

Abstract
N9-substituted 2,4-diaminoquinazolines were synthesized and evaluated as inhibitors of Pneumocystis carinii (pc) and Toxoplasma gondii (tg) dihydrofolate reductase (DHFR). Reduction of commercially available 2,4-diamino-6-nitroquinazoline 14 with Raney nickel afforded 2,4,6-triaminoquinazoline 15. Reductive amination of 15 with the appropriate benzaldehydes or naphthaldehydes, followed by N9-alkylation, afforded the target compounds 5–13. In the 2,5-dimethoxybenzylamino substituted quinazoline analogues, replacement of the N9—CH3 group of 4 with the N9—C2H5 group of 8 resulted in a 9- and 8-fold increase in potency against pcDHFR and tgDHFR, respectively. The N9—C2H5 substituted compound 8 was highly potent, with IC50 values of 9.9 and 3.7 nM against pcDHFR and tgDHFR, respectively. N9-propyl and N9-cyclopropyl methyl substitutions did not afford further increases in potency. This study indicates that the N9-ethyl substitution is optimum for inhibitory activity against pcDHFR and tgDHFR for the 2,4-diaminoquinazolines. Selectivity was unaffected by N9 substitution.
Introduction
Opportunistic infections, such as Pneumocystis pneumonia (PCPa) and toxoplasmosis, remain a major cause of mortality in acquired immunodeficiency syndrome (AIDS) patients.1–3 Trimethoprim (TMP) (Figure 1) is a relatively weak inhibitor (Ki = 25 ± 7 nM) of dihydrofolate reductase (DHFR) from P. jirovecii, the form of Pneumocystis found in humans; TMP is even less effective (Ki = 770 ± 340 nM) against the DHFR found in P. carinii (pc), the organism that infects rats. In humans and in the drug testing model in rats, TMP must be coadministered with sulfonamides to provide synergistic antifolate effects.4,5 Similarly, when pyrimethamine, a rather nonselective DHFR inhibitor, is used to treat toxoplasmosis, it is necessary to combine the weak DHFR inhibitor with a sulfonamide to achieve synergistic effects. In both of these situations, the side effects associated with sulfonamides often result in discontinuation of therapy.6
Figure 1.

Lipophilic, nonclassical antifolates, such as trimetrexate (TMQ) (Figure 1), can passively diffuse into the pathogenic cells, as well as host cells. TMQ is 286-fold and 159-fold more potent against pcDHFR and tgDHFR, respectively, than TMP, but TMQ is not selective for the mammalian DHFR and thus must be coadministered with the classical folate leucovorin (5-formyltetrahydrofolate) due to lack of selectivity.7–9 Host tissues can be selectively rescued because leucovorin is only taken up by the mammalian transport system(s). Drawbacks of the TMQ-leucovorin regimen are the high cost and variable effectiveness of leucovorin.10 Thus, the search for single agents that are potent and selective against pcDHFR and/or tgDHFR continues in several laboratories.
Gangjee et al.,11–16 as well as others,17–21 have reported several structural classes of DHFR inhibitors. Selectivity is reported as the ratio of the IC50 value of mammalian DHFR/IC50 value of pcDHFR or tgDHFR. A series of 2,4-diaminopyrido[2,3-d]pyrimidines were reported by Gangjee et al.9 as two atom-bridged variations of the potent nonclassical antifolate piritrexim (PTX).22 Compound 1 (see Figure 2) showed moderate DHFR inhibitory potency with IC50 values of 3.80 and 0.31 µM against pcDHFR and tgDHFR, respectively. N9-methylation of 1 to afford 2 resulted in a 45-fold and 49-fold increase in potency against pcDHFR (IC50 = 84 nM) and tgDHFR (IC50 = 6.3 nM), respectively. The selectivity ratio for tgDHFR versus recombinant human (rh) DHFR of compound 2 was 304. Thus, N9-methylation in the reverse-bridged analogue significantly increased potency against pcDHFR and tgDHFR, as well as selectivity for tgDHFR versus rhDHFR. However, 2 was 447-times less potent against T. gondii cells in culture compared to isolated tgDHFR. These results suggest inadequate cell penetration.
Figure 2.

To explore the effect on potency, selectivity, and cell penetration, Gangjee et al.23 synthesized a series of analogues of compound 2 with an isosteric replacement of the N8 with a carbon to yield 2,4-diaminoquinazolines with increased lipophilicity. A comparison of the potency and selectivity of the quinazoline analogues with the pyrido[2,3-d]pyrimidines with identical side chain substituents revealed different structure–activity/selectivity relationships for the two classes. Contrary to the pyrido[2,3-d]pyrimidines, in most cases, the N9—H quinazoline analogues showed better selectivity for tgDHFR versus rlDHFR and tgDHFR versus hDHFR than their corresponding N9-methyl analogues. The N9-methyl quinazolines were, however, considerably more potent against both pcDHFR and tgDHFR than the N9—H quinazolines. Three trimethoxyphenyl substituted N9—H quinazolines were selected for evaluation in T. gondii cell culture studies. The cell culture/enzyme ratio (IC50 value in T. gondii cell culture/IC50 value in tgDHFR) ranged from 3 to 142, which was significantly better than the ratio of 447 for compound 2. This increase in inhibition of T. gondii cells in culture was attributed, in part, to the increased lipophilicity of the quinazolines versus the pyrido-[2,3-d]pyrimidines and provided the impetus to explore further modifications of quinazolines to improve selectivity and potency.23
X-ray crystal structures of 5-deaza-5-methyl antifolates show a hydrophobic interaction between the 5-methyl moiety and Val115 in hDHFR.24 In the crystal structure of PTX complexed with pcDHFR, a hydrophobic interaction between the 5-methyl moiety and Ile123 is observed.25 The fact that the 5-methyl moiety of PTX has hydrophobic interactions in both pcDHFR and hDHFR may explain, in part, the lack of selectivity of PTX. The X-ray crystal structures of 2 complexed with hDHFR23 and pcDHFR26 showed that the distance between the N9-methyl moiety of 2 and the Val115 (hDHFR) or Ile123 (pcDHFR)25 was 4.66 Å and 4.20 Å, respectively,27 beyond the range for van der Waals interaction (≤4.00 Å). The Val115 of hDHFR corresponds to Ile123 in pcDHFR. Because the quinazolines demonstrated better T. gondii cell to tgDHFR inhibitory ratios and the N9-methyl moiety of the pyrido[2,3-d]pyrimidines did not interact with Ile123, it was of interest to increase the size of the N9-alkyl substitution in the quinazoline series. It was anticipated that this increase would provide both potent and selective inhibitors.
Thus, the N9-alkyl 2,4-diaminoquinazolines 5–13 were designed and synthesized as analogues of compound 4. Compound 5 was synthesized as a phenyl ring unsubstituted analogue. Compounds 6 and 7 contain α- and β-naphthyl moieties. A naphthyl group has been shown by Gangjee et al.28 to interact with Phe69 in pcDHFR. Compound 9 contains 2,3,5-trichloro substituents to determine the effect of electron withdrawing groups on biological activity. The N9-propyl analogues 10 and 11 and the cyclopropyl methyl analogues 12 and 13 were synthesized to determine the importance of the size of the N9-alkyl moiety on biological activity. The increase in the number of carbons at the N9-position contributes to the lipophilicity of the analogues and also serves as a site for possible hydrophobic interactions with Val115 of hDHFR, Ile123 of pcDHFR, and/or hydrophobic residues in tgDHFR. The N9-ethyl substituted compounds 5–9 were designed to determine whether potency and/or selectivity can be increased by decreasing the distance between the N9-alkyl moiety and the hydrophobic residues in the DHFR binding site. The N9-propyl and -cyclopropyl methyl analogues 10–13 were designed to determine whether the increase in the number of carbons from 2 to 3 or 4 would increase the potential hydrophobic interactions or exceed the optimum distance between the DHFR residues and the N9-moiety.
Chemistry
The synthesis of target compounds 5–13 began with commercially available 2,4-diamino-6-nitroquinazoline 14 and is outlined in Scheme 1.23 Starting material 14 was reduced with hydrogen at 30–35 psi and Raney nickel catalyst for 3 h to afford the key intermediate 2,4,6-triaminoquinazoline 15. To the reaction vessel was added the appropriate benzaldehyde or naphthaldehyde, and the reaction mixture was hydrogenated for an additional 15 h to afford 16–20, the N9—H precursors to the target compounds, in yields ranging from 36% to 82%, depending on the phenyl substitution. Reductive N9-alkylation with acetaldehyde, proprionaldehyde, or cyclopropanecarboxaldehyde using sodium cyanoborohydride afforded the target compounds 5–13 in yields ranging from 9% to 67%, depending on the aldehyde and the phenyl substitution.
Scheme 1a.
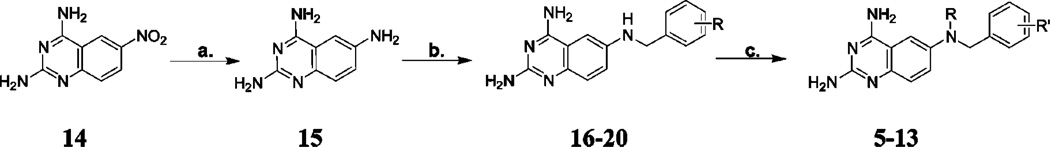
Reagents: (a.) Raney nickel, DMF, H2 at 30–35 psi; (b.) Raney nickel, DMF, CH3COOH, ArCHO, H2 at 30–35 psi; (c.) RCHO, NaCNBH3, HCl, CH3CN.
Biological Activity and Discussion
Compounds 5–13 were evaluated as inhibitors of pcDHFR, tgDHFR, rlDHFR, and Mycobacterium avium (ma) DHFR, and the results are reported in Table 1. In general, the N9-ethyl substituted analogues were more potent than the unsubstituted or N9-methyl substituted compounds against pcDHFR and, in most cases, against tgDHFR as well. Against pcDHFR, the α-naphthyl compound 6 was the most potent. Against tgDHFR, the unsubstituted phenyl analogue 5 and the 2,5-diOCH3 phenyl analogue 8 were most potent. The α-naphthyl analogue 6 and the β-naphthyl analogue 7 were 8-fold and 3-fold more potent against pcDHFR, respectively, than the unsubstituted phenyl analogue 5. This suggests a tolerance for larger side chain rings in the pcDHFR binding site. Against tgDHFR, compounds 6 and 7 were 2-fold and 3-fold less potent, respectively, than 5, which suggests the naphthyl rings bind less favorably than the phenyl ring, contrary to that observed for pcDHFR. Substitution of electron-donating methoxy groups on the side chain phenyl ring in 8 increased potency against pcDHFR relative to 5. Against tgDHFR, 5 and 8 were equipotent. Electron-withdrawing chloro groups on the phenyl ring also resulted in an increase in potency against pcDHFR for compound 9. The fact that methoxy and chloro groups both contributed to increased potency against pcDHFR compared to the unsubstituted phenyl ring suggests that the increase was not due to electronic factors but was more likely due to favorable binding conformations induced by the substitutions on the phenyl ring in compounds 8 and 9 compared to 5. Against tgDHFR, electron-withdrawing chloro substituents on the side chain phenyl ring results in a 5-fold decrease in inhibitory potency compared to the unsubstituted phenyl ring, with IC50 values for compounds 5 and 9 of 3.6 and 19 nM, respectively.
Table 1.
Inhibition Concentrations (IC50, in nM) against DHFR from P. carinii (pc), T. gondii (tg), M. avium (ma), and Rat Liver (rl) and Selectivity Ratiosa
| R | R′ | pcDHFR | rlDHFR | rl/pc | tgDHFR | rl/tg | maDHFR | rl/ma | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | H | 3800 | 350 | 0.09 | 310 | 1.1 | |||
| 2 | CH3 | 84 | 57 | 0.7 | 6.3 | 9.0 | |||
| 3 | H | 4600 | 1100 | 0.24 | 160 | 6.88 | |||
| 4 | CH3 | 87 | 26 | 0.29 | 30 | 0.86 | |||
| 5 | C2H5 | H | 21 | 4.1 | 0.2 | 3.6 | 1.1 | 1.1 | 3.73 |
| 6 | C2H5 | 2,3-C4H4 | 2.5 | 6.5 | 2.6 | 7.8 | 0.8 | 2.9 | 2.24 |
| 7 | C2H5 | 3,4-C4H4 | 7.2 | 14.5 | 2.0 | 12 | 1.2 | 2.3 | 6.30 |
| 8 | C2H5 | 2,5-diOCH3 | 9.9 | 4.8 | 0.5 | 3.7 | 1.3 | 0.98 | 4.90 |
| 9 | C2H5 | 2,3,5-triCl | 6.9 | 19 | 2.8 | 19 | 1.0 | 14 | 1.36 |
| 10 | C3H7 | 2,5-diOCH3 | 38 | 5.3 | 0.1 | 9.5 | 0.6 | 3.3 | 1.63 |
| 11 | C4H7 | 3,4-C4H4 | 27 | 17 | 0.6 | 19 | 0.9 | 13 | 1.31 |
| 12 | C4H7 | 2,5-diOCH3 | 25 | 9.6 | 0.4 | 7.3 | 1.3 | 16 | 0.60 |
| 13 | C3H7 | 3,4-C4H4 | 42 | 16 | 0.4 | 25 | 0.6 | 34 | 0.47 |
| TMPb | 12000 | 133000 | 11.1 | 2,700 | 49 | ||||
| TMQb | 42 | 3.0 | 0.07 | 10 | 0.3 |
These assays were carried out at 37 °C under saturating conditions of substrate (0.092 mM dihydrofolic acid) and cofactor (0.117 mM NADPH); rlDHFR and tgDHFR were assayed in the presence of 150 mM KCl.
These values are from ref 28. Most recent quality control values for rlDHFR are 121400 ± 7500 nM for TMP (R2 = 0.9768), 8.0 ± 0.5 nM (R2 = 0.9561) for TMQ and 4.4 ± 0.4 nM (R2 = 0.9386) for PTX. Most recent quality control values for pcDHFR are 26,820 ± 1700 nM (R2 = 0.995) for TMP, 47 ± 13 nM (R2 = 0.923) for TMQ, and 34.3 ± 10 nM (R2 = 0.9876) for PTX. These quality control assays used recombinant pcDHFR, as did all the assays for experimental compounds shown above. Published data on TMP, TMQ and PTX used native pcDHFR; side by side comparisons show no difference in drug susceptibility between the two forms of pcDHFR.
The effect on inhibitory potency against pcDHFR and tgDHFR of increasing the number of carbons at the N9-position in the quinazoline series can be deduced from a comparison of IC50 values for the 2,5-dimethoxy phenyl substituted analogues 3, 4, 8, 10, and 12. As previously reported,23 the potency against pcDHFR increased 53-fold upon N9-methylation, with IC50 values of 4600 and 87 nM for 3 and 4, respectively. N9-substitution with the homologous ethyl group to yield compound 8 resulted in a 9-fold increase in potency against pcDHFR compared to the N9-methyl analogue 4, with an IC50 value of 9.9 nM for 8. However, propyl substitution at the N9-position caused the inhibitory potency against pcDHFR to decrease. The N9-propyl analogue 10 showed an IC50 value of 38 nM, 4-fold less than 8. The N9-cyclopropyl methyl analogue 12 had a slight increase in potency against pcDHFR compared to 10, with an IC50 value of 25 nM. Despite the N9-cyclopropyl methyl group having one additional carbon compared to the N9-propyl moiety, the similarity in IC50 values for 10 and 12 was consistent with the similarity in carbon chain lengths in the two groups. The increase in inhibitory potency against pcDHFR upon N9-ethylation is also shown in the α-naphthyl quinazoline analogues. The IC50 values for the previously reported N9—H and N9-methyl α-naphthyl quinazolines23 were 720 and 17 nM, respectively, indicating a 42-fold increase in potency against pcDHFR upon N9-methylation. The N9-ethyl analogue 6 showed an IC50 value of 2.5 nM and was 7-fold more potent against pcDHFR than the N9-methyl analogue. Also, as seen in the 2,5-dimethoxy series, in the β-naphthyl quinazoline analogues, replacement of the N9-ethyl group with a propyl or cyclopropyl methyl group resulted in a decrease in potency against pcDHFR. The N9-ethyl analogue 7 was 4-fold and 6-fold more potent than 11 and 13, respectively.
Against tgDHFR, compound 4 was 5-fold more potent than 3, illustrating that just as for pcDHFR, N9-ethylation resulted in an increase in potency against tgDHFR compared to the N9-methyl analogue. Compound 8 was 8-fold more potent than 4 against tgDHFR. However, the N9-propyl homologue 10 showed a 3-fold decrease in potency compared to 8. The N9-cyclopropyl methyl analogue 12 showed a very similar potency against tgDHFR compared to 10. Potency against tgDHFR also increased with N9-ethylation in the α-naphthyl quinazoline analogues. The N9-ethyl analogue 6 showed a 3-fold increase in potency compared to the N9-methyl analogue against tgDHFR. In the β-naphthyl quinazoline analogues 7, 11, and 13 reported herein, the increase in the number of carbons at the N9-position caused about a 2-fold reduction in potency against tgDHFR.
Selectivity for pcDHFR in the quinazoline analogues is generally low. N9-substituents with increasing numbers of carbons did not significantly improve selectivity for pcDHFR. Compounds 3 and 4 both showed increased potency against rlDHFR compared to pcDHFR with selectivity ratios of 0.24 and 0.29, respectively. N9-ethyl, -propyl, or -cyclopropyl methyl substitutions (compounds 8, 10, and 12), in general, did not improve selectivity for pcDHFR. Only compounds 6, 7, and 9 showed greater potency against pcDHFR compared to rlDHFR with marginal increase in selectivity ratios. N9-propylation and N9-cyclopropylmethylation of the β-naphthyl quinazoline resulted in a decrease in selectivity for pcDHFR compared to 7.
Selectivity for tgDHFR in the N9—H quinazoline analogues was generally higher than selectivity for pcDHFR, with selectivity ratios of 7 and 0.2 for tgDHFR and pcDHFR, respectively, for the 2,5-dimethoxyphenyl analogue 3. N9-alkylation resulted in a decrease in selectivity for tgDHFR; compounds 8, 10, and 12 showed selectivity ratios of 1.3, 0.6, and 1.3, respectively, for tgDHFR. The selectivity for tgDHFR of the N9-ethyl, -propyl, and -cyclopropyl analogues was low, indicating a minimal but consistent decrease in selectivity for tgDHFR with a number of carbons greater than one in the N9-position.
Compounds 5–13 were also evaluated as inhibitors of maDHFR. The N9-ethyl dimethoxyphenyl analogue 8 was the most potent with an IC50 value of 0.98 nM. Compound 8 was equipotent compared to the unsubstituted phenyl analogue 5. The α- and β-naphthyl analogues 6 and 7 were slightly less potent. Compound 9, with the electron-withdrawing 2,3,5-trichloro substituents on the side chain phenyl ring, was 13-fold less potent than 5 against maDHFR. N9-propylation of the 2,5-dimethoxy quinazoline resulted in a 3-fold decrease in potency for 10 against maDHFR compared to 8. Cyclopropyl methylation at the N9-position decreased potency against maDHFR by 5-fold for 12 compared to 10. N9-propylation of the β-naphthyl quinazoline resulted in a 6-fold decrease in potency against maDHFR for 11 compared to 7. The N9-cyclopropyl methyl β-naphthyl quinazoline 13 was 3-fold less potent compared to 11.
The unsubstituted phenyl analogue 5 was selective for maDHFR versus rlDHFR with a selectivity ratio of 3.7. The α-naphthyl analogue 6 was less selective compared to 5 with a selectivity ratio of 2.2. However, the β-naphthyl analogue 7 was more selective for maDHFR compared to 5 with a selectivity ratio of 6.3 and was the most selective analogue for maDHFR reported herein. Compound 8, with electron-donating 2,5-dimethoxy substituents on the phenyl ring, was more selective for maDHFR compared to 5. The decrease in selectivity for maDHFR of 9 (selectivity ratio = 1.4), with electron-withdrawing 2,3,5-trichloro substituents on the phenyl ring, suggests that selectivity for maDHFR versus rlDHFR may be affected by the electronic nature of the phenyl ring substitutions. Selectivity for maDHFR was generally greater for the N9-ethyl quinazoline analogues and decreased with an increase in the number of carbons at the N9-position. N9-propyl analogues 10 (selectivity ratio = 1.6) and 11 (selectivity ratio = 1.3) were 3-fold and 5-fold less selective compared to 8 and 7, respectively. N9-cyclopropylmethylation resulted in analogues that were more selective for rlDHFR versus maDHFR with selectivity ratios for 12 and 13 of less than one.
Experimental Section
All evaporations were carried out in vacuo with a rotary evaporator. Analytical samples were dried in vacuo over P2O5. Thin-layer chromatography (TLC) was performed on silica gel plates with fluorescent indicator. Spots were visualized by UV light (254 and 365 nM). All analytical samples were homogeneous on TLC in at least two different solvent systems. Purification by column chromatography was carried out using Merck silica gel 60 (200–400 mesh). The amount of silica gel used for column chromatography was depended upon the chemical nature and amount of the compounds being separated. Columns were usually wet-packed unless otherwise noted. Solvent systems are reported as volume percent mixtures. Melting points were determined on a Mel-Temp II melting point apparatus with a digital thermometer and are uncorrected. 1H NMR spectra were recorded on a Bruker WH-300 (300 MHz)NMR spectrometer. The chemical shift (δ) values are reported as parts per million (ppm) relative to tetramethylsilane as internal standard: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad peak, exch = exchangeable protons by addition of D2O. Elemental analyses were performed by Atlantic Microlabs, Inc., Norcross, GA. Elemental compositions were within ± 0.4% of the calculated values. Fractional moles of water or organic solvents frequently found in some analytical samples of antifolates could not be removed despite 24 h of drying in vacuo and were confirmed, where possible, by their presence in the 1H NMR spectrum. All solvents and chemicals were purchased from Aldrich Chemical Co. and Fisher Scientific Co. and were used as received except where anhydrous solvents were needed, which were freshly dried in the laboratory.
General Procedure for the Synthesis of Compounds 5–13
To the 2,4-diamino[(N-substituted benzyl)amino]quinazoline suspended in CH3CN was added acetaldehyde, proprionaldehyde, or cyclopropanecarboxaldehyde, followed by NaCNBH3. The suspension was pH adjusted to 2–3 by dropwise addition of concentrated HCl. As the mixture was acidified, the suspended starting material began to dissolve into solution, followed by precipitation of the crude product. In some cases, dissolution was not seen but a visible color change occurred. The suspension containing the crude product was stirred for an additional 0.5 h. The crude product was filtered, stirred in 2 N Na2CO3, and washed with H2O, C2H5OH, and C2H5OC2H5. The purity of each product was checked by TLC using a solvent system of 5:2:0.1 (A) or 5:1:0.1 (B) by volume of CHCl3:CH3OH: NH4OH. Crude products were dissolved in CH3OH, pH adjusted to 8 with NH4OH, added to silica gel, followed by the evaporation of the solvent to afford a silica gel plug. The dry plug containing the product was placed on a column of wet silica gel and eluted with solvent B or a solvent system of 10:1 (C) by volume of CHCl3: CH3OH. Pure fractions, identified as single spots by TLC, were collected and the solvent evaporated to yield analytically pure solids.
2,4-Diamino-6-[N-(benzyl)-N-ethylamino]quinazoline 5
Compound 5 was synthesized from 16 (0.20 g, 0.75 mmol), acetaldehyde (0.20 mL, 3.6 mmol), and NaCNBH3 (0.125 g, 2.0 mmol) in 50 mL of CH3CN. Compound 5 was filtered from the reaction mixture to yield an analytically pure bright-yellow solid (0.10 g, 45%): mp 280–282 °C; TLC (solvent A) Rf 0.46. 1H NMR (DMSO-d6) δ 1.11 (t, 3 H, N-CH2-CH3), 3.48 (q, 2 H, N-CH2-CH3), 4.60 (s, 2 H, 10-CH2), 7.24–7.38 (m, 10 H, Ar, NH2), 8.6 (br, 2 H, NH2). Anal. (C17H19N5 • 1.8HCl • 0.6CH3OH) C, H, N.
2,4-Diamino-6-[N-(α-naphthyl)-N-ethylamino]quinazoline 6
Compound 6 was synthesized from 17 (0.47 g, 1.50 mmol), acetaldehyde (0.40 mL, 7.2 mmol), and NaCNBH3 (0.25 g, 4.0 mmol) in 50 mL of CH3CN. Compound 6 was filtered from the reaction mixture to yield an analytically pure solid (0.20 g, 39%): mp 266–267 °C; TLC (solvent B) Rf 0.40. 1H NMR (DMSO-d6) δ 1.16 (t, 3 H, N-CH2-CH3), 3.49–3.56 (q, 2 H, N-CH2-CH3), 5.05 (s, 2 H, 10-CH2), 7.18–7.21 (m, 2 H, Ar), 7.27–7.30 (m, 1 H, Ar), 7.39–7.62 (m, 4 H, Ar, 2 H, NH2), 7.82–7.85 (d, 1 H, Ar), 7.96–7.99 (m, 1 H, Ar), 8.08–8.11 (m, 1 H, Ar), 8.60 (s, 2 H, NH2), 8.92 (s, 2 H, NH2). Anal. (C21H21N5 • 1.5HCl) C, H, N.
2,4-Diamino-6-[N-(β-naphthyl)-N-ethylamino]quinazoline 7
Compound 7 was synthesized from 18 (0.32 g, 1.01 mmol), acetaldehyde (0.27 mL, 4.85 mmol), and NaCNBH3 (0.17 g, 2.7 mmol) in 50 mL of CH3CN. Compound 7 was filtered from the reaction mixture to yield an analytically pure solid (0.23 g, 67%): mp 278–280 °C; TLC (solvent B) Rf 0.45. 1H NMR (DMSO-d6) δ 1.12–1.17 (t, 3 H, N-CH2-CH3), 3.56–3.58 (q, 2 H, N-CH2-CH3) 4.77 (s, 2 H, 10-CH2), 7.27 (s, 2 H, Ar), 7.40–7.50 (m, 4 H, Ar, 2 H, NH2), 7.72 (s, 1 H, Ar), 7.81–7.89 (m, 3 H, Ar), 8.62 (br, 2 H, NH2). Anal. (C21H21N5 • 1.0HCl) C, H, N.
2,4-Diamino-6-[N-(2,5-dimethoxybenzyl)-N-ethylamino]quinazoline 8
Compound 8 was synthesized from 19 (0.24 g, 0.75 mmol), acetaldehyde (0.20 mL, 3.6 mmol), and NaCNBH3 (0.125 g, 2.0 mmol) in 50 mL of CH3CN. Compound 8 was purified by column chromatography to yield an analytically pure bright-yellow solid (0.16 g, 59%): mp 200–204 °C; TLC (solvent B) Rf 0.43. 1H NMR (DMSO-d6) δ 1.05 (br, 3 H, N-CH2-CH3), 3.36–3.38 (br, 2 H, N-CH2-CH3), 3.52 (s, 3 H, OCH3), 3.73 (s 3 H, OCH3), 4.38 (s, 2 H, 10-CH2), 5.66 (br, 2 H, NH2), 6.52 (s, 1 H, Ar), 6.72 (m, 1 H, Ar), 6.86–6.94 (m, 3 H, Ar), 7.02–7.05 (d, 1 H, Ar), 7.12–7.17 (br, 2 H, NH2). Anal. (C19H23N5O2 • 0.7H2O) C, H, N.
2,4-Diamino-6-[N-(2,3,5-trichlorobenzyl)-N-ethylamino]quinazoline 9
Compound 9 was synthesized from 20 (0.20 g, 0.55 mmol), acetaldehyde (0.15 mL, 2.7 mmol), and NaCNBH3 (0.10 g, 1.65 mmol) in 20 mL of CH3CN. Compound 9 was purified by column chromagraphy to yield an analytically pure bright-yellow solid (0.05 g, 21%): mp 222–224 °C; TLC (solvent B) Rf 0.46. 1H NMR (DMSO-d6) δ 1.1–1.14 (t, 3 H, N-CH2-CH3), 3.45–3.49 (q, 2 H, N-CH2-CH3), 4.56 (s, 2 H, 10-CH2), 5.65 (s, 2 H, NH2), 7.05–7.06 (d, 2 H, Ar), 7.11–7.16 (m, 4 H, Ar, NH2), 7.74–7.75 (d, 1 H, Ar). Anal. (C21H21N5Cl3 • 0.3H2O) C, H, N, Cl.
2,4-Diamino-6-[N-(2,5-dimethoxybenzyl)-N-propylamino]quinazoline 10
Compound 10 was synthesized from 19 (0.20 g, 0.62 mmol), propionaldehyde (0.11 g, 1.8 mmol), and NaCNBH3 (0.116 g, 1.8 mmol) in 50 mL of CH3CN. Compound 10 was purified by column chromatography to yield an analytically pure solid (13%): mp 172–173 °C; TLC (solvent B) Rf 0.58. 1H NMR (DMSO-d6) δ 0.86–0.91 (t, 3 H, N-CH2–CH2-CH3), 1.53–1.58 (m, 2 H, N-CH2-CH2-CH3), 3.31 (br, N-CH2-CH2–CH3), 3.56 (s, 3 H, OCH3), 3.78 (3.3 H, OCH3), 4.46 (s, 2 H, 10-CH2), 5.71 (s, 2 H, NH2), 6.52–6.53 (d, 1 H, Ar), 6.72–6.76 (m, 1 H, Ar), 6.90–7.00 (m, 2 H, Ar), 7.07–7.14 (d, 2 H, Ar), 7.23 (br, 2 H, NH2). Anal. (C20H25N5O2 • 0.5H2O) C, H, N.
2,4-Diamino-6-[N-(β-naphthyl)-N-propylamino]quinazoline 11
Compound 11 was synthesized from 18 (0.20 g, 0.63 mmol), propionaldehyde (0.74 g, 12.7 mmol), and NaCNBH3 (0.12 g, 2.0 mmol) in 50 mL of CH3CN. Compound 11 was purified by column chromatography to yield an analytically pure solid (46%): mp 189.0–190.7 °C; TLC (solvent B) Rf 0.42. 1H NMR (DMSO-d6) δ 0.86–0.91 (t, 3 H, N-CH2–CH2-CH3), 1.56–1.66 (t, 2 H, N-CH2-CH2-CH3), 4.71 (s, 2 H, 10-CH2), 5.67 (br, 2 H, NH2), 7.21–7.29 (m, 4 H, Ar, NH2), 7.40–7.46 (m, 3 H, Ar), 7.72 (s, 1 H, Ar), 7.79–7.86 (m, 3 H, Ar). Anal. (C22H23N5 • 0.3H2O) C, H, N.
2,4-Diamino-6-[N-(2,5-dimethoxybenzyl)-N-cyclopropyl methylamino]quinazoline 12
Compound 12 was synthesized from 19 (0.20 g, 0.62 mmol), cyclopropanecarboxaldehyde (0.86, 12.3 mmol), and NaCNBH3 (0.12 g, 1.8 mmol) in 50 mL of CH3CN. Compound 12 was purified by column chromatography to yield an analytically pure solid (21%): mp 122.7–123.5 °C; TLC (solvent B) Rf 0.54. 1H NMR (DMSO-d6) δ 0.18–0.19 (d, 2 H, N-CH2-cyclopropyl), 0.41–0.44 (d, 2 H, N-CH2-cyclopropyl), 1.06–1.08 (m, 1 H, N-CH2-cyclopropyl), 3.29–3.31 (d, 2 H, N-CH2-cyclopropyl), 3.56 (s, 3 H, OCH3), 3.79 (3.3 H, OCH3), 4.56 (s, 2 H, 10-CH2), 5.80 (br, 1 H, NH2), 6.58 (s, 1 H, Ar), 6.72–6.75 (d, 1 H, Ar), 6.90–6.93 (d, 1 H, Ar), 7.0–7.035 (d, 1 H, Ar), 7.08–7.10 (d, 1 H, Ar), 7.24 (s, 1 H, Ar), 7.29 (br, 1 H, NH2). Anal. (C21H25N5O2 • 0.6H2O) C, H, N.
2,4-Diamino-6-[N-(β-naphthyl)-N-cyclopropyl methylamino]quinazoline 13
Compound 13 was synthesized from 18 (0.20 g, 0.63 mmol), cyclopropanecarboxaldehyde (0.89 g, 12.7 mmol), and NaCNBH3 (0.12 g, 2.0 mmol) in 50 mL of CH3CN. Compound 13 was purified by column chromatography to yield an analytically pure solid (9%): mp 136–138 °C (dec); TLC (solvent B) Rf 0.58. 1H NMR (DMSO-d6) δ 0.20–0.21 (d, 2 H, N-CH2-cyclopropyl), 0.42–0.45 (d, 2 H, N-CH2-cyclopropyl), 1.06–1.11 (m, 1 H, N-CH2-cyclopropyl), 4.80 (s, 2 H, 10-CH2), 6.00 (br, 1 H, NH2), 7.08 (d, 1 H, Ar), 7.21 (d, 1 H, Ar), 7.32 (s, 1 H, Ar), 7.43 (m, 4 H, Ar), 7.24 (s, 1 H, Ar), 7.74–8.00 (m, 3 H, Ar, NH2). Anal. (C23H23N5 • 1.1H2O) C, H, N.
Supplementary Material
Acknowledgment
This work was supported in part by grants from the National Institute of Allergy and Infectious Diseases, NIH, AI069966 (AG) and AI41743 (AG).
Footnotes
Abbreviations: pc, Pneumocystis carinii; tg, Toxoplasma gondii; DHFR, dihydrofolate reductase; HIV, human immunodeficiency virus; PCP, Pneumocystis pneumonia; AIDS, acquired immunodeficiency syndrome; TMP, Trimethoprim; PTX, piritrexim; TMQ, trimetrexate; ma, Mycobacterium avium.
Supporting Information Available: Detailed elemental analysis data for compounds 5–13. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Seage GR, Losina E, Goldie SJ, Paltiel AD, Kimmel AD, Freedberg KA. The Relationship of Preventable Opportunistic Infections, HIV-1 RNA, and CD4 Cell Counts to Chronic Mortality. JAIDS, J. Acquired Immune Defic. Syndr. 2002;30:421–428. doi: 10.1097/00042560-200208010-00008. [DOI] [PubMed] [Google Scholar]
- 2.Klepser ME, Klepser TB. Drug Treatment of HIV-Related Opportunistic Infections. Drugs. 1997;53:40–73. doi: 10.2165/00003495-199753010-00004. [DOI] [PubMed] [Google Scholar]
- 3.DeClercq E. Toward Improved Anti-HIV Chemotherapy: Therapeutic Strategies for Intervention with HIV Infections. J. Med. Chem. 1995;38:2491–2517. doi: 10.1021/jm00014a001. [DOI] [PubMed] [Google Scholar]
- 4.Allegra CJ, Kovacs JA, Drake JC, Swan JC, Chabner BA, Masur H. Activity of Antifolates against Pneumocystis carinii Dihydrofolate Reductase and Identification of a Potent New Agent. J. Exp. Med. 1987;165:926–931. doi: 10.1084/jem.165.3.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischl MA, Dickinson GM, LaVoie L. Safety and Efficacy of Sulfamethoxazole and Trimethoprim Chemoprophylaxis for Pneumocystis carinii Pneumonia in AIDS. J. Am. Med. Assoc. 1988;259:1185–1189. doi: 10.1001/jama.259.8.1185. [DOI] [PubMed] [Google Scholar]
- 6.Roudier C, Caumes E, Rogeauz O, Bricaire F, Gentilini M. Adverse Cutaneous Reactions to Trimethoprim-Sulfamethoxazole in Patients with the Acquired Immunodeficiency Syndrome and Pneumocystis carinii Pneumonia. Arch. Dermatol. 1994;30:1383–1386. [PubMed] [Google Scholar]
- 7.Allegra CJ, Chabner BA, Tuazon CU, Ogata-Arakaki D, Baird B, Drake JC, Simmons JT, Lack EE, Shelhamer JH, Balis F, Walker R, Kovacs JA, Lane HC, Masur H. Trimetrexate for the Treatment of Pneumocystis carinii Pneumonia in Patients with the Acquired Immunodeficiency Syndrome. N. Engl. J. Med. 1987;317:978–985. doi: 10.1056/NEJM198710153171602. [DOI] [PubMed] [Google Scholar]
- 8.Masur H, Polis MA, Tuazon CV, Ogota-Arakaki D, Kovacs JA, Katz D, Hilt D, Simmons T, Feuerstein I, Lundgren B, Lane HC, Chabner BA, Allegra CJ. Salvage Trial of Trimetrexate-Leucovorin for the Treatment of Cerebral Toxoplasmosis. J. Infect. Dis. 1993;167:1422–1426. doi: 10.1093/infdis/167.6.1422. [DOI] [PubMed] [Google Scholar]
- 9.Gangjee A, Vasudevan A, Queener SF, Kisliuk RL. 2,4-Diamino-5-Deaza-6-Substituted Pyrido[2,3-d]pyrimidine Antifolates as Potent and Selective Nonclassical Inhibitors of Dihydrofolate Reductases. J. Med. Chem. 1996;39:1438–1446. doi: 10.1021/jm950786p. [DOI] [PubMed] [Google Scholar]
- 10.News. FDA Approves Trimetrexate as Second line Therapy of Pneumocystis carinii Pneumonia. Am. J. Hosp. Pharm. 1994;51:591–592. [PubMed] [Google Scholar]
- 11.Gangjee A, Zeng Y, McGuire JJ, Kisliuk RL. Synthesis of N-[4-[1-Ethyl-2-(2,4-Diaminofuro[2,3-d]pyridimin-5-yl)-ethyl]benzoyl]-l-glutamic Acid as an Antifolate. J. Med. Chem. 2002;45:1942–1948. doi: 10.1021/jm010575m. [DOI] [PubMed] [Google Scholar]
- 12.Gangjee A, Adair O, Queener SF. Synthesis of 2,4-Diamino-6-(thioarylmethyl)pyrido[2,3-d]pyrimidines as Dihydrofolate Reductase Inhibitors. Bioorg. Med. Chem. 2001;9:2929–2935. doi: 10.1016/s0968-0896(01)00223-1. [DOI] [PubMed] [Google Scholar]
- 13.Gangjee A, Yu J, McGuire JJ, Cody V, Galitsky N, Kisliuk RL, Queener SF. Design, Synthesis and X-ray Crystal Structure of a Potent Dual Inhibitor of Thymidylate Synthase and Dihydrofolate Reductase as an Antitumor Agent. J. Med. Chem. 2000;43:3837–3851. doi: 10.1021/jm000200l. [DOI] [PubMed] [Google Scholar]
- 14.Gangjee A, Zeng Y, McGuire JJ, Kisliuk RL. Effect of C-9 Methyl Substitution and C8–C9 Conformational Restriction on Antifolate and Antitumor Activity of Classical 5-Substituted 2,4-Diaminofuro[2,3-d]pyrimidines. J. Med. Chem. 2000;43:3125–3133. doi: 10.1021/jm000130i. [DOI] [PubMed] [Google Scholar]
- 15.Gangjee A, Adair O, Queener SF. Pneumocystis carinii and Toxoplasma gondii Dihydrofolate Reductase Inhibitors and Antitumor Agents: Synthesis and Biological Activities of 2,4-Diamino-5-methyl-6-[(monosubstituted anilino)methyl]-pyrido[2,3-d]pyrimidines. J. Med. Chem. 1999;42:2447–2455. doi: 10.1021/jm990079m. [DOI] [PubMed] [Google Scholar]
- 16.Gangjee A, Zhu Y, Queener SF. 6-Substituted 2,4-Diaminopyrido[3,2-d]pyrimidine Analogues of Piritrexim as Inhibitors of Dihydrofolate Reductase from Rat Liver, Pneumocystis carinii, and Toxoplasma gondii and as Antitumor Agents. J. Med. Chem. 1998;41:4533–4541. doi: 10.1021/jm980206z. [DOI] [PubMed] [Google Scholar]
- 17.Rosowsky A, Papoulis AT, Forsch RA, Queener SF. Synthesis and Antiparasitic and Antitumor Activity of 2,4-Diamino-6-(Arylmethyl)-5,6,7,8-Tetrahydroquinazoline Analogues of Piritrexim. J. Med. Chem. 1999;42:1007–1017. doi: 10.1021/jm980572i. [DOI] [PubMed] [Google Scholar]
- 18.Robson C, Meek MA, Grunwaldt J-D, Lambert PA, Queener SF, Schmidt D, Griffin RJ. Nonclassical 2,4-Diamino-5-aryl-6-ethylpyrimidine Antifolates: Activity as Inhibitors of Dihydrofolate Reductase from Pneumocystis carinii and Toxoplasma gondii and as Antitumor Agents. J. Med. Chem. 1997;40:3040–3048. doi: 10.1021/jm970055k. [DOI] [PubMed] [Google Scholar]
- 19.Stevens MFG, Phillip KS, Rathbone DL, O’Shea DM, Queener SF, Schwalbe CH, Lambert PA. Structural Studies on Bioactive Compounds. 28. Selective Activity of Triazenyl-Substituted Pyrimethamine Derivatives against Pneumocystis carinii Dihydrofolate Reductase. J. Med. Chem. 1997;40:1886–1893. doi: 10.1021/jm970050n. [DOI] [PubMed] [Google Scholar]
- 20.Piper JR, Ramamurthy B, Johnson CA, Otter GM, Sirotnak FM. Analogues of 10-Deazaaminopterin and 5-Alkyl-5,10-dideazaaminopterin with the 4-Substituted 1-Naphthoyl Group in the Place of 4-Substituted Benzoyl. J. Med. Chem. 1996;39:614–618. doi: 10.1021/jm9506940. [DOI] [PubMed] [Google Scholar]
- 21.Kuyper LF, Baccanari DP, Jones ML, Hunter RN, Tansik RL, Joyner SS, Boytos CM, Rudolph SK, Knick B, Wilson HR, Caddell JM, Friedman HS, Comley JCW, Stables JN. High-Affinity Inhibitors of Dihydrofolate Reductase: Antimicrobial and Anticancer Activities of 7,8-Dialkyl-1,3-diaminopyrrolo[3,2-f]quinazolines with Small Molecular Size. J. Med. Chem. 1996;39:892–903. doi: 10.1021/jm9505122. [DOI] [PubMed] [Google Scholar]
- 22.Kovacs JA, Allegra CJ, Swan JC, Parillo JE, Chabner BA, Masur H. Potent Antipneumocystis and Antitoxoplasma Activities of Piritrexim, a Lipid Soluble Antifolate. Antimicrob. Agents Chemother. 1988;32:430–433. doi: 10.1128/aac.32.4.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gangjee A, Vidwans AP, Vasudevan A, Queener SF, Kisliuk RL, Cody V, Li R, Galitsky N, Luft JR, Pangborn W. Structure-Based Design and Synthesis of Lipophilic 2,4-Diamino-6-Substituted Quinazolines and Their Evaluation as Inhibitors of Dihydrofolate Reductases and Potential Antitumor Agents. J. Med. Chem. 1998;41:3426–3434. doi: 10.1021/jm980081y. [DOI] [PubMed] [Google Scholar]
- 24.Cody V, Wojtczak A, Kalman TI, Friesheim JH, Blakley RL. Conformational Analysis of Human Dihydrofolate Reductase Inhibitor Complexes: Crystal Structure Determination of Wild-Type and F31 Mutant Binary and Ternary Inhibitor Complexes. Adv. Exp. Med. Biol. 1993;338:481–486. doi: 10.1007/978-1-4615-2960-6_97. [DOI] [PubMed] [Google Scholar]
- 25.Champness JN, Achari A, Ballantine SP, Bryant PIK, Delves CJ, Stammers DK. The Structure of Pneumocystis carinii Dihydrofolate Reductase to 1.9 A Resolution. Structure. 1994;2:915–924. doi: 10.1016/s0969-2126(94)00093-x. [DOI] [PubMed] [Google Scholar]
- 26.Cody V, Galitsky N, Luft JR, Pangborn W, Queener SF, Gangjee A. Analysis of Quinazoline and Pyrido[2,3-d]pyrimidine N9–C10 Reversed-bridged Antifolates in Complex with NAPD+ and Pneumocystis carinii Dihydrofolate Reductase. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002;58:1393–1399. doi: 10.1107/S0907444902010442. [DOI] [PubMed] [Google Scholar]
- 27.Molecular Distance Measurements with SYBYL 6.3. St. Louis, MO: Tripos Associates, Inc.: 1699 S. Hanely Road, Suite 303; 63144 pp. [Google Scholar]
- 28.Gangjee A, Guo X, Queener SF, Cody V, Galitsky N, Luft JR, Pangborn W. Selective Pneumocystis carinii Dihydrofolate Reductase Inhibitors: Design, Synthesis and Biological Evaluation of New 2,4-Diamino-5-substituted-furo[2,3-d]pyrimidines. J. Med. Chem. 1998;41:1263–1271. doi: 10.1021/jm970537w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.