

Abstract
Herein, we describe the enantioselective construction of the 12-membered macrocyclic pyrrole core 4 of marineosin A in 5.1% overall yield from (S)-propylene oxide. The route features a key Stetter reaction to install a 1,4-diketone, which is then subjected to Paal-Knorr pyrrole synthesis and ring closing metathesis (RCM) to afford macrocycle 4. A divergence point in the synthetic scheme also enabled access to a highly functionalized spiroaminal model system 8 via an acid-mediated hydroxyketoamide cyclization strategy.
Keywords: marineosin A, pyrrole, alkaloid, metathesis, Stetter
Introduction
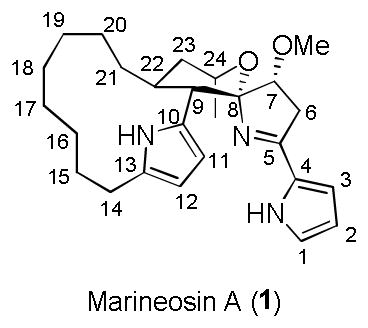
Marineosin A (1) is a novel macrocyclic spirolactam spiroiminal alkaloid isolated in 2008 from a marine-derived Streptomyces-related actinomycete,[1] and is structurally related to the prodigiosin family.[2] Marineosin A displayed potent inhibition against human colon carcinoma cell growth, with an IC50 of 0.5 μM in HCT-116 cells.[1] In the isolation paper, Fenical proposed a biosynthesis of 1 employing an inverse-electron demand hetero Diels-Alder reaction; however, we were unable to demonstrate this proposal synthetically.[1,3] Shortly thereafter, Snider proposed an alternative, prodigiosin-inspired biosynthesis requiring only a two-electron oxidation, which he then validated in a model system of the spiroiminal moeity of 1.[4] Thus, despite significant interest from the synthetic community, limited synthetic efforts have been reported en route to a total synthesis of 1.
In light of our longstanding interest in the synthesis of alkaloids,[5] and 1 in particular,[2,6] we recently reported the synthesis of an unfunctionalized spiroaminal model system of 1 that enabled late stage introduction of the C1–C4 pyrrole moiety.[6] This was a significant development, as synthetic efforts with early stage pyrrole incorporation uniformly led to compound instability and poor results that forced routes to be abandoned. Herein, we describe the enantioselective construction of the macrocyclic pyrrole core 4, and a highly functionalized spiroaminal 8 en route to the total synthesis of 1.
Results and Discussion
Our retrosynthetic analysis for marineosin A (1) is shown in Scheme 1, and relies on late stage introduction of the C1–C4 pyrrole. Spiroaminal 2 can be formed by acid-mediated cyclization of advanced intermediate 3. A Paal-Knorr pyrrole synthesis and ring closing metathesis (RCM) allow for the formation of macrocycle 4 from 1,4-diketone 5. The Stetter reaction will be employed as a key carbon-carbon bond forming reaction en route to 5 from 6. Poly-functionalized 6 is a critical intermediate derived ultimately from Evans’ auxiliary phosphonate 7, vinyl magnesium bromide and (S)-propylene oxide, which sets the stereochemistry. Intermediate 6 also serves as a divergence point to validate the acid-mediated cyclization strategy for 2, and provides access to model system 8.
Scheme 1.

Retrosynthesis of marineosin A (1) and a functionalized spiroaminal lactam 8. TBDPS = tert-butyldiphenylsilyl, TIPS = triisopropylsilyl, Piv = pivalate, Bn = benzyl.
The synthesis of 2 started from commercial (S)-propylene oxide 9 (Scheme 2). Opening of the epoxide with a copper-catalyzed Grignard addition, and in situ silylation of the resulting secondary alcohol provided olefin 10 in 81% yield for the two steps. Ozonolysis afforded aldehyde 11 in 84% yield. The required Evans’ auxiliary phosphonate 7 was prepared in two steps from (R)-oxazolidinone 12 in 73% yield for the two steps. Aldehyde 11 then underwent a Horner-Wadsworth-Emmons olefination with Evans’ auxiliary phosphonate 7 to provide acyloxazolidinone 14 in 75% yield. A copper-catalyzed conjugate addition with allyl magnesium bromide delivered 15 in 81% yield and >20:1 dr.
Scheme 2.

Synthesis of advanced intermediate 15. a) i. 20 mol% CuI, vinyl magnesium bromide, THF, −20 °C, 20 h; ii. TBDPSCl, ImH, CH2Cl2, rt, 4 h, 81% over two steps; b) i. O3, CH2Cl2, 1h, −78 °C; ii. PPh3, rt, 1h, 84%; c) α-bromo acetylbromide, THF, −78 °C to rt, 89%; d) P(OEt)3, 100 °C, 2 h, 82%; e) 7 + 11, NaH, THF, −78 °C to 0 °C, 2 h, 75%; f) 20 mol% CuBr-DMS, allyl magnesium bromide, THF:SMe2 (2:1), −78 °C, 81% (>20:1 dr). TBDPS = tert-butyldiphenylsilyl, ImH = imidazole, Bn = benzyl.
With intermediate 15 in hand, we then prepared the necessary aldehyde 18 to access key intermediate 6 (Scheme 3). Commercial cis-butene-1,4-diol 16 was mono-PMB protected to provide alcohol 17, and subsequently oxidized with MnO2 to afford the aldehyde 18 in 25% yield for the two steps. Intermediate 15 was treated with TiCl4, followed by introduction of aldehyde 18 to facilitate an Aldol reaction under Crimmins’ conditions delivering the Evans’ syn product 19 in 65% yield and 10:1 dr.[7,8] Hydrolysis of the auxiliary with LiBH4 in MeOH liberated the primary alcohol in low yield (44%, a survey of multiple reactions conditions failed to improve the yield), which was immediately protected as the TIPS silyl ether 20 in 82% yield. A hydroxyl-directed VO(acac)2-mediated epoxidation produced oxirane 21 as a single stereoisomer in 95% yield.[9] Protection of the secondary hydroxyl as a benzyl ether and subsequent DDQ-mediated removal of the PMB group led to primary alcohol 22 in 62% yield for the two steps. Directed opening of the epoxide with Red-Al afforded the desired 1,3-diol 23 in 79% yield and >20:1 ratio over the 1,2-diol congener.[10] Finally, protection of the primary hydroxyl as a pivalate and conversion of the secondary alcohol to a methyl ether afforded the key intermediate 6 in 75% yield over the two steps. With 6 in hand, we were poised to evaluate our synthetic approach to access both the macrocyclic pyrrole core 4 as well as the functionalized spiroaminal model system 8.
Scheme 3.

Synthesis of key intermediate 6. a) NaH, TBAI, PMBCl, THF, 0 °C, rt, 12 h, 68%; b) MnO2, CH2Cl2, rt, 36 h, 36%; c) i. TiCl4, DIPEA, NMP, CH2Cl2, 0 °C, ii. 18, CH2Cl2, 0 °C, 65% (10:1 dr); d) i. LiBH4, MeOH, THF, 0 °C, 6 h, 44%; ii. TIPSCl, ImH, CH2Cl2, rt, 4 h, 82%; e) VO(acac)2, TBHP, CH2Cl2, 0 °C, 3 h, 95%; f) i. BnBr, NaH, TBAI, THF, 0 °C, 4 h, 85%; ii. DDQ, CH2Cl2, pH 7 buffer, 0 °C, 14 h, 73%; g) Red-Al, THF, 0 °C to rt, 4 h, 79% (>20:1, 1,3:1,2); h) i. PivCl, pyr, CH2Cl2, rt, 3h; ii. Me3OBF4, CH2Cl2, rt, 1.5 hr, 75% over two steps. PMB = para-methoxybenzyl, TBAI, tetrabutyl ammonium iodide, TBDPS = tert-butyldiphenylsilyl, TIPS = triisopropylsilyl, ImH = imidazole, TBHP = tert-butyl hydrogen peroxide, Bn = benzyl, Piv = pivalate, pyr = pyridine.
Our next goal was to prepare 4 from 6. Towards this end, we deprotected the TIPS ether with BF3·Et2O, the only conditions identified for chemoselective removal, to provide primary alcohol 24 in 94% yield (Scheme 4). The primary hydroxyl was oxidized using Parikh-Doering conditions to give aldehyde 25 in 75% yield.[11] Then a two-step sequence involving the addition of vinyl Grignard followed by a Dess Martin Periodinane oxidation generated the α,β-unsaturated ketone 26 in 68% yield over two steps. Application of a Stetter reaction employing 6-heptenal afforded the RCM substrate 27 in 80% yield.[12] We surveyed a number of RCM catalysts and conditions to form the 13-membered macrocycle, and found that Grubbs II provided primarily cross metathesis; however, 30 mol% Grubbs I under dilute conditions in refluxing DCM provided the desired RCM product 28 in 84% yield. Similarly, a diverse array of Paal-Knorr conditions were explored to facilitate pyrrole construction from 1,4-diketone 28, but all classical conditions provided low yields or decomposition.[13,14] Ultimately, we developed microwave-assisted conditions (NH4OAc, MeOH, 120 °C, 20 min) that delivered the desired macrocyclic pyrrole 4 in 78% yield. Thus, our retrosynthetic strategy granted access to the advanced macrocyclic pyrrole moiety of marineosin A (1) in 5.1% overall yield from (S)-propylene oxide.
Scheme 4.

Synthesis of the macrocyclic pyrrole 4 of marineosin A (1). a) BF3OEt2, CH2Cl2, 0.5 h, rt, 94%; b) SO3-Pyr, Et3N, DMSO, CH2Cl2, 6 h, rt, 75%; c) i. vinyl magnesium bromide, THF, −78 °C, 1h; ii. Dess Martin Periodinane, CH2Cl2, 0.75 h, rt, 68% over two steps; d) 6-heptenal, Et3N, thiazolium salt. 1,4-dioxane, 70 °C, 12 h, 80%; e) 30 mol% Grubbs I, CH2Cl2 (0.0005 M), 9 h, 40 °C, 84%; f) NH4OAc, MeOH, 120 °C, mw, 20 min, 78%.
In order to complete the total synthesis of marineosin A, we wanted to ensure that our strategy to form the spiroaminal lactam 2 from 3 through an acid-mediated cyclization strategy was viable. With intermediate 6 in hand, we thought it prudent to explore this chemistry on a properly functionalized, yet minimized model system, before committing advanced material. Reductive removal of the pivalate 6 with DIBAL-H proceeded smoothly to give primary alcohol 29 in 93% yield, which was then oxidized under Parikh-Doering conditions to provide aldehyde 30 in 93% yield (Scheme 5).[11] Pinnick oxidation rapidly afforded the corresponding carboxylic acid which was coupled to ammonium chloride under standard coupling conditions to deliver the primary amide 31 in 78% yield for the two steps.[15,16] Hydrogenolysis of the benzyl ether and concomitant hydrogenation of the olefin provided a secondary alcohol that was immediately oxidized under Ley conditions to the ketone 32 in 84% yield over the two steps. After surveying a number of conditions, we found that 0.01 M HCl in MeOH affected the acid-mediated cyclization strategy providing 8 in 82% yield as a single diastereomer. Interestingly, the TIPS ether was hydrolysed under these conditions and intercepted by MeOH to afford the methyl ether, which was confirmed by 1D and 2D NMR studies. Efforts are under way to more thoroughly study this transformation. Extensive analysis of HMBC, HSQC, COSY and NOESY NMR experiments confirmed the absolute structure of 8 as the marineosin A isomer based on the stereochemistry relative to the axial methyl group within the pyran ring, which was set by the (S)-methyl oxirane at the outset of the synthesis. From 6, overall yield to 8 was 44.7%. Thus, advanced intermediate 6 provided access to both the macrocyclic pyran core 4 of marineosin A (1), as well as validated our acid-mediated cyclization strategy of a hydroxyketoamide into the spiroaminal lactam model 8.
Scheme 5.

Synthesis of the spiroaminal lactam 8 of marineosin A (1). a) DIBAL-H, CH2Cl2, −78 °C, 1h, 93%; b) SO3-pyr, Et3N, DMSO, CH2Cl2, 0 °C, 1h, 93%; c) i. NaO2Cl, NaH2PO4, 2-methyl-2-butene, 0 °C, 0.5 h; ii. EDC, HOBt, DIPEA, NH4Cl, DMF, rt, 78% over two steps; d) i. Pd/C, H2, EtOAc, rt, 8 h; ii. TPAP, NMO, CH2Cl2, 0 °C to rt, 2 h, 84% over two steps; e) 0.01 M HCl, MeOH, rt, 10 h, 82%. EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, HOBt = N-hydroxybenzotriazole, DIPEA, diisopropylethyl amine, TPAP = tetrapropylammonium perruthenate, NMO = N-methyl morpholine oxide.
Conclusions
In summary, we have developed an enantioselective synthetic route from a chiral pool starting material, (S)-propylene oxide, to access the 12-membered macrocyclic pyrrole core 4 of marineosin A (1), as well as a highly functionalized spiroaminal lactam 8 derived from the acid-mediated cyclization of a hydroxyketoamide. These efforts validate our retrosynthetic approach for the total synthesis of marineosin A, and efforts are underway to complete the total synthesis employing these tactics and strategies with final step pyrrole incorporation. Further refinements, applications to the related alkaloids, and biological investigations are in progress and will be reported in due course.
Experimental Section
Please see the Supporting Information Section for full experimental details
Supplementary Material
Acknowledgments
The authors acknowledge the Vanderbilt Department of Pharmacology, VUMC and the NIH for funding our research, andin particular the MLPCN (U54MH084659) for the Vanderbilt Specialized Chemistry Center. The financial support from the Warren Foundation in the form of the William K. Warren, Jr. Endowed Chair is gratefully acknoweldged.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Boonlarppadab C, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2008;10:5505–5508. doi: 10.1021/ol8020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fürstner A. Angew Chem Int Ed. 2003;42:3582–3603. doi: 10.1002/anie.200300582. [DOI] [PubMed] [Google Scholar]
- 3.Aldrich LN, Dawson ES, Lindsley CW. Org Lett. 2010;12:1048–1051. doi: 10.1021/ol100034p. [DOI] [PubMed] [Google Scholar]
- 4.Cai XC, Wu X, Snider BB. Org Lett. 2010;12:1600–1603. doi: 10.1021/ol100333d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected recent examples see: Panarese JD, Lindsley CW. Org Lett. 2012;14:5808–5801. doi: 10.1021/ol3024665.Senter TJ, Fadeyi OO, Lindsley CW. Org Lett. 2012;14:1869–1871. doi: 10.1021/ol300466a.Fadeyi OO, Senter TJ, Hahn KN, Lindsley CW. Chem Eur J. 2012;18:5826–5831. doi: 10.1002/chem.201200629.Hahn KN, Fadeyi OO, Cho HP, Lindsley CW. Tetrahedron Lett. 2012;53:3577–3580. doi: 10.1016/j.tetlet.2012.05.009.Brogan JT, Stoops SL, Lindsley CW. ACS Chem Neurosci. 2012;3:658–664. doi: 10.1021/cn300064r.
- 6.Panarese JD, Konkol LC, Berry CB, Bates BS, Aldrich LN, Lindsley CW. Tetrahedron Lett. 2013;54:2231–2234. doi: 10.1016/j.tetlet.2013.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans DA, Bartoli J, Shih TL. J Am Chem Soc. 1981;103:2127–2129. [Google Scholar]
- 8.Crimmins MT, She J. Synlett. 2004;8:1371–1374. [Google Scholar]
- 9.Katsuki T, Sharpless KB. J Am Chem Soc. 1980;102:5974–5976. [Google Scholar]
- 10.Finan JM, Kishi Y. Tet Lett. 1982;23:2719–2722. [Google Scholar]
- 11.Parikh JR, Doering WE. J Am Chem Soc. 1967;89:5505–5507. [Google Scholar]
- 12.Stetter H. Angew Chem Int Eng Ed. 1976;15:639–647. [Google Scholar]
- 13.Paal C. Berichte der deutschen chemischen Gesellschaft. 1884;17:2756. [Google Scholar]
- 14.Knorr L. Berichte der deutschen chemischen. Gesellschaft. 1884;17:2863. [Google Scholar]
- 15.Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 16.Wang W, McMurray JS. Tet Lett. 1999;40:2501–2504. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.