

Abstract
Key Clinical Message
A patient with syndromic Duane retraction syndrome harbors a chromosome 811.1q13.2 inversion and 8p11.1-q12.3 marker chromosome containing subregions with differing mosaicism and allele frequencies. This case highlights the potential requirement for multiple genetic methods to gain insight into genotype–phenotype correlation, and ultimately into molecular mechanisms that underlie human disease.
Keywords: 8q12 microduplication syndrome, copy number variation, cytogenetics, Duane retraction syndrome, DURS1
Introduction
Duane retraction syndrome (DRS) occurs in approximately 1 in 1000 individuals and most commonly manifests as limited abduction with globe retraction on attempted adduction. It is believed to result from errors in the development of the abducens nucleus or nerve, and aberrant innervation of the lateral rectus muscle by axons of the oculomotor nerve. While dominant DRS pedigrees can harbor mutations in alpha-chimerin (CHN1) [1] or Sal-like protein 4 (SALL4), [2,3] most cases of DRS are simplex and genetically undefined. For almost two decades, rare patients with simplex, syndromic DRS have been reported to harbor cytogenetic abnormalities in the chromosomal region 8q12-8q13 that define the DURS1 locus, as summarized below and in Figure 1.
Figure 1.
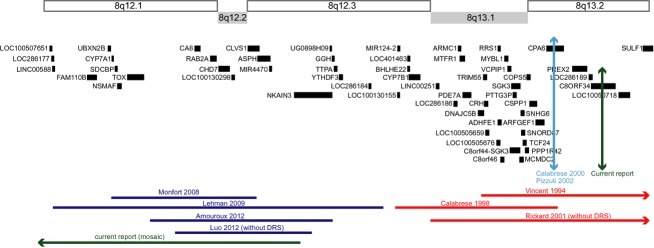
Schematic of the DURS1 region. Horizontal lines at the top of the page indicate cytogenetic bands 8q12.1-8q13.2. Under these bands are genes in the region as per the UCSC Genome Browser hg 19 (genome.ucsc.edu). Previous reports of duplications (blue) and deletions (red) are indicated by horizontal lines at the bottom of the figure, and labeled according to the first author and year of the corresponding report. The previously reported translocation breakpoint disrupting CPA6 is denoted by a vertical light blue line. The mosaic duplication and the translocation breakpoint found in the patient in the current report are denoted by a green horizontal and green vertical line, respectively. An arrow at the end of a horizontal line denotes that the deletion or duplication extends further in the indicated direction.
The initial three patients that defined the DURS1 locus harbored a deletion or had a translocation breakpoint at 8q13. The first patient had DRS, branchiootorenal syndrome, hydrocephalus, trapezius muscle aplasia, and a large de novo interstitial deletion del(8)(q13.1-q21.11) originating on the paternal allele [4]. The second patient had bilateral DRS type 1, severe intellectual disabilities, microcephaly, dysmorphisms, brachydactyly and left club foot, and harbored an insertion of 8q11.2-q13 into 6q25 with a deletion, del(8)(q12.3q13.2) [5]. The third patient had DRS, dysgenetic gonads, hypoplastic external genitalia and glandular hypospadias, and a de novo reciprocal translocation t(6;8)(q26;q13) with the chromosome 8q13 translocation breakpoint located within intron 1 of carboxypeptidase A6 (CPA6) [6,7]. Notably, a fourth patient with branchiootorenal syndrome but not DRS is reported to harbor a deletion from distal 8q13.1 through 8q21.13 [8].
Recently, three patients with DRS were reported to harbor 8q12 microduplications [9–11]. Their phenotypes included DRS, sensorineural deafness, intellectual disabilities, hypotonia, dysmorphisms, and congenital heart and kidney defects [9–11]. The three patients share a 1.2 Mb duplicated region encompassing carbonic anhydrase VIII (CA8), RAS-associated protein RAB2 (RAB2A), chromodomain helicase DNA binding protein 7 (CHD7), and clavesin 1 (CLVS1) (Fig.1). A fourth patient harboring a 2.7 Mb 8q12 microduplication also encompassing these four genes had dysmorphic features, congenital heart defect, and torticollis, but did not exhibit DRS [12].
In this study, we describe a boy with syndromic DRS and complex structural variations involving both 8q12 and 8q13.
Material and Methods
Participant enrollment
The proband and his maternal grandmother participated in an ongoing genetic study of DRS at Boston Children's Hospital, and provided written informed consent to a protocol conforming to the Declaration of Helsinki and approved by Boston Children's Hospital institutional review board. The parents of the proband were not available for participation. Medical and ophthalmologic history and physical examination findings were obtained from medical records. Both participants provided a blood sample for DNA extraction, and the proband also provided a sample for cell line generation.
Cell line generation
Epstein-Barr virus transformation was performed by the Biosample Services Facility at Partners Center for Personalized Genetic Medicine, Cambridge MA, for initiation of a lymphoblastoid cell line.
Probe preparation for fluorescence in situ hybridization
Bacterial artificial chromosome (BAC) clones were selected using University of California, Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu; hg19) and obtained from Children's Hospital Oakland Research Institute (CHORI, Oakland, CA). BAC DNA was isolated using standard protocols and labeled directly with either SpectrumGreen- or SpectrumOrange-conjugated dUTP following the manufacturer's instructions (Nick Translation Kit, catalog no.: 32-801300; Abbott Laboratories. IL). Cot-I DNA (10 μL) was added for every 1 μg of labeled probe to suppress repetitive sequences, and probes were ethanol precipitated and resuspended in 50% Hybrisol (50% formamide, 2×saline-sodium citrate [SSC], 10% dextran sulfate) (Abbott Laboratories).
Fluorescence in situ hybridization
Metaphase chromosomes were prepared using standard cytogenetic protocols [13]. Fluorescence in situ hybridization (FISH) was performed with direct-labeled BAC probes to map each inversion breakpoint. Probes were hybridized in differentially labeled pairs (SpectrumGreen and SpectrumOrange [Vysis, Abbott Laboratories]). The telomeric inversion breakpoint was mapped using BAC clones RP11-89A16 (8q12.3-8q13.1), RP11-282D10 (8q13.1), RP11-212P10 (8q13.1), RP11-271O1 (8q13.1), RP11-343B22 (8q13.2), and RP11-131P18 (8q13.2), and refined using 8q13.2 BAC clones RP11-396J6, RP11-566L6, RP11-664D7, 349K17, RP11-159C14, RP11-50A22, RP11-779P1, and RP11-939K17. The centromeric inversion breakpoint was mapped using RP11-726G23 (8p11.21-8p11.1), RP11-8790P20 (8q11.21), RP11-598P20 (8p11.21), RP11-1031I13 (8q11.1), and 1102L10 and 1130I3 (8q11.21).
Probes and chromosomes were codenatured at 72°C for 2 min and hybridized overnight at 37°C in a HYBrite apparatus (Abbott Molecular/Vysis). Slides were washed in 50% formamide/2×SSC at 37°C for 20 min and 2×SSC at 37°C for 20 min. 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI) was used as counterstain. Hybridization results were assessed with a Zeiss Axioskop 2 epifluorescence microscope (Thornwood, NY) or an Olympus BX51 microscope (Center Valley, PA), and images were acquired with an Applied Imaging CytoVision cytogenetics workstation (Santa Clara, CA). A minimum of 10 metaphases was scored per hybridization.
Chromosomal microarray analysis
Chromosomal microarray analysis (CMA) studies were performed to detect copy number variation (CNV) using two different platforms. A custom high-resolution microarray was designed to target the DURS1 region (hg19; chr8: 41,880,843-74,837,446). Overlapping probes of 50–60 bases in length were tiled across the DURS1 region beginning every ∼10 bases (8p11.21-8q13.3) (Roche Nimblegen, Madison, WI). The experiment was performed twice, using standard dye swap.
An Illumina HumanOmniExpress BeadChip array composed of ∼730 K single nucleotide polymorphisms (SNPs) (Illumina, San Diego, CA) was performed following the manufacturer's directions. Data were evaluated and analyzed using Illumina's GenomeStudio v2011.1 and Nexus CN 7.0 Standard Edition software (updated on 19 April 2012). The Nexus analysis settings used for reporting CNV(s) were as follows: SNP-FASST2 segmentation; significance threshold = 1.0E−9; max contiguous probe spacing (Kbp) = 1000.0; min number of probes per segment = 15; Log-R thresholds were high gain = 0.41; gain = 0.13; loss = −0.23; big loss = −1.1; sex chromosome gain (3:1) = 1.2; sex chromosome gain (4:1) = 1.7; homozygous frequency threshold = 0.95; homozygous value threshold = 0.8; heterozygous imbalance threshold = 0.4; minimum SNP probe density (Probes/MB) = 0.0; regions minimum size (Kbp) = 50. The HumanOmniExpress BeadChip SNP CMA experiment was carried out twice with similar results.
Results
Clinical history and examination
The proband was evaluated at 12.5 years of age. He was born at term to a 15-year-old mother, with birth weight of 3266 g (25–50 percentile) and length of 53 cm (75–90 percentile). He had neonatal apnea that resolved without treatment and an otherwise unremarkable neonatal course. On initial hearing evaluation left conductive hearing loss was reported, but repeat testing was normal. The patient had numerous ear infections, frequent respiratory infections, and asthma. Gastroesophageal reflux had been diagnosed by pH probe. He underwent correction surgeries for right esotropia and cleft uvula and submucous cleft palate.
Developmental testing revealed learning disabilities, fine and gross motor delays, and articulation difficulties. His WISC-III full-scale IQ was 90 when tested at 9 years and at 11 years of age. He had been diagnosed with panic disorder, anxiety disorder, attention deficit hyperactivity disorder, and adjustment disorder, and subsequently treated with Paroxetine and Methylphenidate. Additional clinical investigations had included magnetic resonance imaging of the brain, electroencephalogram, sleep study, and abdominal sonogram, all reported as normal. Echocardiography at age 11 was normal except for false tendons in the left ventricle. DNA testing for Fragile X and FISH for chromosome 22q11.2 microdeletion associated with velocardiofacial syndrome were normal.
The biological mother was of Middle Eastern and Irish ancestry. She completed 10th grade, obtained a general education diploma, and was reported to have normal cognition. Both she and her maternal half-sister were reported to have pectus carinatum and leg length discrepancies. The boy's biological father was of Puerto Rican ancestry. Several paternal half-siblings were reported to have motor delays but no additional details are available. No relative was known to have DRS, cleft palate, or dysmorphic features.
On examination at 12.5 years of age, height was 163.25 cm (95th percentile), weight was 47.25 kg (75th percentile), and head circumference was 53.9 cm (50th percentile). Bilateral DRS was noted and was more severe on the right (Fig.2A and B). Dysmorphic features included synophrys, almond-shaped palpebral fissures, flat midface, high nasal bridge, malar hypoplasia, and inverted W-shaped posterior hairline (Fig.2A). Ears were prominent and measured 6.9 cm (90th percentile). Palm length was 10.2 cm (85th percentile) and middle finger length was 8.2 cm (97th percentile). He had slight asymmetric pectus carinatum with hypoplastic right first rib noted on radiograph, mild metatarsus adductus, flat feet, and wide gap between the first and second toes. Pubic hair was Tanner II, with testes measuring 5 mL.
Figure 2.

Photographs of the proband. (A) Dysmorphic facial features included synophrys (which has been shaved), almond-shaped palpebral fissures, flat midface, high nasal bridge, and malar hypoplasia. (A) and (B) Primary positions of gaze reveal bilateral DRS, more pronounced in the right eye. Note relatively well aligned central gaze (A), with limited abduction of the right > left eye and narrowing of the right palpebral fissure on attempted adduction (B). Up and downgaze are relatively preserved (B).
Karyotype reveals a complex chromosome 8 inversion and marker chromosome
Chromosome analysis revealed a pericentric inversion of chromosome 8 between the centromere and the long arm and mosaicism for a supernumerary marker chromosome: 47,XY,inv(8)(p11.1q13.2),+mar[11]/46,XY,inv(8)(p11.1q13.2)[9] (Fig.3A). Multiplex fluorescence in situ hybridization (M-FISH) confirmed that the marker chromosome was derived from chromosome 8 (Fig.3B). The parents were not available for participation and, thus, their karyotypes are not known.
Figure 3.
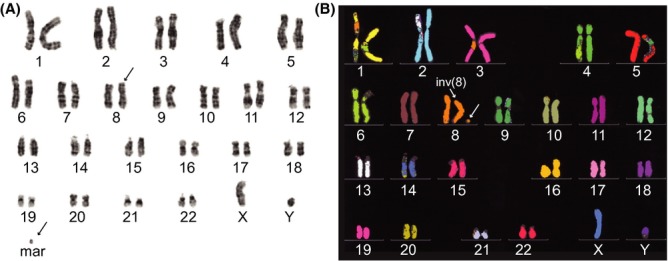
Cytogenetic analysis of proband peripheral blood lymphocyte chromosomes. (A) GTG-banded karyotype revealed 47,XY,inv(8)(p11.1q13.2),+mar[11]/46,XY,inv(8)(p11.1q13.2)[9]. (B) M-FISH confirms that the marker chromosome is derived from chromosome 8.
Telomeric breakpoint reveals an intragenic rearrangement of the chromosome 8 open reading frame 34 gene (C8ORF34)
The proband's transformed lymphoblasts were analyzed by FISH to define the chromosome 8 inversion breakpoints. The 8p11.1 breakpoint was confirmed through successive BAC hybridizations; the centromere marker, CEP8, but no BAC clone was disrupted in mutant cells, consistent with the original karyotype (Fig.4A–C). The 8q13.2 inversion breakpoint was defined by the inversion of probe RP11-50A22 at chr8:69,471,542-69,634,621 (Fig.4A and B) but not of the more telomeric probes RP11-779P1 at chr8:69,621,417-69,803,905 (Fig.4D, E) and RP11-865I6.2 at chr8:69,760,977-69,764,998 (Fig.4F). Although inverted probe RP11-50A22 overlaps with noninverted probe RP11-779P1 by ∼13 kb, we did not visualize a split of either BAC, suggesting the split occurs within or near the region of overlap. In a structurally normal chromosome 8, C8ORF34 maps to 8q13.2 and its 14 exons are transcribed in a centromeric (5′) to telomeric (3′) direction. The inverted BAC probe RP11-50A22 includes C8ORF34 exons 8–10, while the noninverted BAC probe RP11-779P1 includes C8ORF34 exons 10–14. Thus, these data support an intragenic breakpoint of C8ORF34 between exons 7–14, and map the telomeric breakpoint maximum critical region to 293 kb between hg19: chr8:69,471,542 and 69,764,998 defined by the start of RP11-50A22 and the end of RP11-865I6.2 (Fig.4F).
Figure 4.

FISH analysis of the proband's lymphoblasts. FISH from two cells with cohybridization of CEP8 centromere probe (green), a telomeric BAC probe for chromosome 8 (tel. marker; red) and BAC probes RP11-50A22 (red, left photo) or RP11-779P1 (red, right photo). Chromosomes A, B, D, and E are enlarged and accompanied by schematics in the upper aspect of the figure. (Chromosome A) A normal chromosome 8 (chr.8) shows the expected signal pattern for RP11-50A22. (Chromosome B) An inv(8) chromosome shows a split CEP8 signal (green) with the RP11-50A22 signal (red) falling between the split CEP8 green signals, consistent with its inversion. This places the BAC centromeric to the inversion breakpoint. (Chromosomes C) The mosaic chromosome 8 marker in each cell contains a CEP8 signal. (Chromosome D) A normal chromosome 8 (chr.8) shows the expected signal pattern for RP11-779P1. (Chromosome E) An inv(8) shows the split CEP8 signal (green) and an intact red signal for RP11-779P1 telomeric to the split CEP8 signal, consistent with its normal orientation. (F) Schematic of the location of the BAC probes along 8q13.2. BAC probes denoted in blue were inverted, while BAC probes denoted in orange had a normal orientation. Thus, the translocation breakpoint falls within the region defined by the start of RP11-50A22 and the end of RP11-865I6.2. This critical region (∼293 Kb) encompasses C8ORF34 on 8q13.2.
Chromosomal microarray analysis demonstrates a complex mosaic duplication of chromosome 8p11.1-q12.3
To define further the boundaries of the mosaic duplication arising from the marker chromosome, we undertook CMA of the proband's DNA. CMA analysis using the custom comparative genomic hybridization (CGH) oligonucleotide based microarray shows a 9 Mb copy gain spanning 8q11.2-q12.1 (hg19: chr8:51,488, 197-60,554,196); the duplicated region contains 38 genes but excludes CA8, RAB2A, CHD7, and CLVS1 (Fig.5A).
Figure 5.

CMA results of chromosome 8 pericentromeric region and q arm using two different array platforms. (A) Nimblegen custom CGH oligonucleotide microarray reveals a copy gain spanning 8q11.2-q12.1 containing 38 genes (hg19: chr8:51,490,197-60,554,196). This duplication does not include the genes involved in the critical region of 8q12 microduplication syndrome CA8, RAB2A, CHD7, and CLVS1 highlighted in red. (B) CMA result of chromosome 8q11.21-q12.3 using the Illumina HumanOmniExpress SNP-based array as interpreted by Nexus 7.0. (B-i) Schematic of the chromosomal region annotated from top to bottom as follows: blue bar denotes the region shaded blue in the Log R ratio plot in (B-ii); schematic of chromosomal banding; yellow and purple zygosity bars denote the regions shaded yellow and purple in the B-allele frequency (BAF) plot in (B-iii–v); schematic of genes within the region with CA1, RAB2A, CHD7, and CLVS1 boxed in red; pink bars denote common CNVs. (B-ii) Log R ratio plot in which each black dot represents the log intensity of the corresponding SNP. The region highlighted in blue is consistent with a copy gain. (B-iii–v) BAF plot in which the black dots represent the genotype calls of the SNPs in (B-ii). SNPs that plot at either 0 or 1 are homozygous, SNPs that plot at 0.5 are heterozygous, while SNPs that plot at 0.33 and 0.66 have an allelic imbalance. (B-iii) Yellow rectangle highlights SNPs from 43.5 to 51 Mb with LOH (aside from five SNPs showing the pattern of allelic imbalance). The combined information from the overlapping Log-R-ratio and BAF plots is atypical for copy gains and might reflect a UPD. (B-iv and v) Purple rectangles highlight SNPs from 51 to 63.5 Mb with allelic imbalance which is characteristic of a copy gain. The drop in the Log R ratio between B-iv and B-v may reflect a reduction in the level of mosaicism.
CMA using the SNP-based array revealed a larger and more complex duplication pattern which encompasses the region identified by the oligonucleotide array; the region spans 8p11.1-q12.3 and contains 56 genes (hg19: chr8:43,460,491-63,696,218) including CA8, RAB2A, CHD7, and CLVS1 (Fig.5B-i–v). Notably, this region contains several contiguous but distinctive patterns of duplication. The pericentromeric area, highlighted in yellow (Fig.5B-iii), has a Log R ratio of 0.39 within the smaller region on 8p11.1, but a ratio of only 0.15 within the larger region on 8q11.1-8q11.21. More remarkably, this region does not harbor the allelic imbalance predicted within a region of duplication, but instead reveals loss of heterozygosity (LOH). In contrast, the allele frequencies within the regions highlighted in purple (Fig.5B-iv, v) harbor the anticipated allelic imbalance. In addition, the first purple region (Fig.5B-iv) has a Log R ratio of 0.22 and corresponds closely to the region of duplication detected by the oligonucleotide array (Fig.5A). Within the second region highlighted in purple (Fig.5B-v) the Log R ratio falls to 0.15. Thus, the regions labeled 5B-ii and 5B-v both have Log R ratios very close to minimum threshold for copy number gain set at 0.13, and this might account for why the mosaicism was not detected by the oligonucleotide array.
Discussion
The 8q12-8q13 DURS1 locus is defined by two patients with syndromic DRS harboring deletions beginning at 8q12 and extending in the telomeric direction [4,5] and one patient with a reciprocal translocation disrupting CPA6 on 8q13.2 [6,7]. While all three patients had DRS, their accompanying syndromic features were quite variable. The definition of the DURS1 locus was then expanded by reports of three patients with DRS and 8q12 microduplications who shared syndromic features of DRS, dysmorphism, neonatal hypotonia, and motor developmental delay [9–11]. Analyses of studies performed to date (Fig.1) reveal that the deleted and the duplicated chromosomal regions within the DURS1 locus are nonoverlapping.
Herein, we report a 12.5-year-old boy with syndromic DRS whose analysis further highlights the complexity of cytogenetic abnormalities that can occur at the DURS1-DRS locus. We find that he has a unique constellation of features associated with DRS, and has both a chromosome 8 inversion that transposes highly repetitive centromeric DNA and multiple 8q genes (8p11.1-8q13.2), and a complex mosaic supernumerary marker chromosome containing 8p11.1-8q12.3 material.
Using FISH, we successfully mapped the telomeric inversion breakpoint to a 293 Kb interval within C8ORF34. C8ORF34 is a cDNA isolated from a human vestibular library which encodes an uncharacterized protein containing a putative cAMP-dependent protein kinase regulatory subunit expressed in adult brain, eye, ear, pituitary gland, thymus, kidney, and stomach (UCSC genome browser http://genome.ucsc.edu and Stanford SOURCE search gene report, http://source.stanford.edu). Thus, alteration or loss of C8ORF34 function in brain and eye could potentially contribute to the patient's DRS and intellectual and social disabilities. The translocation disrupting CPA6 [7] and the inversion disrupting C8ORF34 (this report) support disruption of these genes, or regulatory elements, in DURS1-DRS. These genes are also deleted in the patient reported with branchiootorenal syndrome without DRS [8], however, suggesting that simply deleting these genes is not adequate to cause DRS, or that DRS is not fully penetrant. The chromosome 8 inversion also transposes highly repetitive centromeric DNA to the long arm of chromosome 8. We are not aware of phenotypic sequelae from germline changes in centromeric repetitive DNA sequence. There is, however, data suggesting that repetitive elements can have epigenetic influences on gene expression [14,15]. Thus, transposition of centromeric DNA may also contribute to the proband's phenotype.
Concordant with our findings from the karyotype and FISH, interpretation of data from the two different CMA platforms is not straightforward, highlighting the complexity of the molecular mechanisms that have resulted in the apparent chromosomal rearrangement. While the custom CGH microarray demonstrates an 8q11.2-q12.1 duplication that does not include the four genes found to be in the 8q12 microduplication syndrome critical region, the SNP-based array reveals a larger region of duplication that includes these, as well as many additional genes. It has been reported that SNP-based arrays can detect subtle changes, such as low level mosaicism, that are missed on CGH [16], and thus we are confident that the extended duplicated region encompassing the 8q12 microduplication region in this patient is real. However, we cannot provide an explanation for the decrease in the level of mosaicism within the most telomeric portion of the duplicated region, nor for the LOH within the pericentromeric region. LOH regions identified from SNP-based arrays usually indicate consanguinity, uniparental disomy (UPD), or true copy number loss. We are unaware of a family history compatible with consanguinity, and no excess of LOH regions were observed in the CMA at the whole genome level. Notably, however, it has been suggested that at least one third of UPD cases emerge in connection with or due to a chromosomal rearrangement [17]. As the LOH region identified in the q arm shows a borderline Log R ratio for a copy gain, it is possible that the region is actually in a euploid state.
The proband shares some dysmorphic features and motor developmental delay with patients previously described with the germline 8q12 microduplication syndrome, suggesting that duplication of CA8, RAB2A, CHD7, and/or CLVS1 may contribute to DRS. He does not, however, share their heart and kidney malformations, he has less severe intellectual disabilities, and he has additional dysmorphisms, including a submucous cleft palate, not present in the previously described patients. These differences may reflect the low level of mosaic duplication in the patient or effects from the additional regions of duplication and inversion. Moreover, a fifth patient with an 8q12 microduplication encompassing these genes was reported to not have DRS [12], Thus, DRS may not be a fully penetrant feature of the 8q12 microduplication syndrome, or may arise from duplication of the 572 Kb region identified in the four patients with DRS but not in the patient without DRS (Fig1). This region (hg19: chr8:60,219,746-60,792,079) is currently annotated by the UCSC Genome Browser to contain spliced ESTs and long noncoding RNAs but no protein-coding genes.
In summary, these data suggest that the DURS1 locus could result in DRS by dosage effect in the region of 8q1, through deletion on 8q13 and/or a duplication of 8q12, or through alterations in gene expression arising from the inversion breakpoints or transposition of repetitive centromeric sequence. Thus, this case highlights the complexity of human disorders, and the potential requirement for multiple methods (including cytogenetics and different chromosomal microarray platforms) to gain insight into genotype–phenotype correlation, and ultimately into molecular mechanisms that underlie human disease.
Acknowledgments
We thank the family for participating in the study. This study was supported by the National Institutes of Health (R01 EY15298). E. C. E. is a Howard Hughes Medical Institute Investigator.
Conflict of Interest
None declared.
References
- 1.Miyake N, Chilton J, Psatha M, Cheng L, Andrews C, Chan WM, et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane's retraction syndrome. Science. 2008;321:839–843. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Baradie R, Yamada K, St Hilaire C, Chan WM, Andrews C, McIntosh N, et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 2002;71:1195–1199. doi: 10.1086/343821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohlhase J, Heinrich M, Schubert L, Liebers M, Kispert A, Laccone F, et al. Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 2002;11:2979–2987. doi: 10.1093/hmg/11.23.2979. [DOI] [PubMed] [Google Scholar]
- 4.Vincent C, Kalatzis V, Compain S, Levillers J, Slim R, Graia F, et al. A proposed new contiguous gene syndrome on 8q consists of branchio-oto-renal (BOR) syndrome, Duane syndrome, a dominant form of hydrocephalus and trapeze aplasia; implications for the mapping of the BOR gene. Hum. Mol. Genet. 1994;3:1859–1866. doi: 10.1093/hmg/3.10.1859. [DOI] [PubMed] [Google Scholar]
- 5.Calabrese G, Stuppia L, Morizio E, Guanciali F, Pompetti F, Mingarelli R, et al. Detection of an insertion deletion of region 8q13-q21.2 in a patient with Duane syndrome: implications for mapping and cloning a Duane gene. Enuropean. J. Hum. Genet. 1998;6:187–193. doi: 10.1038/sj.ejhg.5200173. [DOI] [PubMed] [Google Scholar]
- 6.Calabrese G, Telvi L, Capodiferro F, Morizio E, Pizzuti A, Stuppia L, et al. Narrowing the Duane syndrome critical region at chromosome 8q13 down to 40 kb. Eur. J. Hum. Genet. 2000;8:319–324. doi: 10.1038/sj.ejhg.5200461. [DOI] [PubMed] [Google Scholar]
- 7.Pizzuti A, Calabrese G, Bozzali M, Telvi L, Morizio E, Guida V, et al. A peptidase gene in chromosome 8q is disrupted by a balanced translocation in a duane syndrome patient. Invest. Ophthalmol. Vis. Sci. 2002;43:3609–3612. [PubMed] [Google Scholar]
- 8.Rickard S, Parker M, van't Hoff W, Barnicoat A, Russell-Eggitt I, Winter RM, et al. Oto-facio-cervical (OFC) syndrome is a contiguous gene deletion syndrome involving EYA1: molecular analysis confirms allelism with BOR syndrome and further narrows the Duane syndrome critical region to 1 cM. Hum. Genet. 2001;108:398–403. doi: 10.1007/s004390100495. [DOI] [PubMed] [Google Scholar]
- 9.Monfort S, Rosello M, Orellana C, Oltra S, Blesa D, Kok K, et al. Detection of known and novel genomic rearrangements by array based comparative genomic hybridisation: deletion of ZNF533 and duplication of CHARGE syndrome genes. J. Med. Genet. 2008;45:432–437. doi: 10.1136/jmg.2008.057596. [DOI] [PubMed] [Google Scholar]
- 10.Lehman AM, Friedman JM, Chai D, Zahir FR, Marra MA, Prisman L, et al. A characteristic syndrome associated with microduplication of 8q12, inclusive of CHD7. Eur. J. Med. Genet. 2009;52:436–439. doi: 10.1016/j.ejmg.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Amouroux C, Vincent M, Blanchet P, Puechberty J, Schneider A, Chaze AM, et al. Duplication 8q12: confirmation of a novel recognizable phenotype with duane retraction syndrome and developmental delay. Eur. J. Hum. Genet. 2012;20:580–583. doi: 10.1038/ejhg.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo H, Xie L, Wang SZ, Chen JL, Huang C, Wang J, et al. Duplication of 8q12 encompassing CHD7 is associated with a distinct phenotype but without duane anomaly. Eur. J. Med. Genet. 2012;55:646–649. doi: 10.1016/j.ejmg.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Ney PA, Andrews NC, Jane SM, Safer B, Purucker ME, Weremowicz S, et al. Purification of the human NF-E2 complex: cDNA cloning of the hematopoietic cell-specific subunit and evidence for an associated partner. Mol. Cell. Biol. 1993;13:5604–5612. doi: 10.1128/mcb.13.9.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haider S, Cordeddu L, Robinson E, Movassagh M, Siggens L, Vujic A, et al. The landscape of DNA repeat elements in human heart failure. Genome Biol. 2012;13:R90. doi: 10.1186/gb-2012-13-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Widschwendter M, Jiang G, Woods C, Muller HM, Fiegl H, Goebel G, et al. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004;64:4472–4480. doi: 10.1158/0008-5472.CAN-04-0238. [DOI] [PubMed] [Google Scholar]
- 16.Schaaf CP, Wiszniewska J, Beaudet AL. Copy number and SNP arrays in clinical diagnostics. Annu. Rev. Genomics Hum. Genet. 2011;12:25–51. doi: 10.1146/annurev-genom-092010-110715. [DOI] [PubMed] [Google Scholar]
- 17.Liehr T. Cytogenetic contribution to uniparental disomy (UPD) Mol. Cytogenet. 2010;3:8. doi: 10.1186/1755-8166-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]