

Abstract
This report describes that protein kinase C delta (PKCδ) overexpression prevents TRAIL-induced apoptosis in breast tumor cells; however, the regulatory mechanism(s) involved in this phenomenon is (are) incompletely understood. In this study, we have shown that TRAIL-induced apoptosis was significantly inhibited in PKCδ overexpressing MCF-7 (MCF7/PKCδ) cells. Our data reveal that PKCδ inhibits caspase-8 activation, a first step in TRAIL-induced apoptosis, thus preventing TRAIL-induced apoptosis. Inhibition of PKCδ using rottlerin or PKCδ siRNA reverses the inhibitory effect of PKCδ on caspase-8 activation leading to TRAIL-induced apoptosis. To determine if caspase-3-induced PKCδ cleavage reverses its inhibition on caspase-8, we developed stable cell lines that either expresses wild-type PKCδ (MCF-7/cas-3/PKCδ) or caspase-3 cleavage-resistant PKCδ mutant (MCF-7/cas-3/PKCδ mut) utilizing MCF-7 cells expressing caspase-3. Cells that overexpress caspase-3 cleavage-resistant PKCδ mutant (MCF-7/cas-3/PKCδmut) significantly inhibited TRAIL-induced apoptosis when compared to wild-type PKCδ (MCF-7/cas-3/PKCδ) expressing cells. In MCF-7/cas-3/PKCδmut cells, TRAIL-induced caspase-8 activation was blocked leading to inhibition of apoptosis when compared to wild-type PKCδ (MCF-7/cas-3/PKCδ) expressing cells. Together, these results strongly suggest that overexpression of PKCδ inhibits caspase-8 activation leading to inhibition of TRAIL-induced apoptosis and its inhibition by rottlerin, siRNA, or cleavage by caspase-3 sensitizes cells to TRAIL-induced apoptosis. Clinically, PKCδ overexpressing tumors can be treated with a combination of PKCδ inhibitor(s) and TRAIL as a new treatment strategy.
Keywords: PKC-δ, CASPASE-3, APOPTOSIS, TRAIL, BREAST CANCER
The protein kinase C (PKC) family of serine/threonine kinases regulates many cellular functions including proliferation and survival [Newton, 1995; Mackay and Twelves, 2007]. PKCδ is activated by numerous apoptotic stimuli and is emerging as an important modulator of apoptosis in human cancers [Brodie and Blumberg, 2003; Jackson and Foster, 2004]. Most studies reported a pro-apoptotic function for PKCδ in response to various stimuli, such as H2O2 [Kaul et al., 2005], tumor necrosis factor-α [Emoto et al., 1995], etoposide [Blass et al., 2002], cisplatin [Basu et al., 2001], and UV radiation [Chen et al., 1999]. On the other hand, studies by Humphries et al. [2006] demonstrated that apoptosis was suppressed in irradiated salivary glands in PKCδ-deficient mice, suggesting that PKCδ plays a major role in the regulation of apoptosis both in vitro and in vivo. Different apoptotic stimuli have been shown to induce caspase-dependent cleavage of PKCδ, resulting in the generation of a constitutively active catalytic fragment [Ghayur et al., 1996; D’Costa and Denning, 2005]. The cleavage of PKCδ has been implicated in pro-apoptotic function [Ghayur et al., 1996; DeVries et al., 2002; D’Costa and Denning, 2005; DeVries-Seimon et al., 2007] while PKCδ has been shown to negatively regulate apoptosis. We have shown that PKCδ overexpression contributes to antiestrogen resistance in mammary tumor cells [Nabha et al., 2005], in non-small cell lung cancer cells [Clark et al., 2003] it promotes survival and chemotherapeutic drug resistance. The mechanisms by which PKCδ regulates pro- and anti-apoptosis remain unclear.
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a novel member of TNF family. Ashkenazi and Dixit [1998] demonstrated that TRAIL ligand has selective toxicity on cancer cells, but not normal cells suggesting the potential as a therapeutic agent.
The binding of TRAIL to DR5 and DR4 receptors leads to the formation of a death-inducing signaling complex (DISC) and this rapidly association with the adaptor protein Fas-associated death domain (FADD). This then conjugates with the initiator caspase-8, mediating apoptosis either by direct activation of downstream effectors caspase-3, −6, −7 cascade or by the cleavage of apoptotic molecules, such as Bid and Bcl-xl leading to activation of the caspase-9 complex [Herr and Debatin, 2001]. The caspases function to cleave cellular proteins critical for life, and the cleavage of caspase substrates sets the stage for the morphological and biochemical changes that are hallmarks of apoptosis [Earnshaw et al., 1999].
In this study, we report the role of PKCδ in TRAIL-induced apoptosis using MCF-7 breast cells and analyzed the molecular mechanism(s) that regulates pro- and anti-apoptotic function in breast cells. Our data clearly demonstrate that overexpression PKCδ in MCF-7 cells (MCF-7/PKCδ cells) increases the expression of death receptors DR4 and DR5 in comparison to vector transfected MCF-7 cells but does not increase TRAIL-induced apoptosis. We show that expression of caspase-3 cleavage-resistant PKCδ (MCF-7/cas-3/PKCδmut) protects the MCF-7 cells from TRAIL-induced apoptosis by inhibiting caspase-8 activation in response to TRAIL treatment leading to inhibition of both death receptors (extrinsic pathway) and mitochondria (intrinsic pathway). However, overexpression of wild-type PKCδ (MCF-7/cas-3/PKCδ) leads to cleavage and activation of PKCδ (enhances apoptosis). This cleavage of PKCδ by caspase-3 reverses the PKCδ-induced casapase-8 inhibition and significantly enhances TRAIL-induced apoptosis. Additionally, TRAIL-induced apoptosis can be reversed by blocking caspase-3 suggesting that caspase-3 induced PKCδ cleavage could play a major role in regulating TRAIL-induced apoptosis.
MATERIALS AND METHODS
CELL LINES AND REAGENTS
The human breast carcinoma cell lines MCF-7 and T47D were obtained from the American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% FCS, 10 μg/ml insulin (Sigma), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Gibco). Caspase-3 stably transfected MCF-7 (MCF-7/Cas-3) cells were generously provided by Dr. Xiaohe Yang (University of Oklahoma Health Sciences Center, OK, USA). The plasmids of full-length PKCδ (PKCδ), caspase-3 cleavage-resistant PKCδ D327A mut (PKCδmut), and control vector were generously provided by Dr. C. Brodie (Henry Ford Hospital, MI, USA). DR4/TRAILR1 monoclonal antibody and DR5/TRAILR2 polyclonal antibody was purchased from Imgenex (San Diego, CA). Monoclonal antibodies of PKCδ, PARP, Bid, caspase-8 were purchased from BD Biosciences (San Diego, CA). Akt, phospho-Akt, Caspase-3, and caspase-9 antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-glyceraldehyde-3-phosphate dehydrogenase (G3PDH) rabbit polyclonal was obtained from Trevigen (Gaithersburg, MD). Rottlerin was purchased from Calbiochem (San Diego, CA), TRAIL and caspase-3 inhibitor was purchased from R&D Systems (Minneapolis, MN). Cell Death Detection ELISAPLUS was purchased from Roche (Indianapolis, IN). Reagents for protein concentration analysis and protein gel electrophoresis were obtained from Bio-Rad (Hercules, CA). Mammalian PKCδ siRNA expression plasmid (pKD-PKCδ-v2) and random siRNA (pKD-NegCon-v1) were purchased from Upstate (Temecula, CA). Tunicamycin and all other chemicals, unless otherwise specified, were obtained from Sigma in the highest suitable purities.
ISOLATION OF STABLE TRANSFECTANTS
MCF-7 or MCF-7/cas-3 cells were transfected with PKCδ, PKCδmut, or control vector using Lipofectamine™ 2000 Reagent (Invitrogen). Stable transfectants were selected in the presence of 500 μg/ml Geneticin (G418; Gibco) after 2–3 weeks. Individual antibiotic-resistant colonies were isolated and screened for the expression of the corresponding protein by Western blot analysis using anti-PKCδ antibody (BD Biosciences). The pools made by mix six positive clones were named MCF-7/Vector, MCF-7/PKCδ, MCF-7/Cas-3/Vector, MCF-7/Cas-3/PKCδ, and MCF-7/Cas-3/PKCδmut cell lines were developed and used for experiments. All cell lines were routinely tested for mycoplasma contamination and found to be negative.
IMMUNOHISTOCHEMICAL ANALYSIS (IHC)
The specimens used for this study were collected under an IRB approved protocol (HIC#113003MP4E) such that the specimens could not be linked to the human subject and were considered exempt. Immunohistochemistry (IHC) was performed on tumor tissue sections placed on glass slides using the standard laboratory protocols as previously described [Visscher et al., 1997]. Briefly, after deparaffinizing and hydrating with phosphate-buffered saline (PBS) buffer (pH 7.4), tissue sections were pretreated with hydrogen peroxide (3%) for 10 min to remove endogenous peroxidase, followed by antigen retrieval via steam bath for 20 min in EDTA. A primary antibody was applied, followed by washing and incubation with the biotinylated secondary antibody for 30 min at room temperature. Detection was performed with diaminobenzidine (DAB) and counterstained with Mayer hematoxylin followed by dehydration and mounting. A score of zero indicates no staining relative to background, 1+= weak staining, 2+= moderate staining, and 3+= strong staining. For comparison of staining among tissues, the results were quantified by calculation of a complete H-score which considers both staining intensity and the percentage of cells stained at a specific range of intensities. A complete H-score was calculated by summing the product of the percentage cells stained (0–100%) and staining intensity (0–3) according to Kerfoot et al. [2004]. Statistical analysis of the complete H-scores obtained for the normal tissue and re-occurring breast tumors was carried out by using the two-tailed Student’s t-test with unpaired data of equal variance.
WESTERN IMMUNOBLOT ANALYSIS
MCF-7 and T47D cells were grown in DMEM with 5% or 10% fetal bovine serum, to near confluence in the presence or absence of various treatments. Cells were lysed and Western blotting was performed as described previously [Nabha et al., 2005] using a standard protocol. In brief, cell extracts were obtained by lysing the cells in RIPA buffer (20 mM HEPES, 100 mM NaCl, 0.1% SDS, 1% Nonidet P-40, 1% deoxycholate, 1 mM Na3VO4, 1 mM EGTA, 50 mM NaF, 10% glycerol, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 1× protease inhibitor mixture). Samples containing 100 μg of total protein were resolved on 10% or 15% SDS–polyacrylamide gels and proteins were electrophoretically transferred onto a PVDF membrane. Membranes were probed with antibodies as indicated, followed by HRP-conjugated mouse or rabbit secondary antibodies (Amersham). Anti-G3PDH was used for loading controls.
RNA INTERFERENCE ASSAY
Cells were plated in six-well tissue culture plates, at a density of 3 × 105/well, in DMEM containing 5% FBS. After 24 h, cells were transfected with 4 μg pKD-PKCδ plasmid or pKD-NegCon-v1 (non-specific siRNA with scrambled sequence) plasmid using Lipofecta-mine transfection reagent according to the manufacturer’s instructions. After 72 h of transfection, cells were treated with or without TRAIL for an additional 4 h at 37°C. Cells were harvested and extracts prepared for Western blot analysis.
APOPTOSIS ASSAY
Apoptosis was assessed using the Cell Death Detection ELISAPLUS kit (Roche) according to the manufacturer’s instructions. This kit quantitatively detected cytosolic histone-associated DNA fragments. In brief, 5 × 105 cells were grown in six-well tissue culture plates for 24 h at 37°C in CO2 incubator. After 24 h, cells were pretreated with either rottlerin (3 μM) or caspase-3 inhibitor (10 μm) for 1 h and then TRAIL (10 ng/ml) was added for 4 h. Cells were harvested and apoptosis measured according to the manufacturer’s instructions. Apoptosis was quantified by ELISA and normalized to values measured in untreated cells. Data are mean ± SE of triplicate determinations.
RESULTS
PKCδ IS OVEREXPRESSED IN HUMAN BREAST TUMORS
We examined PKCδ expression in re-occurring human and compared with normal adjacent breast tissue using IHC. Interpretation of slides was performed by microscopic examination by a pathologist. Staining patterns of tumor at various stages of tumor development are shown in Figure 1. Results indicate a strong level of PKCδ expression in ductal carcinoma in situ (DCIS) and a moderate level of expression in invasive breast tissue when compared to surrounding normal breast tissue. The staining intensity of each specimen was judged relative to the intensity of a control slide containing an adjacent normal section. The staining of the section labeled with the negative reagent control was considered background. The summary of PKCδ staining intensity in re-occurring human breast cancers and adjoining normal breast tissue is given in Table I. The data suggest that re-occurring human breast tumors over-express PKCδ compared to surrounding normal breast tissue.
Fig. 1.

PKCδ immunohistochemical staining in breast tissue specimens (40×). A: Normal adjacent breast epithelia shows mild, (B) DCIS breast cancer shows strong staining, and (C,D) invasive breast cancer showed moderate staining.
TABLE I.
Summary of PKCδ Immunohistochemical Staining Intensity in Normal and Cancer Breast Tissue
| Breast tumors | n | Staining intensity
|
Mean complete H-score | |||
|---|---|---|---|---|---|---|
| 1+ | 2+ | 3+ | ||||
| Normal tissue | 6 | 3 | 3 | 0 | 95 | P <0.01* |
| Breast tumors (re-occurring) | 7 | 0 | 6 | 1 | 161.4 | |
PKCδ OVEREXPRESSION PROTECTS CELLS FROM TRAIL-INDUCED APOPTOSIS
To determine the mechanism by which PKCδ regulates both pro- and anti-apoptotic process in breast tumor cells, we stably transfected PKCδ cDNA into MCF-7 cells. MCF-7 cells normally express low levels of PKCδ (Supplementary Fig. 1). We have previously demonstrated that PKCδ overexpression does not alter other PKC isoforms in MCF-7 cells [Nabha et al., 2005]. Overexpression of PKCδ in MCF-7 (MCF-7/PKCδ) cells was associated with increased expression of death receptors DR4 and DR5 when compared to vector transfected (MCF-7/vector) control cells (Fig. 2A). In addition, we utilized an endoplasmic reticulum stress inducer tunicamycin (10 μg/ml) to upregulate the expression of death receptors. Increased DR4 and DR5 receptors do not increase TRAIL-induced apoptosis in PKCδ overexpressing MCF-7/PKCδ cells (Fig. 2B), suggesting the possibility that PKCδ overexpression protects MCF-7 cells from TRAIL-induced apoptosis. To confirm that PKCδ overexpression protects TRAIL-induced apoptosis in MCF-7 cells, we inhibited PKCδ with rottlerin (3 μM), a pharmacologic inhibitor of PKCδ. Our data show that rottlerin significantly inhibits PKCδ levels and TRAIL had minimal effect on its expression in MCF-7/PKCδ cells (Fig. 2A). The combination of rottlerin and TRAIL significantly reduced PKCδ levels and enhanced apoptosis as shown in Figure 2A,B. Currently, the use of rottlerin as a PKCδ inhibitor remains controversial. To further confirm the role of PKCδ in TRAIL-induced apoptosis, we knocked down PKCδ using PKCδ siRNA in MCF-7/PKCδ cells. Inhibition of PKCδ by siRNA in the presence of TRAIL significantly reduced phosphorylated Akt (p-Akt), while increasing both Bid and PARP cleavage when compared to random siRNA-treated cells (Fig. 3A) and increased apoptotic activity. These results suggest that PKCδ overexpression protects MCF-7 cells from TRAIL-induced apoptosis and this inhibition enhances apoptosis (Fig. 3B).
Fig. 2.

A: PKCδ overexpression protects the MCF-7 cells from TRAIL-induced apoptosis. MCF-7 cells overexpressing PKCδ (MCF-7/PKCδ) or vector alone (MCF-7/vector) were treated with TRAIL (10 ng/ml), rottlerin (3 μM), tunicamycin (10 μg/ml) alone, or in combination with TRAIL for 4 h. After 4 h PKCδ, DR4, DR5, and PARP cleavage were evaluated using specific antibodies by Western blots. MCF-7/PKCδ cells showed enhanced DR4 and DR5 expression in comparison to vector-transfected control cells. However, addition of TRAIL was unable to induce a significant increase in PARP cleavage in comparison to controls. Combination of TRAIL and rottlerin (PKCδ inhibitor) significantly enhanced PARP cleavage. Equivalent protein loading was assessed by stripping the membrane and re-probing with G3PDH antibody. Arrow indicates cleaved fragment. B: PKCδ inhibition enhances TRAIL-induced apoptosis in MCF-7 cells. Inhibition of PKCδ by rottlerin (3 μM) enhances TRAIL-induced apoptosis in MCF-7 cells. Apoptosis was quantified by ELISA and normalized to values measured in untreated cells. MCF-7/PKCδ cell showed a significant increase in TRAIL-induced apoptosis in the presence of rottlerin in comparison to untreated cells ((P <0.001). Data are mean +SE of triplicate determinations.
Fig. 3.

PKCδ siRNA enhances TRAIL-induced apoptosis: MCF-7/PKCδ cells were transfected with PKCδ siRNA or random siRNA alone for 72 h and then cells were treated with TRAIL for 4 h. A: Western blot shows PKCδ siRNA significantly reduces PKCδ, p-Akt levels, and enhances Bid cleavage, and PARP cleavage. Equivalent protein loading was assessed by stripping the membrane and re-probing with G3PDH antibody. Arrow indicates cleaved fragment. B: PKCδ inhibition by siRNA enhances TRAIL-induced apoptosis compared to random transfected cells in the presence of TRAIL.
ROLE OF CASPASE-3 IN PKCδ-INDUCED PRO- AND ANTI-APOPTOSIS
Caspase-3 is a member of the cysteine protease family, which plays a crucial role in the apoptotic pathway by cleaving a variety of key cellular proteins including PKCδ. Previous studies suggest that pro-apoptotic function of PKCδ is typically associated with its cleavage by caspase-3 in the hinge region, resulting in a phospholipid-independent activation of the enzyme [Emoto et al., 1995; Ghayur et al., 1996; Janicke et al., 1998; Okhrimenko et al., 2005]. Devarajan et al. [2002] showed that 75% of human breast tumors lack caspase-3 transcripts, but other caspases, such as caspase-8 and −9 were normal. MCF-7 cells were ideally suited for these proposed studies because they lack caspase-3 expression as a result of a functional deletion mutation in the casp-3 gene, while expression of other caspases-8 and −9 remains normal. In order to determine if restoration of caspase-3 enhances TRAIL-induced apoptosis in breast tumors, we developed stably transfected MCF-7 cell lines that express both PKCδ and caspase-3 (MCF-7/cas-3/PKCδ), and evaluated the role of caspase-3 in PKCδ and TRAIL-induced apoptosis. The introduction of caspase-3 gene in PKCδ over-expressing cells (MCF-7/cas-3/PKCδ) significantly enhanced the cleavage of the PKCδ, caspase-3, and PARP cleavage in the presence of TRAIL when compared to untreated cells (Fig. 4A). Similar results were observed in PKCδ/T47D cells (Supplementary Fig. 2). Inhibition of caspase-3 activity by its inhibitor Z-DEVD-fmk reversed PKCδ and PARP cleavage and blocked TRAIL-induced apoptosis in MCF-7/cas-3/PKCδ cells (Fig. 4B). These data suggest that caspase-3-induced cleavage of PKCδ may play a significant role in enhancing TRAIL-induced apoptosis in breast tumor cells.
Fig. 4.
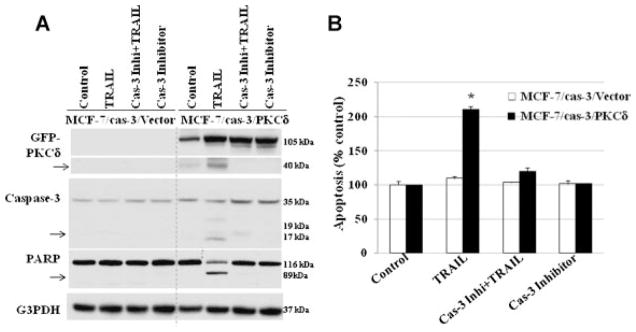
Caspase-3 is essential for TRAIL-induced apoptosis. A: Effect of TRAIL on MCF-7/Cas-3/PKCδ expressing and control cells. Addition of TRAIL enhances PKCδ cleavage (activated), caspase-3 cleavage (activated), and PARP cleavage compared to untreated cells. Inhibition of caspase-3 by Z-DEVD-fmk reverses TRAIL-induced biochemical changes and apoptosis (A,B). Equivalent protein loading was assessed by stripping the membrane and re-probing with G3PDH antibody. Arrow indicates cleaved fragment.
PKCδ REGULATES TRAIL-INDUCED PRO- AND ANTI-APOPTOSIS
Studies were undertaken to determine the mechanism(s) by which PKCδ overexpression regulates TRAIL-induced pro- and anti-apoptosis. For this stable MCF-7 cell lines that express wild-type PKCδ (MCF-7/cas-3/PKCδ cells) or caspase-3 cleavage-resistant PKCδmut (PKCδD327A) (MCF-7/cas-3/PKCδmut cells) expressing cell were developed utilizing MCF-7 cells expressing caspase-3. Cells that overexpress caspase-3 cleavage-resistant PKCδ mutant (MCF-7/cas-3/PKCδmut) significantly inhibited TRAIL-induced apoptosis when compared to wild-type PKCδ (MCF-7/cas-3/PKCδ) expressing cells. Western blot analysis shows that in MCF-7/cas-3/PKCδmut cells, TRAIL-induced caspase-8 activation was blocked leading to inhibition of downstream signaling molecules such as caspase-3, −9, Bid, and PARP cleavage (Fig. 5A) and apoptosis (Fig. 5B) compared to wild-type PKCδ (MCF-7/cas-3/PKCδ) expressing cells. In addition, our data also show that MCF-7/cas-3/PKCδmut cells have significantly reduced intracellular FADD levels when compared to wild-type PKCδ expressing cells (Fig. 5B), suggesting a role for PKCδ upstream of caspase-8 processing and activation. These data suggest that overexpression of PKCδ protects these cells from TRAIL-induced apoptosis by inhibiting caspase-8 activation and its cleavage by caspase-3 reverses this inhibition and enhances TRAIL-induced apoptosis.
Fig. 5.

Caspase-3-resistant PKCδ reverses TRAIL-induced apoptosis. We used three MCF-7 cell lines: (i) caspase-3 expressing MCF-7 cells (MCF-7/cas-3/vector), (ii) caspase-3 and PKCδ expressing MCF-7 cells (MCF-7/cas-3/PKCδ), and (iii) caspase-3 and PKCδ mutant that prevents caspase-3-induced PKCδ cleavage (MCF-7/cas-3/PKCδmut) in the presence or absence of TRAIL. In MCF-7/cas-3/PKCδ cells TRAIL significantly induces Bid cleavage, caspase-8, caspase-3, caspase-9 activation and PARP cleavage in comparison to control cells. However, MCF-7/cas-3/PKCδmut cells blocked TRAIL-induced pro-apoptotic biochemical changes. B: MCF-7/cas-3/PKCδmut cells show a significant reduction in FADD in the presence of TRAIL in comparison to untreated cells. C: TRAIL-induced apoptosis is blocked in MCF-7/cas-3/PKCδmut cells. MCF-7/cas-3/PKCδ cell showed a significant increase in TRAIL-induced apoptosis compared to untreated cells ((P <0.001). However, MCF-7/cas-3/PKCδmut cells blocked TRAIL-induced apoptosis suggesting PKCδ cleavage plays a major role in TRAIL-induced apoptosis. Bars: mean +SE of triplicate determinations.
DISCUSSION
In this study, we explored the signaling pathway(s) by which PKCδ regulates TRAIL-induced apoptosis in human breast cancer cells. Despite the potential of TRAIL as a useful tool in anti-cancer therapy, very little remains known about the mechanisms regulating the sensitivity to TRAIL-induced apoptosis in PKCδ overexpressing breast tumor cells. It has been suggested that the decoy receptors TRAIL-R3 and -R4 may regulate the sensitivity of cells to TRAIL [Sheridan et al., 1997; Pan et al., 1997a]. However, analyses of TRAIL receptor expression in a number of human tumor cell lines have indicated no correlation between TRAIL sensitivity and decoy receptor TRAIL-R3 and -R4 mRNA expression [Griffith and Lynch, 1998]. There is a possibility that resistance to TRAIL-induced apoptosis may be mediated by intracellular pro- and anti-apoptotic molecules such as PKCδ, Akt, and caspases that can block or activate apoptotic signaling. In this study, we demonstrated that over-expression of PKCδ in MCF-7 (MCF-7/PKCδ) protects these cells from TRAIL-induced apoptosis. Inhibition of PKCδ by rottlerin or PKCδ siRNA sensitized the cells to TRAIL-induced apoptosis suggesting PKCδ overexpression protects the MCF-7 cells from TRAIL-induced apoptosis. Some of the mechanisms by which PKCδ inhibition increases TRAIL-induced DNA damage and apoptosis in MCF-7 cells which lack caspase-3 expression as a result of mutation in caspase-3 gene [Janicke et al., 1998] may be associated with: (i) although classic apoptotic models are involved in the activation of caspase-3, evidence indicates that apoptosis can proceed independently of caspase-3 through cathepsin B, etc. [Nylandsted et al., 2000; Foghsgaard et al., 2001; Lin et al., 2006] and (ii) PKCδ was also shown to induce ornithine decarboxylase in response to oxidation damage induced by hydrogen peroxide. The impairment of an ornithine decarboxylase response to DNA damage can explain the persistent DNA fragmentation observed in cells treated with PKCδ antisense oligonucleotides [Otieno and Kensler, 2000; McCracken et al., 2003]. In addition, there are data to suggest that rottlerin suppresses the NF-κB and enhances the caspase-processing leading to apoptosis [Zhang et al., 2005]. Rottlerin was also shown to affect the mitochondrial function independent of PKCδ, thereby sensitizing the cells to TRAIL-induced apoptosis [Tillman et al., 2003]. The mechanisms that are involved in the protective effect of PKCδ in human breast tumors remain unclear. Other studies have shown that caspase-3-dependent cleavage of PKCδ leads to the generation of a constitutively active catalytic fragment that is associated with PKCδ apoptotic function [Ghayur et al., 1996; Basu et al., 2001; Blass et al., 2002]. In an attempt to determine the role of caspase-3 cleavage on PKCδ and TRAIL-induced apoptosis, we expressed wild-type or caspase-3 cleavage-resistant PKCδmut in caspase-3 expressing MCF-7 cells. Our data clearly show that overexpression of caspase-3 cleavage-resistant PKCδ mutant (MCF-7/cas-3/PKCδmut) prevents caspase-8 activation, a first step in TRAIL-induced apoptosis, thereby blocking TRAIL-induced apoptosis. In contrast, overexpression of wild-type PKCδ (MCF-7/cas-3/PKCδ) leads to caspase-8 activation and PKCδ, Bid, caspase-3, and −9, and PARP cleavage resulting in a significant increase in apoptosis (Fig. 5). The above data suggest that PKCδ overexpression protects the cells from TRAIL-induced apoptosis and its inhibition by rottlerin, siRNA, or cleavage by caspase-3 reverses PKCδ-induced caspase-8 inhibition which then leads to enhanced TRAIL-induced apoptosis.
These data show that caspase-3 mRNA expression levels in breast tumors were at least 10–50 times lower than those in normal breast tissues [Devarajan et al., 2002] representing a clinically significant observation which has the potential to prevent TRAIL-induced apoptosis in PKCδ overexpressing breast tumors. Table I shows that overexpression of PKCδ was statistically significant in re-occurring breast tumors when compared to the adjacent normal breast tissue. Previously, we showed that tamoxifen-resistant breast tumor cells overexpress PKCδ [Nabha et al., 2005], suggesting that PKCδ plays a major role in a subset of breast tumors and these tumors do not respond to TRAIL and other drug treatments.
The involvement of the death adaptor protein FADD and apoptosis-initiating caspase-8 in death signaling by DR4 and DR5 are controversial. Some studies suggest that dominant-negative FADD inhibited TRAIL-induced apoptosis [Chaudhary et al., 1997; Schneider et al., 1997; Sprick et al., 2000]. Our studies also show significant reduction of FADD levels in MCF-7/cas-3/PKCδmut cells in the presence of TRAIL. In other studies, overexpression of dominant-negative FADD did not prevent TRAIL-induced apoptosis [MacFarlane et al., 1997; Pan et al., 1997a,b]. Co-immunoprecipitation of overexpressed cytoplasmic domains of DR4 and DR5 with FADD support a role for FADD in TRAIL-induced apoptosis [Chaudhary et al., 1997; Schneider et al., 1997]. Murine embryonic fibroblasts (MEF) from FADD-deficient mice underwent apoptosis upon expression of human DR4 [Yeh et al., 1998]. Thus, the role of FADD in TRAIL-induced apoptosis in PKCδ overexpressing cells needs further studies.
The ability of cells to evade apoptosis is one of the essential hallmarks of cancer cells. Thus, loss or reduction of caspase-3 and/or other caspases may serve as an important step in the survival of tumor cells. For example, complete inactivation of caspase-8 gene in neuroblastomas was reported and these cells were resistant to doxorubicin- and death receptor-induced apoptosis [Teitz et al., 2000]. In addition, there are data to suggest that caspase-3 mRNA were undetectable in breast and cervical cancers and substantially decreased in ovarian cancer [Devarajan et al., 2002]. Similarly, 80% of prostate tumors were shown to have lost caspase-1 protein expression and reduced levels of caspase-3 expression [Winter et al., 2001]. Taken together these results suggest that a lack of caspases can attenuate TRAIL-induced apoptosis and this may represent an important mechanism of cell survival.
In conclusion, our results demonstrate that PKCδ overexpression protects breast tumor cells by inhibiting caspase-8 activation, a first step in TRAIL-induced apoptosis, thus preventing TRAIL-induced apoptosis. Our experimental evidence suggests that PKCδ over-expression protects the cells from TRAIL-induced apoptosis and its inhibition by rottlerin, siRNA, or its cleavage by caspase-3 reverses PKCδ-induced inhibition of caspase-8 activation leading to enhanced TRAIL-induced apoptosis. Clinically, PKCδ overexpressing tumors can be treated with a combination of PKCδ inhibitor(s) and TRAIL as a potential cancer treatment strategy.
Supplementary Material
Acknowledgments
Grant sponsor: National Institute of Health; Grant number: RO1 CA 118089.
Abbreviations used
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- DR4
death receptor 4
- DR5
death receptor 5
- PKC-δ
protein kinase C delta
- PARP
poly(ADP-ribose) polymerase
- FADD
Fas-associated protein with death domain
- Cas-3
caspase-3
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Basu A, Woolard MD, Johnson CL. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. Cell Death Differ. 2001;8:899–908. doi: 10.1038/sj.cdd.4400885. [DOI] [PubMed] [Google Scholar]
- Blass M, Kronfeld I, Kazimirsky G, Blumberg PM, Brodie C. Tyrosine phosphorylation of protein kinase Cdelta is essential for its apoptotic effect in response to etoposide. Mol Cell Biol. 2002;22:182–195. doi: 10.1128/MCB.22.1.182-195.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- Chen N, Ma W, Huang C, Dong Z. Translocation of protein kinase Cepsilon and protein kinase Cdelta to membrane is required for ultraviolet B-induced activation of mitogen-activated protein kinases and apoptosis. J Biol Chem. 1999;274:15389–15394. doi: 10.1074/jbc.274.22.15389. [DOI] [PubMed] [Google Scholar]
- Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003;63:780–786. [PubMed] [Google Scholar]
- D’Costa AM, Denning MF. A caspase-resistant mutant of PKC-delta protects keratinocytes from UV-induced apoptosis. Cell Death Differ. 2005;12:224–232. doi: 10.1038/sj.cdd.4401558. [DOI] [PubMed] [Google Scholar]
- Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH, Tora AD, Mehta K. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene. 2002;21:8843–8851. doi: 10.1038/sj.onc.1206044. [DOI] [PubMed] [Google Scholar]
- DeVries TA, Neville MC, Reyland ME. Nuclear import of PKCdelta is required for apoptosis: Identification of a novel nuclear import sequence. EMBO J. 2002;21:6050–6060. doi: 10.1093/emboj/cdf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries-Seimon TA, Ohm AM, Humphries MJ, Reyland ME. Induction of apoptosis is driven by nuclear retention of protein kinase C delta. J Biol Chem. 2007;282:22307–22314. doi: 10.1074/jbc.M703661200. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M, Elling F, Leist M, Jaattela M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol. 2001;153:999–1010. doi: 10.1083/jcb.153.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang Y, Kharbanda S, Allen H, Kamen R, Wong W, Kufe D. Proteolytic activation of protein kinase C delta by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med. 1996;184:2399–2404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith TS, Lynch DH. TRAIL: A molecule with multiple receptors and control mechanisms. Curr Opin Immunol. 1998;10:559–563. doi: 10.1016/s0952-7915(98)80224-0. [DOI] [PubMed] [Google Scholar]
- Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98:2603–2614. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- Humphries MJ, Limesand KH, Schneider JC, Nakayama KI, Anderson SM, Reyland ME. Suppression of apoptosis in the protein kinase Cdelta null mouse in vivo. J Biol Chem. 2006;281:9728–9737. doi: 10.1074/jbc.M507851200. [DOI] [PubMed] [Google Scholar]
- Jackson DN, Foster DA. The enigmatic protein kinase Cdelta: Complex roles in cell proliferation and survival. FASEB J. 2004;18:627–636. doi: 10.1096/fj.03-0979rev. [DOI] [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Kaul S, Anantharam V, Yang Y, Choi CJ, Kanthasamy A, Kanthasamy AG. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J Biol Chem. 2005;280:28721–28730. doi: 10.1074/jbc.M501092200. [DOI] [PubMed] [Google Scholar]
- Kerfoot C, Huang W, Rotenberg SA. Immunohistochemical analysis of advanced human breast carcinomas reveals downregulation of protein kinase C alpha. J Histochem Cytochem. 2004;52:419–422. doi: 10.1177/002215540405200314. [DOI] [PubMed] [Google Scholar]
- Lin J, Zhang Z, Zeng S, Zhou S, Liu BF, Liu Q, Yang J, Luo Q. TRAIL-induced apoptosis proceeding from caspase-3-dependent and -independent pathways in distinct HeLa cells. Biochem Biophys Res Commun. 2006;346:1136–1141. doi: 10.1016/j.bbrc.2006.05.209. [DOI] [PubMed] [Google Scholar]
- MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem. 1997;272:25417–25420. doi: 10.1074/jbc.272.41.25417. [DOI] [PubMed] [Google Scholar]
- Mackay HJ, Twelves CJ. Targeting the protein kinase C family: Are we there yet? Nat Rev Cancer. 2007;7:554–562. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- McCracken MA, Miraglia LJ, McKay RA, Strobl JS. Protein kinase C delta is a prosurvival factor in human breast tumor cell lines. Mol Cancer Ther. 2003;2:273–281. [PubMed] [Google Scholar]
- Nabha SM, Glaros S, Hong M, Lykkesfeldt AE, Schiff R, Osborne K, Reddy KB. Upregulation of PKC-delta contributes to antiestrogen resistance in mammary tumor cells. Oncogene. 2005;24:3166–3176. doi: 10.1038/sj.onc.1208502. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: Structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- Nylandsted J, Rohde M, Brand K, Bastholm L, Elling F, Jaattela M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc Natl Acad Sci USA. 2000;97:7871–7876. doi: 10.1073/pnas.97.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okhrimenko H, Lu W, Xiang C, Ju D, Blumberg PM, Gomel R, Kazimirsky G, Brodie C. Roles of tyrosine phosphorylation and cleavage of protein kinase Cdelta in its protective effect against tumor necrosis factor-related apoptosis inducing ligand-induced apoptosis. J Biol Chem. 2005;280:23643–23652. doi: 10.1074/jbc.M501374200. [DOI] [PubMed] [Google Scholar]
- Otieno MA, Kensler TW. A role for protein kinase C-delta in the regulation of ornithine decarboxylase expression by oxidative stress. Cancer Res. 2000;60:4391–4396. [PubMed] [Google Scholar]
- Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997a;277:815–818. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997b;276:111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, Holler N, Tschopp J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity. 1997;7:831–836. doi: 10.1016/s1074-7613(00)80401-x. [DOI] [PubMed] [Google Scholar]
- Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, Krammer PH, Walczak H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12:599–609. doi: 10.1016/s1074-7613(00)80211-3. [DOI] [PubMed] [Google Scholar]
- Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- Tillman DM, Izeradjene K, Szucs KS, Douglas L, Houghton JA. Rottlerin sensitizes colon carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis via uncoupling of the mitochondria independent of protein kinase C. Cancer Res. 2003;63:5118–5125. [PubMed] [Google Scholar]
- Visscher DW, Sarkar FH, Kasunic TC, Reddy KB. Clinicopathologic analysis of amphiregulin and heregulin immunostaining in breast neoplasia. Breast Cancer Res Treat. 1997;45:75–80. doi: 10.1023/a:1005845512804. [DOI] [PubMed] [Google Scholar]
- Winter RN, Kramer A, Borkowski A, Kyprianou N. Loss of caspase-1 and caspase-3 protein expression in human prostate cancer. Cancer Res. 2001;61:1227–1232. [PubMed] [Google Scholar]
- Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu N, Zhang J, Liu S, Liu Y, Zheng D. PKCdelta protects human breast tumor MCF-7 cells against tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis. J Cell Biochem. 2005;96:522–532. doi: 10.1002/jcb.20535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.