg JC, Kimberly RP. Cross-linking of Fc gamma receptor IIa and Fc gamma receptor IIIb induces different proadhesive phenotypes on human neutrophils. J Immunol. 1997;159:3940–3948. [PubMed] [Google Scholar]
- 31.Tax WJ, Hermes FF, Willems RW, Capel PJ, Koene RA. Fc receptors for mouse IgG1 on human monocytes: polymorphism and role in antibody-induced T cell proliferation. J Immunol. 1984;133:1185–1189. [PubMed] [Google Scholar]
- 32.Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25:1369–1372. doi: 10.1038/nbt1207-1369. [DOI] [PubMed] [Google Scholar]
- 33.Hamaguchi Y. Molecular mechanisms of B lymphocyte depletion by CD20 immunotherapy. Nihon Rinsho Meneki Gakkai Kaishi. 2009;32:29–34. doi: 10.2177/jsci.32.29. [DOI] [PubMed] [Google Scholar]
- 34.Saylor CA, Dadachova E, Casadevall A. Murine IgG1 and IgG3 isotype switch variants promote phagocytosis of Cryptococcus neoformans through different receptors. J Immunol. 2010;184:336–343. doi: 10.4049/jimmunol.0902752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song X, Shapiro S, Goldman DL, Casadevall A, Scharff M, Lee SC. Fcgamma receptor I- and III-mediated macrophage inflammatory protein 1alpha induction in primary human and murine microglia. Infect Immun. 2002;70:5177–5184. doi: 10.1128/IAI.70.9.5177-5184.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran TM, Ivanyi P, Hilgert I, Brdicka T, Pla M, Breur B, Flieger M, Ivaskova E, Horejsi V. The epitope recognized by pan-HLA class I-reactive monoclonal antibody W6/32 and its relationship to unusual stability of the HLA-B27/beta2-microglobulin complex. Immunogenetics. 2001;53:440–446. doi: 10.1007/s002510100353. [DOI] [PubMed] [Google Scholar]
- 37.Mulder A, Kardol MJ, Arn JS, Eijsink C, Franke ME, Schreuder GM, Haasnoot GW, Doxiadis II, Sachs DH, Smith DM, Claas FH. Human monoclonal HLA antibodies reveal interspecies crossreactive swine MHC class I epitopes relevant for xenotransplantation. Mol Immunol. 2010;47:809–815. doi: 10.1016/j.molimm.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Zoet YM, Eijsink C, Kardol MJ, Franke-van Dijk MEI, Wilson GL, de Paus R, Mickelson E, Heemskerk M, van den Elsen PJ, Claas FHJ, Mulder A, Doxiadis IIN. The Single Antigen expressing Lines (SALs) Concept: An Excellent Tool for Screening for HLA-Specific Antibodies. Human Immunology. 2005;66:519–525. doi: 10.1016/j.humimm.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 39.Erl W, Weber C, Wardemann C, Weber PC. Adhesion properties of Mono Mac 6, a monocytic cell line with characteristics of mature human monocytes. Atherosclerosis. 1995;113:99–107. doi: 10.1016/0021-9150(94)05434-k. [DOI] [PubMed] [Google Scholar]
- 40.Erl W, Weber PC, Weber C. Monocytic cell adhesion to endothelial cells stimulated by oxidized low density lipoprotein is mediated by distinct endothelial ligands. Atherosclerosis. 1998;136:297–303. doi: 10.1016/s0021-9150(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 41.Gao Zh, McAlister VC, Wright JR, McAlister CC, Peltekian K, MacDonald AS. Immunoglobulin-G subclass antidonor reactivity in transplant recipients. Liver Transpl. 2004;10:1055–1059. doi: 10.1002/lt.20154. [DOI] [PubMed] [Google Scholar]
- 42.Griffiths EJ, Nelson RE, Dupont PJ, Warrens AN. Skewing of pretransplant anti-HLA class I antibodies of immunoglobulin G isotype solely toward immunoglobulin G1 subclass is associated with poorer renal allograft survival. Transplantation. 2004;77:1771–1773. doi: 10.1097/01.tp.0000129408.07168.40. [DOI] [PubMed] [Google Scholar]
- 43.Honger G, Hopfer H, Arnold ML, Spriewald BM, Schaub S, Amico P. Pretransplant IgG subclasses of donor-specific human leukocyte antigen antibodies and development of antibody-mediated rejection. Transplantation. 2011;92:41–47. doi: 10.1097/TP.0b013e31821cdf0d. [DOI] [PubMed] [Google Scholar]
- 44.Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. 2005;115:2914–2923. doi: 10.1172/JCI24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tse WY, Abadeh S, McTiernan A, Jefferis R, Savage CO, Adu D. No association between neutrophil FcgammaRIIa allelic polymorphism and anti-neutrophil cytoplasmic antibody (ANCA)-positive systemic vasculitis. Clin Exp Immunol. 1999;117:198–205. doi: 10.1046/j.1365-2249.1999.00960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warmerdam PA, van de Winkel JG, Gosselin EJ, Capel PJ. Molecular basis for a polymorphism of human Fc gamma receptor II (CD32) The Journal of Experimental Medicine. 1990;172:19–25. doi: 10.1084/jem.172.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, Eddy SM, Ward PA. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. 1994;94:1147–1155. doi: 10.1172/JCI117430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas JM, Chakraborty A, Sharp MK, Berson RE. Spatial and temporal resolution of shear in an orbiting petri dish. Biotechnol Prog. 2011;27:460–465. doi: 10.1002/btpr.507. [DOI] [PubMed] [Google Scholar]
- 50.Dardik A, Chen L, Frattini J, Asada H, Aziz F, Kudo FA, Sumpio BE. Differential effects of orbital and laminar shear stress on endothelial cells. J Vasc Surg. 2005;41:869–880. doi: 10.1016/j.jvs.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 51.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. Journal of Leukocyte Biology. 2007;81:584–592. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 52.van de Winkel JG, Tax WJ, Jacobs CW, Huizinga TW, Willems PH. Cross-linking of both types of IgG Fc receptors, Fc gamma RI and Fc gamma RII, enhances intracellular free Ca2+ in the monocytic cell line U937. Scand J Immunol. 1990;31:315–325. doi: 10.1111/j.1365-3083.1990.tb02774.x. [DOI] [PubMed] [Google Scholar]
- 53.Van de Winkel JG, Tax WJ, Van Bruggen MC, Van Roozendaal CE, Willems HW, Vlug A, Capel PJ, Koene RA. Characterization of two Fc receptors for mouse immunoglobulins on human monocytes and cell lines. Scand J Immunol. 1987;26:663–672. doi: 10.1111/j.1365-3083.1987.tb02302.x. [DOI] [PubMed] [Google Scholar]
- 54.van der Poel CE, Karssemeijer RA, Boross P, van der Linden JA, Blokland M, van de Winkel JG, Leusen JH. Cytokine-induced immune complex binding to the high-affinity IgG receptor, FcgammaRI, in the presence of monomeric IgG. Blood. 2010;116:5327–5333. doi: 10.1182/blood-2010-04-280214. [DOI] [PubMed] [Google Scholar]
- 55.Wang R, Stephens J, Lacy MJ. Characterization of monoclonal antibody HTA125 with specificity for human TLR4. Hybrid Hybridomics. 2003;22:357–365. doi: 10.1089/153685903771797057. [DOI] [PubMed] [Google Scholar]
- 56.Gosselin EJ, Brown MF, Anderson CL, Zipf TF, Guyre PM. The monoclonal antibody 41H16 detects the Leu 4 responder form of human Fc gamma RII. J Immunol. 1990;144:1817–1822. [PubMed] [Google Scholar]
- 57.Parren PW, Warmerdam PA, Boeije LC, Arts J, Westerdaal NA, Vlug A, Capel PJ, Aarden LA, van de Winkel JG. On the interaction of IgG subclasses with the low affinity Fc gamma RIIa (CD32) on human monocytes, neutrophils, and platelets. Analysis of a functional polymorphism to human IgG2. The Journal of Clinical Investigation. 1992;90:1537–1546. doi: 10.1172/JCI116022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones DH, Looney RJ, Anderson CL. Two distinct classes of IgG Fc receptors on a human monocyte line (U937) defined by differences in binding of murine IgG subclasses at low ionic strength. J Immunol. 1985;135:3348–3353. [PubMed] [Google Scholar]
- 59.Burns AR, Bowden RA, Abe Y, Walker DC, Simon SI, Entman ML, Smith CW. P-selectin mediates neutrophil adhesion to endothelial cell borders. J Leukoc Biol. 1999;65:299–306. doi: 10.1002/jlb.65.3.299. [DOI] [PubMed] [Google Scholar]
- 60.Bian H, Reed EF. Anti-HLA class I antibodies transduce signals in endothelial cells resulting in FGF receptor translocation, down-regulation of ICAM-1 and cell proliferation. Transplantation Proceedings. 2001;33:311. doi: 10.1016/s0041-1345(00)02022-4. [DOI] [PubMed] [Google Scholar]
- 61.Panes J, Perry MA, Anderson DC, Manning A, Leone B, Cepinskas G, Rosenbloom CL, Miyasaka M, Kvietys PR, Granger DN. Regional differences in constitutive and induced ICAM-1 expression in vivo. American Journal of Physiology - Heart and Circulatory Physiology. 1995;269:H1955–H1964. doi: 10.1152/ajpheart.1995.269.6.H1955. [DOI] [PubMed] [Google Scholar]
- 62.Bian H, Harris PE, Reed EF. Ligation of HLA class I molecules on smooth muscle cells with anti-HLA antibodies induces tyrosine phosphorylation, fibroblast growth factor receptor expression and cell proliferation. International Immunology. 1998;10:1315–1323. doi: 10.1093/intimm/10.9.1315. [DOI] [PubMed] [Google Scholar]
- 63.Jin YP, Jindra PT, Gong KW, Lepin EJ, Reed EF. Anti-HLA class I antibodies activate endothelial cells and promote chronic rejection. Transplantation. 2005;79:S19–21. doi: 10.1097/01.tp.0000153293.39132.44. [DOI] [PubMed] [Google Scholar]
- 64.Jindra PT, Hsueh A, Hong L, Gjertson D, Shen XD, Gao F, Dang J, Mischel PS, Baldwin WM, 3rd, Fishbein MC, Kupiec-Weglinski JW, Reed EF. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol. 2008;180:2214–2224. doi: 10.4049/jimmunol.180.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wagner DD, Olmsted JB, Marder VJ. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J Cell Biol. 1982;95:355–360. doi: 10.1083/jcb.95.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamamoto K, de Waard V, Fearns C, Loskutoff DJ. Tissue distribution and regulation of murine von Willebrand factor gene expression in vivo. Blood. 1998;92:2791–2801. [PubMed] [Google Scholar]
- 67.Oynebraten I, Bakke O, Brandtzaeg P, Johansen FE, Haraldsen G. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood. 2004;104:314–320. doi: 10.1182/blood-2003-08-2891. [DOI] [PubMed] [Google Scholar]
- 68.Fishbein GA, Fishbein MC. Morphologic and immunohistochemical findings in antibody-mediated rejection of the cardiac allograft. Hum Immunol. 2012 doi: 10.1016/j.humimm.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 69.Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, Wagner DD, Furie B. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59:305–312. doi: 10.1016/0092-8674(89)90292-4. [DOI] [PubMed] [Google Scholar]
- 70.Morrell CN, Murata K, Swaim AM, Mason E, Martin TV, Thompson LE, Ballard M, Fox-Talbot K, Wasowska B, Baldwin WM., 3rd In vivo platelet-endothelial cell interactions in response to major histocompatibility complex alloantibody. Circ Res. 2008;102:777–785. doi: 10.1161/CIRCRESAHA.107.170332. [DOI] [PubMed] [Google Scholar]
- 71.Takahashi D, Fujihara M, Azuma H, Wakamoto S, Sato S, Kato T, Ikeda H. Adhesive interaction between peripheral blood mononuclear cells and activated platelets in the presence of anti-human leukocyte antigen Class I alloantibody causes production of IL-1beta and IL-8. Vox Sang. 2012;102:250–257. doi: 10.1111/j.1423-0410.2011.01549.x. [DOI] [PubMed] [Google Scholar]
- 72.Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, Cotran RS, Mayadas TN. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186:1853–1863. doi: 10.1084/jem.186.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pendu R, Terraube V, Christophe OD, Gahmberg CG, de Groot PG, Lenting PJ, Denis CV. P-selectin glycoprotein ligand 1 and beta2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108:3746–3752. doi: 10.1182/blood-2006-03-010322. [DOI] [PubMed] [Google Scholar]
- 74.Tsuboi N, Asano K, Lauterbach M, Mayadas TN. Human neutrophil Fcgamma receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. 2008;28:833–846. doi: 10.1016/j.immuni.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsuboi N, Ernandez T, Li X, Nishi H, Cullere X, Mekala D, Hazen M, Kohl J, Lee DM, Mayadas TN. Regulation of human neutrophil Fcgamma receptor IIa by C5a receptor promotes inflammatory arthritis in mice. Arthritis Rheum. 2011;63:467–478. doi: 10.1002/art.30141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pankhurst T, Nash G, Williams J, Colman R, Hussain A, Savage C. Immunoglobulin subclass determines ability of immunoglobulin (Ig)G to capture and activate neutrophils presented as normal human IgG or disease-associated anti-neutrophil cytoplasm antibody (ANCA)-IgG. Clin Exp Immunol. 2011;164:218–226. doi: 10.1111/j.1365-2249.2011.04367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stokol T, O'Donnell P, Xiao L, Knight S, Stavrakis G, Botto M, von Andrian UH, Mayadas TN. C1q governs deposition of circulating immune complexes and leukocyte Fcgamma receptors mediate subsequent neutrophil recruitment. J Exp Med. 2004;200:835–846. doi: 10.1084/jem.20040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pan XQ, Darby C, Indik ZK, Schreiber AD. Activation of three classes of nonreceptor tyrosine kinases following Fc gamma receptor crosslinking in human monocytes. Clin Immunol. 1999;90:55–64. doi: 10.1006/clim.1998.4644. [DOI] [PubMed] [Google Scholar]
- 79.Smith DF, Galkina E, Ley K, Huo Y. GRO family chemokines are specialized for monocyte arrest from flow. Am J Physiol Heart Circ Physiol. 2005;289:H1976–1984. doi: 10.1152/ajpheart.00153.2005. [DOI] [PubMed] [Google Scholar]
- 80.Jones SL, Knaus UG, Bokoch GM, Brown EJ. Two signaling mechanisms for activation of alphaM beta2 avidity in polymorphonuclear neutrophils. J Biol Chem. 1998;273:10556–10566. doi: 10.1074/jbc.273.17.10556. [DOI] [PubMed] [Google Scholar]
- 81.Ortiz-Stern A, Rosales C. Cross-talk between Fc receptors and integrins. Immunol Lett. 2003;90:137–143. doi: 10.1016/j.imlet.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 82.Xiong Y, Cao C, Makarova A, Hyman B, Zhang L. Mac-1 promotes FcgammaRIIA-dependent cell spreading and migration on immune complexes. Biochemistry. 2006;45:8721–8731. doi: 10.1021/bi060529u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shashidharamurthy R, Zhang F, Amano A, Kamat A, Panchanathan R, Ezekwudo D, Zhu C, Selvaraj P. Dynamics of the interaction of human IgG subtype immune complexes with cells expressing R and H allelic forms of a low-affinity Fc gamma receptor CD32A. J Immunol. 2009;183:8216–8224. doi: 10.4049/jimmunol.0902550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Endeman H, Cornips MC, Grutters JC, van den Bosch JM, Ruven HJ, van Velzen-Blad H, Rijkers GT, Biesma DH. The Fcgamma receptor IIA-R/R131 genotype is associated with severe sepsis in community-acquired pneumonia. Clin Vaccine Immunol. 2009;16:1087–1090. doi: 10.1128/CVI.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Norsworthy P, Theodoridis E, Botto M, Athanassiou P, Beynon H, Gordon C, Isenberg D, Walport MJ, Davies KA. Overrepresentation of the Fcgamma receptor type IIA R131/R131 genotype in caucasoid systemic lupus erythematosus patients with autoantibodies to C1q and glomerulonephritis. Arthritis Rheum. 1999;42:1828–1832. doi: 10.1002/1529-0131(199909)42:9<1828::AID-ANR6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 86.Tebo AE, Kremsner PG, Luty AJ. Fcgamma receptor-mediated phagocytosis of Plasmodium falciparum-infected erythrocytes in vitro. Clin Exp Immunol. 2002;130:300–306. doi: 10.1046/j.1365-2249.2002.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heinemann FM, Roth I, Rebmann V, Arnold ML, Spriewald BM, Grosse-Wilde H. Characterization of anti-HLA antibodies eluted from explanted renal allografts. Clin Transpl. 2006:371–378. [PubMed] [Google Scholar]
- 88.Chen G, Sequeira F, Tyan DB. Novel C1q assay reveals a clinically relevant subset of human leukocyte antigen antibodies independent of immunoglobulin G strength on single antigen beads. Hum Immunol. 2011;72:849–858. doi: 10.1016/j.humimm.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 89.Sutherland SM, Chen G, Sequeira FA, Lou CD, Alexander SR, Tyan DB. Complement-fixing donor-specific antibodies identified by a novel C1q assay are associated with allograft loss. Pediatr Transplant. 2012;16:12–17. doi: 10.1111/j.1399-3046.2011.01599.x. [DOI] [PubMed] [Google Scholar]
- 90.Honger G, Wahrmann M, Amico P, Hopfer H, Bohmig GA, Schaub S. C4d-fixing capability of low-level donor-specific HLA antibodies is not predictive for early antibody-mediated rejection. Transplantation. 2010;89:1471–1475. doi: 10.1097/TP.0b013e3181dc13e7. [DOI] [PubMed] [Google Scholar]
- 91.Hirohashi T, Uehara S, Chase CM, DellaPelle P, Madsen JC, Russell PS, Colvin RB. Complement independent antibody-mediated endarteritis and transplant arteriopathy in mice. Am J Transplant. 2010;10:510–517. doi: 10.1111/j.1600-6143.2009.02958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hirohashi T, Chase CM, Della Pelle P, Sebastian D, Alessandrini A, Madsen JC, Russell PS, Colvin RB. A novel pathway of chronic allograft rejection mediated by NK cells and alloantibody. Am J Transplant. 2012;12:313–321. doi: 10.1111/j.1600-6143.2011.03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fleck RA, Athwal H, Bygraves JA, Hockley DJ, Feavers IM, Stacey GN. Optimization of nb-4 and hl-60 differentiation for use in opsonophagocytosis assays. In Vitro Cell Dev Biol Anim. 2003;39:235–242. doi: 10.1290/1543-706X(2003)039<0235:OONAHD>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 94.Fleck RA, Romero-Steiner S, Nahm MH. Use of HL-60 cell line to measure opsonic capacity of pneumococcal antibodies. Clin Diagn Lab Immunol. 2005;12:19–27. doi: 10.1128/CDLI.12.1.19-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.