

Abstract
Currently, one of the most prominent methods used to impart biocompatibility to aqueous-in-oil droplets is to synthesize a triblock copolymer surfactant composed of perfluoropolyether and polyether blocks. The resulting surfactants (EA surfactant, KryJeffa, etc.) allow generation of highly biocompatible droplet surfaces while maintaining the heat stability of the starting material. However, production of these surfactants requires expertise in synthetic organic chemistry, creating a barrier to widespread adoption in the field. Herein we describe a simple alternative to synthetic modification of surfactants to impart biocompatibility. We have observed that aqueous-in-oil droplet surfaces can be made biocompatible and heat stable by merely exploiting binding interactions between polyetherdiamine additives in the aqueous phase and carboxylated perfluorocarbon surfactants in the oil phase. Droplets formed under these conditions are shown to possess biocompatible surfaces capable of supporting picoliter-scale protein assays, droplet polymerase chain reaction (PCR), and droplet DNA amplification with isothermal recombinase polymerase amplification (RPA). Droplets formed with polyetherdiamine aqueous additives are stable enough to withstand temperature cycling during PCR (30–40 cycles at 60–94 °C) while maintaining biocompatibility, and the reaction efficiency of RPA is shown to be similar to that with a covalently-modified surfactant (KryJeffa). The binding interaction was confirmed with various methods, including FT-IR spectroscopy, NMR spectroscopy, ESI-MS, and fluorescence microscopy. Overall, our results suggest that by simply introducing a commercially–available, polyetherdiamine additive (Jeffamine ED-900) to the aqueous phase, researchers can avoid synthetic methods in generating biocompatible droplet surfaces capable of supporting DNA and protein analysis at the sub-nanoliter scale.
Keywords: microfluidics, droplet fluidics, block copolymer, surfactant, Jeffamine, Krytox, PCR, RPA, pFRET
Introduction
Based on cellular compartmentalization in nature, in vitro compartmentalization (IVC) is the utilization of water-in-oil emulsion droplets of fL – nL volume to perform > 1010 reactions simultaneously in just 1 mL of sample volume.1,2 This capability can greatly reduce assay cost and time and can open the door to a variety of high-throughput experiments, such as directed evolution of proteins and RNAs,1,3,4 screening large libraries for rare mutations,2,3 and fluorescence-activated cell sorting (FACS) of emulsion-induced products.3,5,6 In recent years, droplet microfluidics has emerged to reinvigorate the IVC field, providing more flexibility, lab portability, and increased likelihood of automation.7–11
One major advantage gained by droplet microfluidics is the ability to produce highly monodispersed emulsions. These emulsion-generating devices typically employ hydrocarbon or fluorocarbon oils for aqueous droplet encapsulation. Since emulsion formation is thermodynamically unfavorable,12 biological experiments requiring in-droplet incubation—particularly those at elevated temperature—will generally need an oil/surfactant mixture. The surfactant molecules assemble at the oil-water interface and act to lower surface tension, prevent adsorption to the surface, and counteract coalescence by creating an energy barrier.12 The goal is to find a surfactant/oil combination that achieves the delicate balance between droplet stability and biocompatibility. Nonionic fluorosurfactants are ideal for this purpose, allowing use of hydrophobic/lipophobic fluorocarbon oils13–15 with the biologically inert interior that nonionic headgroups provide through minimization of charge interactions.12 There are a few fluorosurfactants available commercially, but these either have an ionic headgroup or do not maintain maximum droplet stability for certain applications.12,16,17
Others have noted the apparent lack of commercially-available, nonionic fluorosurfactants and opted to synthesize their own block copolymer surfactants. After several synthetic steps, an amide bond is formed between a carboxylated, perfluorocarbon chain and a polyetherdiamine.16,18 This perfluoropolyether-polyethelene glycol triblock copolymer (PFPE-PEG-PFPE) surfactant, termed “EA-surfactant” or “KryJeffa” in subsequent uses, has emerged as one of the most effective biocompatible surfactants for generating aqueous-in-fluorocarbon oil droplets, particularly with microfluidic emulsion generators.16,19,20 However, this surfactant is not commercially available, and the multi-step synthetic route required serves as a barrier to widespread adoption by others.
In this work, we show that synthesis of a biocompatible surfactant is unnecessary for certain biological applications. The Weber group has thoroughly studied the noncovalent complex formation between a carboxlyated perfluoropolyether (Krytox 157 FSH) and heterocyclic, nitrogenous bases in flourous solvents.21–23 Depending on solvent properties, the two can associate through a mixture of both hydrogen and ionic bonding. We rely on a similar property and show evidence that the carboxylated PFPE, Krytox 157 FSH (referred to herein as “Krytox”), will bind directly to primary amines in a similar fashion. A polyetherdiamine (PED), Jeffamine ED900 (referred to herein as “Jeffamine”), is shown to bind with Krytox to form a noncovalent structure similar to synthetic nonionic fluorosurfactants (PFPE/PED or PFPE/PED/PFPE), thereby imparting biocompatibility to aqueous/perfluorocarbon oil interfaces. FT-IR and NMR spectroscopy clearly show that Jeffamine binds with Krytox. Additional ESI mass spectrometry (ESI-MS) and FT-IR evidence, akin to results from the Weber group,21–23 indicates that the binding can facilitate extraction of Jeffamine from aqueous solution into perfluorocarbon oil. Other primary amines, such as ethylenediamine or amine-labeled DNA, show evidence of binding to Krytox as well. Using Jeffamine as an aqueous additive and Krytox in perfluorocarbon oil, biocompatible droplets of picoliter volume were formed that were stable at 95 °C and useful for DNA amplification (PCR and RPA) and homogeneous immunoassays. Since our approach utilizes commercially-available reagents and does not involve synthetic steps or separation/purification steps, it should allow users without expertise in synthetic chemistry to generate biocompatible droplet surfaces for a multitude of applications.
Experimental
Materials
Krytox 157 FSH was obtained from DuPont, and Jeffamine ED900 and ethylenediaminetetraacetic acid (EDTA) were obtained from Sigma. NH4OH, NaH2PO4 · H2O, Na2HPO4 · 7H2O, NaCl, methanol, acetone, acetone-d6, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), CaCl2, NaOH, HCl, NaCl, Corning plate reader assay plates (96-well, flat bottom, black polystyrene), Whatman filter papers (55-mm circles), Acrodisc syringe filters (0.2 μm HT Tuffryn), mineral oil, half-skirt 96-well PCR plates, a 100-mL glass syringe (SGE), glass slides, PCR tubes, and BSA were obtained from VWR. Novec 7500 Engineered Fluid (HFE-7500) was obtained from 3M. RPA enzyme kits and reagents were obtained from TwistDx, with probe DNA from Biosearch Technologies. Unless otherwise specified, all other DNA was obtained from Integrated DNA Technologies (IDT), with sequences included in Supporting Information (SI). Fluo-4 pentapotassium salt (cell-impermeant), Platinum Tfi DNA Polymerase with reaction buffer MgCl2, and sulforhodamine 101 were obtained from Life Technologies. Additional specialty materials, along with a detailed methods section, are included in SI.
Methods
FT-IR Spectrometry
1.8% w/w Krytox carboxylate salt in HFE 7500 oil was added to a 50 mL centrifuge tube. Jeffamine solution (75 mg/mL in PBS) was layered onto the oil/surfactant mixture, avoiding emulsion formation. The layers were incubated and rocked overnight at RT, then the aqueous phase was removed. ~400 μL of each surfactant/oil combination were added to a KBr demountable cell. After sample loading, the FT-IR system (IR Prestige-21, Shimadzu) was flushed with N2 for 20 min before scanning in % transmittance mode (16 scans, 2 cm−1 resolution, 2000–1500 cm−1 range, square triangle apodization). Pure HFE 7500 oil was used to rinse the cell between scans and for background spectra.
Mass Spectrometry
Binding of Krytox to Jeffamine was performed in pure HFE 7500 oil or 1.8% w/w Krytox in HFE 7500 oil. Each sample was diluted in acetone, then analyzed by LC/MS (Waters Acquity UPLC and Q-Tof Premier) using positive electrospray ionization.
NMR Spectroscopy to Assay Binding
For 19F NMR, Jeffamine and Krytox were mixed directly at a 1:10 ratio by weight in acetone-d6, incubated overnight, then analyzed. For proton NMR, 1.8 % w/w Jeffamine-bound Krytox in HFE 7500 was dried (rotary evaporator, Heidolph), then centrifuged for 50 min at 15000 rcf. ~3 drops of the top layer were dissolved in acetone-d6 then analyzed. A 250 MHz NMR spectrometer (Bruker) was used, with 64 scans for proton NMR and 3500 scans for 19F NMR.
DNA/Surfactant Binding Experiment
Using microfluidics (SI, Fig. S-2), droplets were generated with 1.8% Krytox in HFE 7500 oil and a mixture of DNA (600 nM amine-modified primer, 500 nM FAM-labeled anti-primer) as the aqueous phase. After mixing with control droplets (only FAM-labeled anti-primer), fluorescence microscopy (470 ± 20 nm ex.; 525 ± 25 nm em.) was used for spatial analysis.
Droplet PCR
Aqueous droplets were filled with PCR amplification mixture (D.I. H2O, 0.75% w/v Jeffamine, 1% BSA, 50 nM probe, 0.2 μM primers, 1.5 mM MgCl2, 1X Platinum Tfi Reaction Buffer, 0.2 mM dNTPs, 0.1 U/μL Platinum Tfi Polymerase) and either zero, 160 fM, or 16 pM template using separate microfluidic devices (Fig. S-2). Negative controls contained 16 pM template without Jeffamine additive. Emulsion PCR was carried out on a CFX96 qPCR system (BioRad). Droplets were imaged in a microfluidic channel via confocal fluorescence microscopy along with reference droplets (50 nM ROX-labeled DNA, 50 nM probe; 1% BSA; 1X buffer). Additional details are included in SI.
Droplet RPA
RPA solutions were prepared and used to dissolve a lyophilized enzyme mixture (TwistDx). To avoid premature reaction, sample and activation solutions were mixed on ice, then droplets were generated using a microfluidic device (Fig. S-2) on ice. For end-point measurements, droplets were collected for 1 hour, incubated off chip at 37 °C for 30 min, then placed back on ice prior to imaging at room temperature in a microfluidic channel. Further RPA details are included in SI.
Results and Discussion
Evidence of Krytox-Diamine Binding
The proposed direct binding of a polyetherdiamine to a carboxylated perfluorocarbon surfactant is depicted in Figure 1. The amphipathic nature of surfactants allows them to prevent coalescence and reduce surface tension as they collect at oil-water interfaces, where their hydrophilic moiety is available to bind to aqueous additives. The water-soluble portion of Krytox is the carboxylate headgroup, and the oil-soluble portion is the perfluoropolyether backbone, having a similar structure to that of the perfluorocarbon oil. As depicted in Figure 1, a polyetherdiamine (PED), Jeffamine, is added to aqueous solution, and an emulsion is formed by using a microfluidic emulsion generator. Our evidence shows that the Jeffamine additive binds to the perfluoropolyether (PFPE), Krytox, at the oil-water interface, rendering the droplet surfaces biocompatible toward processes such as DNA amplification and protein sensing. Evidence shown herein and in the literature21–23 suggests that the interaction could be taking place through a mixture of ionic and hydrogen bonding. This system is favorable in that it allows biocompatible, heat-stable droplets to be formed without requiring synthetic chemistry to modify the surfactant headgroup.
Figure 1.
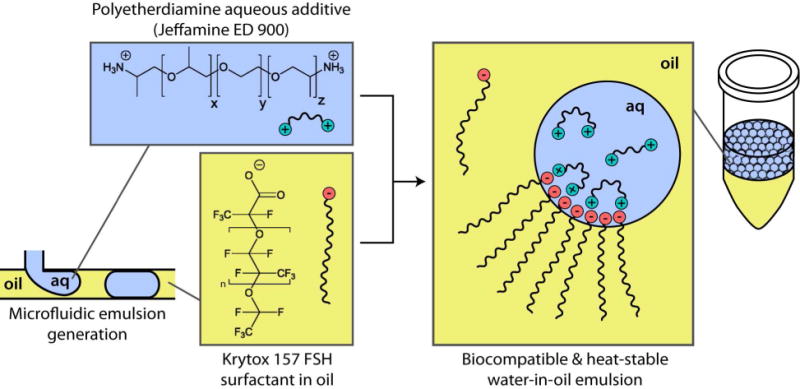
Direct binding of surfactant to a polyetherdiamine. The unmodified surfactant, a carboxylated perfluoropolyether called Krytox, is dissolved in perfluorocarbon oil (HFE 7500). In emulsions, the anionic headgroup moves to the oil-water interface and binds to the polyetherdiamine aqueous additive (Jeffamine). This provides a biocompatible interface for biological processes and assays, without requiring synthetic chemistry.
Several techniques were used to show evidence of the binding interaction between Krytox and Jeffamine. Since binding likely occurs between the carboxylic acid group of Krytox and the primary amine groups of Jeffamine, we first chose FT-IR spectroscopy to monitor Krytox carbonyl stretches that would change upon binding. Figure 2A illustrates the changes that the carbonyl stretch undergoes upon binding of the carboxylic acid to the primary amine. Peak assignments for asymmetric CO2 stretches of many carboxylic acids and carboxylates appear in the range from 1700–1540 cm−1, and several studies have shown that perfluorinated carboxylic acid stretches are shifted to higher energy.24,25 For comparison, we synthesized KryJeffa16,18 (covalently linked Krytox and Jeffamine), and its FT-IR spectrum (black trace) matches well with that of previous reports,24,25 showing the product peak at 1729 cm−1 and remaining Krytox carboxylic acid at 1770 cm−1. In contrast, the “Krytox carboxylate,” which refers to the ammonium carboxylate salt of Krytox, has a carbonyl stretch that appears at 1685 cm−1 (blue trace). With the addition of Jeffamine (blue-green trace), we observed a Krytox carbonyl peak shift to 1702 cm−1 likely due to a binding interaction, as well as a broad signature of primary amine in the oil at 1626 cm−1. This amine signature increased with Jeffamine concentration and was not present in the oil blank (red trace) or the Krytox-free oil exposed to Jeffamine overnight (orange trace), indicating that Jeffamine binding and extraction into the oil is dependent upon Krytox. The Jeffamine-bound Krytox sample showed a carbonyl stretch at a distinctly different energy than the Krytox or the KryJeffa product. This suggests that Krytox and Jeffamine participate in a binding interaction that is possibly ionic in nature.
Figure 2.
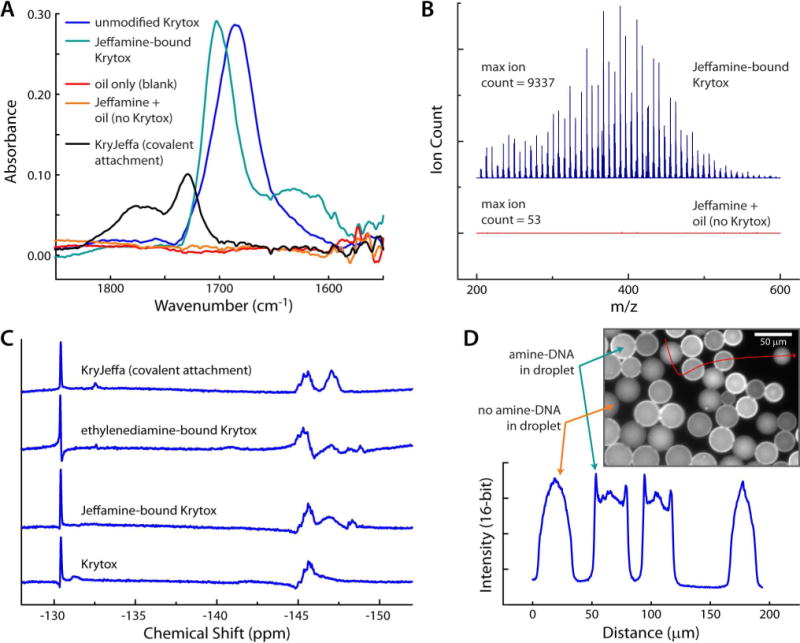
Evidence of direct binding. (A) FT-IR evidence of Jeffamine/Krytox interaction and comparison to the covalently-modified product, KryJeffa (black trace). (B) ESI-MS showed that Jeffamine extraction into HFE 7500 oil was dependent upon the presence of Krytox in the oil. (C) NMR spectra of the starting material (Krytox; bottom trace), Jeffamine-bound Krytox (second from bottom), ethylenediamine-bound Krytox (third from bottom), and the covalently-modified surfactant, KryJeffa (top trace). Primary amine bound Krytox spectra show distinct patterns different from both the starting material and the covalent product. (D) Fluorescence microscopy shows that amine-labeled DNA collects at the oil-water interface, while non-amine-labeled DNA is evenly dispersed.
We then used an LC/MS system with positive ESI to measure the relative amounts of Jeffamine extracted into HFE 7500 oil, in the presence or absence of Krytox surfactant (Figure 2B). The spectrum of oil containing Krytox (blue trace) shows strong PEG and PPG signatures from the Jeffamine with a maximum ion count of 9337, while there is no evidence of these patterns in the pure HFE 7500 oil spectrum (orange trace) with a maximum ion count of only 53. This supports the Krytox-mediated dissolution of Jeffamine into HFE 7500 oil. NMR spectroscopy also suggests a binding interaction. After mixing Jeffamine and Krytox acid at a 10:1 ratio in deuterated solvent and leaving the mixture to sit overnight, we were left with a clear solution ready for NMR analysis. 19F was used to determine if interactions between Jeffamine and Krytox (Figure 2C; second trace from bottom) would show a clear spectral change compared to Krytox alone (bottom trace). The original structure of Krytox was used to assign peaks (Figure 1). The NMR data shows clear changes signified by the disappearance of a peak at −132 ppm and the appearance of a sharp peak near −148 ppm. In addition, a peak at −146 ppm is changed from a singlet to a doublet. Based on results reported in the literature,26 these peaks can be assigned to the ~CF group in closest proximity to the carboxylic acid (−132 ppm) and its movement upfield upon the interaction of Krytox with a primary amine (−148 ppm). A peak corresponding to the ~CF group inside the repeating unit of Krytox (−146 ppm) is also changed into a doublet upon binding to a primary amine.26 To confirm the interaction chemistry, we mixed a smaller diamine, ethylenediamine, with Krytox and collected the 19F NMR spectrum of the mixture (Figure 2C; third trace from bottom). This data shows a similar signature to that of Jeffamine-bound Krytox, suggesting that the binding chemistry occurs in the same fashion for other diamines. The spectrum of the covalently-modified Krytox, KryJeffa (Figure 2C; top trace),16,18 showed clear differences from the starting material spectrum (Krytox) and from the diamine-bound spectra, with the ~CF group in closest promimity to the carboxylic acid only moving slightly upfield (−133 ppm), and the appearance of the ~CF doublet peak being much sharper (−147 ppm). A peak that remains constant in all spectra is the presence of a sharp peak at −131 ppm. This corresponds to a ~CF2 group along the perfluorocarbon backbone that is further away from the carboxylic acid headgroup.26 Because of its distance away, it feels little or no effect after a binding interaction has taken place. Collectively, the FT-IR, MS, and NMR data strongly suggests that Jeffamine and Krytox participate in a binding interaction, and prior work suggests that this could be a combination of charge-based and hydrogen bonding interactions.21–23
Fluorescence microscopy provided a final confirmation of the binding interaction. We generated emulsions containing amine-labeled ssDNA and its fluorescently-labeled complementary sequence on a microfluidic device (SI, Fig. S-2). Using fluorescence microscopy, it was possible to determine the position of the amine-DNA within the droplets using its labeled complementary sequence. Figure 2D shows a mixture of droplets containing the double-stranded fluorescent DNA/amine-DNA complex with droplets containing only the fluorescent complementary sequence (no amine-DNA). The control droplets (no amine-DNA) gave a spherically symmetric fluorescence image, while amine-DNA containing droplets showed clear evidence of surface fluorescence. A plot of fluorescence intensity versus distance (Figure 2D, lower trace) shows sharp intensity spikes at the edges of the amine-DNA containing droplets and no intensity spikes in the control droplets, suggesting again that the surfactant interacts with primary amines. This test confirms our prior findings and shows that Krytox is capable of binding to different classes of primary amines, opening the door to a variety of possibilities in surface-based biological assays within picoliter volume droplets.
Emulsion Stability, Surface Biocompatibility, and pH Effects
Several qualitative and quantitative tests were developed to investigate the stability of Jeffamine/Krytox stabilized water-in-oil emulsions. First, a heat and ionic strength stability test was developed to mimic PCR conditions. The table in Figure 3A shows that emulsions made with 1.8% w/w Krytox remain stable after incubation at 95 °C when either 0.75% w/v Jeffamine or 1.5 mM MgCl2 is added at all ionic strengths tested. The fact that the emulsions remain stable after incubation at 95 °C makes the Jeffamine-bound surfactant an attractive alternative for droplet PCR and digital droplet PCR. Since it could be perceived advantageous to pre-saturate the Krytox with Jeffamine to avoid aqueous additives, the same tests were carried out on Jeffamine-saturated Krytox in oil (overnight incubation). Interestingly, we found that emulsions were destabilized at the two highest ionic strengths. Upon adding 1.5 mM MgCl2, however, some (but not all) of the stability was recovered at the second-highest ionic strength; this result is reasonable since divalent cations stabilize PEG-like structures in aqueous solution.27 We speculate that the Jeffamine-saturated Krytox, once fully dissolved in perfluorocarbon oil, is less likely to move to the interface and serve as a surfactant during droplet formation; although the system could be better understood in future studies with more quantitative measurements and a wider range of conditions. It appears that Jeffamine-saturated Krytox in oil is non-ideal for use with droplet PCR conditions, while the aqueous Jeffamine additive is promising for use in droplet PCR. Since the dynamics of surfactant mobility have not been well-studied for this particular system, determination of the time scales of adsorption to the interface for both Krytox and Jeffamine in future work could provide valuable information that could help to explain the observed trends seen in Figure 3A and to further optimize this system.
Figure 3.
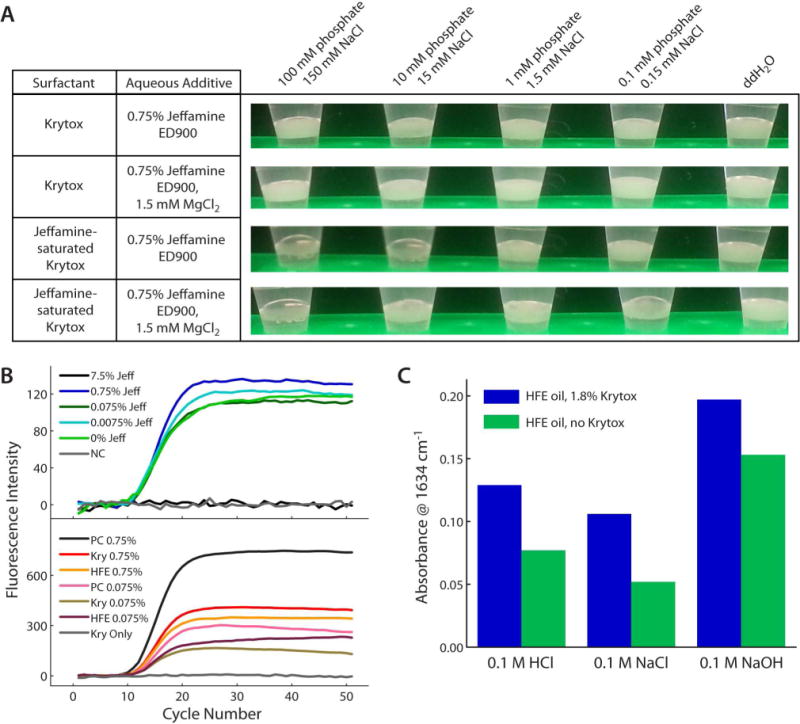
Ionic strength, pH, and heat stability of Jeffamine-bound Krytox emulsions. (A) Heat stabilities of emulsions of varying aqueous ionic strengths with Krytox (upper 2 rows) and with Jeffamine-saturated Krytox (lower 2 rows). (B) Real-time PCR tests showed that all concentrations of Jeffamine at or below 0.75% w/v were PCR compatible. 7.5% Jeffamine (black trace) inhibited PCR (upper data set). In real-time PCR “float tests”, Krytox surfactant at the oil-water interface (gray trace, lower data set) inhibited PCR, and 0.75% w/v Jeffamine additive (red trace) sufficiently rescued the biocompatibility in the presence of Krytox. (C) Jeffamine extracttion into HFE 7500 oil at different aqueous pH values was monitored by FT-IR. Results are consistent with an ionic interaction between positively charged Jeffamine and negatively charged Krytox.
Since aqueous Jeffamine additives did not interfere with heat stability, it was necessary to determine the biocompatibility of this approach, for which we employed real-time PCR. First, a titration experiment was conducted to determine if any concentration of Jeffamine additive would interfere with PCR efficiency (Figure 3B, top). At a concentration of 7.5% (black trace), PCR is inhibited entirely, with a profile matching that of the no-template negative control (NC, gray trace). However, with Jeffamine additives at or below 0.75% w/v, the threshold cycles (Ct) were essentially equal to that of the positive control (0% Jeffamine, green trace). From this, we concluded that Jeffamine could be used as a PCR additive at a concentration less than or equal to 0.75% w/v. A second PCR test was performed to determine if the Jeffamine additive could now impart biocompatibility between Krytox and the PCR assay (Figure 3B, bottom). In this test, the PCR amplification mixture was not formed into an emulsion, but was thermally cycled as it floated atop HFE 7500 oil (density ~1.6 g/mL); oil was either pure or contained 1.8% w/w Krytox. The results showed that at Jeffamine additive concentrations of both 0.75% w/v (red trace) and 0.075% w/v (gold trace) PCR was successful in the presence of Krytox. Without Jeffamine additive, Krytox inhibited PCR (gray trace). Since neither oil nor surfactant were present in the positive control (PC, black trace), the real-time PCR optics registered higher fluorescence intensity for the positive control due to the difference in solution position. In emulsion PCR, there will be higher surface-area-to-volume ratios in droplets; these results suggest that for optimal droplet stability and biocompatibility, unmodified Krytox in HFE 7500 oil should be used with the highest allowable percentage (0.75% w/v) of Jeffamine as an aqueous additive.
Finally, to determine the role of ionic interactions in the Jeffamine/Krytox binding, we evaluated the pH-dependence of the interaction using FT-IR (Figure 3C). Jeffamine was bound directly to Krytox by overnight saturation of 1.8% Krytox in HFE 7500 with Jeffamine-containing aqueous solutions of pH 2, 7, and 12 (equal ionic strengths). FT-IR spectroscopy was used to monitor the change in absorbance for the amine peak at approximately 1634 cm−1 (see Figure 2A, blue-green trace), thus determining the extent of extraction of Jeffamine into HFE 7500 by Krytox. In all cases, the presence of Krytox significantly increased the amount of Jeffamine found in the oil. As expected for a charge-based interaction, the amount of extracted Jeffamine decreased from pH 2 to pH 7, as there will be reduced amount of positively charged Jeffamine at pH 7. Note that the amount of negatively-charged Krytox should be essentially equal at all pH values tested, since the pKa of carboxylated perfluorocarbons typically decreases as the number and proximity of fluorine atoms to the carboxylic acid increases. Based on its structure, Krytox should have a negative pKa.28,29 At pH 12, the results show that more Jeffamine was extracted into the oil, even when Krytox was not present. Because Jeffamine will be predominantly neutral at this pH, the Jeffamine solubility in the oil may be increased due to the lack of charge and/or due to hydrogen bonding interactions with negatively charged Krytox.21–23 An alternative interpretation would be that Jeffamine-bound Krytox could form micelles in the HFE-7500 oil, thus the extraction of Jeffamine would be highly dependent upon the critical micelle concentration (CMC) of the system. Since Jeffamine was maintained at a rather high concentration in the studies in Figure 3, micelle formation could certainly play a role, along with charged interactions, hydrogen bonding21–23, or other factors. This level of complexity warrants further study of the system and is beyond the scope of the current work.
Biocompatibility within Picoliter Droplets
To demonstrate biocompatibility at higher surface area-to-volume ratios, we have used the Jeffamine aqueous additive to rescue the efficiency of biochemical reactions within Krytox stabilized picoliter droplets. One application involves our novel proximity FRET (pFRET) protein assay, which is similar in structure to the proximity ligation assay30 and similar in readout to the molecular pincer assay.31 Two antibody-oligonucleotide probes assemble only in the presence of the target protein (insulin), thereby quenching fluorescence in a highly selective way. As seen in Figure 4A, in a float test (aqueous solution floating on perfluorocarbon oil) without added surfactant in the oil phase (HFE 7500 only, blue bars), the assay responded nicely to the presence of 5.0 nM insulin (+ins). However, when exposed to 1.8% Krytox in the HFE 7500 oil, no significant assay response was observed (orange bars). Upon inclusion of the Jeffamine aqueous additive, even in the presence of 1.8% Krytox in HFE 7500, the assay performance was rescued (green bars) to a similar sensitivity level as in the control. These results support the hypothesis that Jeffamine additives can impart biocompatibility to Krytox-stabilized oil-water interfaces.
Figure 4.
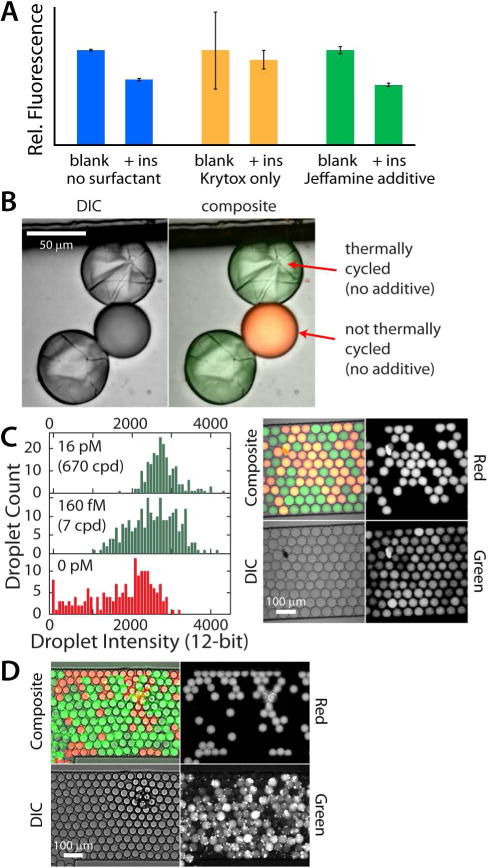
Biocompatibility of Jeffamine-bound surfactant with three biochemical assays. (A) Homogeneous pFRET assay response to insulin (blue bars) was inhibited by Krytox (orange bars), then rescued by the Jeffamine additive (green bars). (B) DIC and composite images of droplets made with Krytox surfactant and no Jeffamine aqueous additive. Thermally cycled droplets show evidence of reagent precipitation at the oil-water interface. (C) Droplet PCR with varying template DNA concentrations of 0, 160 fM, or 16 pM (0, 7, or 670 copies/droplet). Control droplets (red in image) contained background levels of probe and were not thermally cycled. (D) Isothermal droplet RPA with 160 fM (7 copies/droplet) template (37 °C). Control droplets (red) were subjected to incubation with RPA reagents, but contained no template DNA. The abbreviation “cpd” refers to DNA copies per droplet.
A second biocompatibility test employed droplet PCR. Since digital PCR and droplet digital PCR (ddPCR) exploded onto the scene, it has been successfully used to achieve limits of detection down to only a few copies of DNA, for quantifying DNA copy number without the need for references or standards, and for detecting the presence of rare mutations in a high background of other DNA.32–35 This robust technique has seen widespread recent application, and it requires the use of a biocompatible surfactant. Working toward ddPCR, we have demonstrated successful droplet PCR using our Jeffamine aqueous additive to create heat-stable, biocompatible droplet interfaces (Figure 4B–C). The importance of creating biocompatible surfaces is highlighted in Figure 4B. All droplets shown here were made with Krytox as the surfactant, yet they contained no Jeffamine aqueous additive. The green and red confocal fluorescence image was overlayed with the transmission image to create a composite for droplet identification. The red and green dye labeled droplet (middle droplet) was not thermally cycled, while the droplets labeled with only green dye were cycled through a typical PCR thermal program. It is clear from the laser transmission image (Figure 4B, left, grayscale) that thermally cycling in the presence of Krytox surfactant (without Jeffamine additive) causes PCR reagents (likely enzymes) to crash out of solution at the oil-water interface.
To perform droplet PCR at different template concentrations, we first used a passively-operated microfluidic device (SI, Fig. S-2), with 1.8% w/w Krytox surfactant as the carrier phase, to rapidly create aqueous-in-oil droplets containing 0.75% w/v Jeffamine as a PCR additive. Template DNA concentrations of 0, 160 fM, or 16 pM (0, 7, or 670 copies/droplet) were added to the inlet of the droplet generating device. After emulsions were collected and transferred to individual PCR tubes and thermally cycled, DNA products within individual droplets were identified via confocal fluorescence imaging (Figure 4C). As shown in the image, droplets exhibited uniform size with no evidence of coalescence after thermally cycling then mixing with reference droplets. The reference droplets included Taqman probes and all reagents other than enzyme. These droplets were labeled with ROX-labeled DNA (“Red”), and they exhibited lower average intensity than the sample droplets with template DNA. The histograms of droplet count versus green fluorescence intensity (left) showed a significant increase in intensity in proportion to template DNA concentration, suggesting that PCR was functional within the Jeffamine-stabilized droplets. With the current optical setup, it was not possible to image enough droplets to obtain the precision necessary for ddPCR, but positive results at only 7 copies/droplet suggest that ddPCR should be possible when using Jeffamine as an aqueous additive.
To provide a third test for biocompatibility of our approach, we successfully carried out isothermal DNA amplification using a multi-enzyme mixture for recombinase polymerase amplification (RPA)36,37 in picoliter-volume droplets. RPA allows exponential amplification of DNA, similar to PCR, but does not require an initial heating step or thermal cycling. This method has recently been shown capable of digital nucleic acid quantitation in small, droplet-like microfluidic compartments,38 but has not been carried out in aqueous-in-oil droplets, to our knowledge. Figure 4D shows a mixture of test and control droplets after incubation for 30 min at 37 °C. The monodispersed emulsion was created with our passively-controlled microfluidic device (SI, Fig. S-2) and 1.8% Krytox surfactant in HFE 7500 oil. Using 160 fM template concentration (7 copies/droplet) and the Jeffamine aqueous additive, all sample droplets appeared to be positive (high green signal), while the reference droplets (red-labeled) show very little signal in the green channel. This result again supported the biocompatibility of Jeffamine-bound Krytox at the droplet surfaces.
As a final test, we monitored RPA in real time within picoliter droplets to assay the efficiency of a biochemical reaction within Jeffamine/Krytox stabilized droplets (Figure 5). The images in Figure 5A show the end result of approximately 10 min of RPA-based amplification from a starting concentration of 16 pM template DNA (670 copies/droplet). The reference droplets (no template DNA) showed essentially no amplification, evidenced by the absence of yellow color in the overlayed image (R + G). Snapshots in time are shown in the rightmost three images, where a template-containing droplet is outlined in green and a control droplet (no template) is outlined in red. Within only 8 min, amplification reached > 95% maximum signal, indicating that RPA was highly efficient within droplets containing the Jeffamine additive. Figure 5B shows a comparison of real-time RPA with “unmodified” surfactant (Krytox only, black trace), Jeffamine-bound surfactant (blue trace), and KryJeffa covalent product (orange trace). Curves depict the average of three droplets of each type, with standard deviations shown as error bars without caps (for clarity). Although the reaction was eventually functional within Krytox-stabilized droplets (no additive), the efficiency was significantly reduced by the presence of Krytox alone. The reagents specific to RPA, which differ greatly from those used in PCR, may contain molecules capable of eventually passivating the interface much like the Jeffamine additive. However, Jeffamine additive seems to preferentially migrate to the interface over these reagents, which is supported by the visible increase in efficiency once Jeffamine is added to the system. With these realtime analyses, there was no observable difference in RPA efficiency within Jeffamine-bound Krytox stabilized droplets and KryJeffa stabilized droplets. By analyzing end-point images (approximately 10 min after reaction), it appeared that KryJeffa promoted a slightly higher efficiency than Jeffamine-bound Krytox (Figure 5C), although both showed significantly higher RPA efficiencies than Krytox alone. Interestingly, neither Jeffamine-bound Krytox nor KryJeffa stabilized droplets allow for 100 % efficiency. This could be a result of a digital effect, where some droplets contained no template, but further investigation was deemed unwarranted. These RPA efficiency comparisons suggested that a simple polyetherdiamine aqueous additive (Jeffamine) provides a significantly simpler alternative to impart biocompatibility to droplets, compared to the synthetic modification of Krytox that is in common practice currently (e.g. EA surfactant, KryJeffa).
Figure 5.
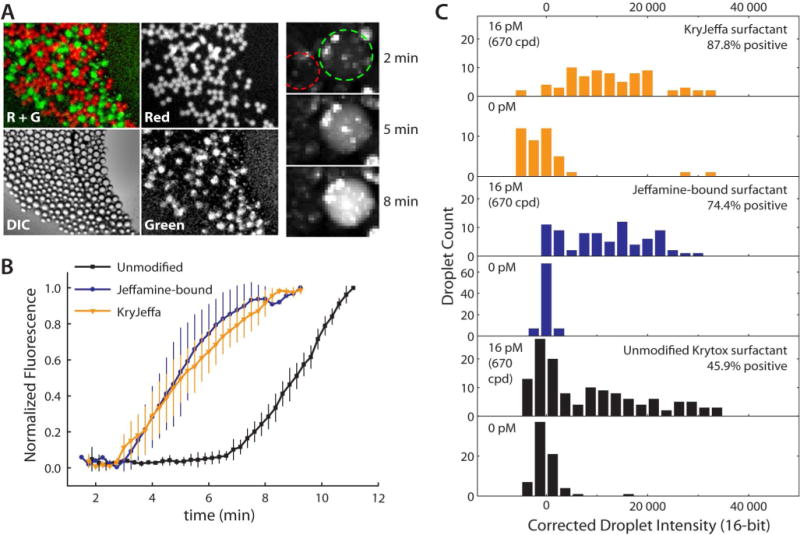
Analysis of biological reaction efficiency in picoliter droplets. (A) Fluorescence images of droplet RPA using the Jeffamine aqueous additive with unmodified Krytox surfactant. Leftmost images show grayscale (and composite) images taken post-RPA using the Red, Transmission (DIC), and Green imaging filters. Red droplets represent zero template controls; all other droplets contained 16 pM template (670 copies/droplet). Composite image (R + G) shows that none of the control droplets generated products. Rightmost image shows snapshots of the real-time RPA reaction; control droplet outlined in red; template-containing droplet outlined in green. (B) RPA reaction kinetics monitored in the presence of unmodified surfactant with no additive (black), Jeffamine-bound surfactant (blue), and KryJeffa surfactant (orange). Average traces from 3 representative droplets are shown with standard deviations. (C) Following droplet RPA with 0 or 16 pM template, droplet population intensities were counted with each treatment, indicating similar efficiency with KryJeffa and Jeffamine-bound surfactants. The abbreviation “cpd” refers to DNA copies per droplet.
Conclusions
Significant evidence was provided to support a direct binding interaction between a carboxylated perfluoropolyether (Krytox) and a polyetherdiamine (Jeffamine) at the interface of oil and aqueous solution. Evidence of the binding interaction was supported by FT-IR, mass spectrometry, fluorescence microscopy, and NMR analyses; and biocompatibility was confirmed with homogeneous protein assays, droplet PCR, and droplet RPA. For further confirmation, the efficiency of real-time RPA within Jeffamine additive stabilized droplets compared favorably with that of the KryJeffa stabilized droplets, even starting with <10 template copies per droplet.
To achieve maximum performance, it appears that Jeffamine should be added directly to the aqueous solution at a concentration of 0.75% w/v. Preparing an oil-soluble, Jeffamine-saturated surfactant in advance actually sacrifices droplet stability at higher ionic strengths, thus this method is discouraged. To further optimize this system, future studies should involve determination of surfactant CMC and determination of the timescales of interfacial adsorption for both surfactant and aqueous additive. Overall, our current results suggest that commercially-available reagents can be used to impart biocompatibility to droplet surfaces. A simple polyetherdiamine (Jeffamine) aqueous additive provides a dynamic shielding effect at droplet surfaces, an attractive alternative to covalent modification of Krytox for droplet-based biochemical reactions.
In addition, we have further improved our understanding of the binding of primary amines to Krytox. This interaction is likely a major factor in the incompatibility of Krytox with enzyme reactions, as proteins typically contain a number of primary amines; and this study should give further insight into interaction mechanisms with other additives39–40 or surfactants.41 By expanding upon these studies in the future, it is feasible that one could spatially direct various biomolecules—such as DNA—to the oil-water interfaces of droplets to participate in surface-based biochemical assays.
Supplementary Material
Acknowledgments
Support for this work was provided by the National Institutes of Health (R01DK093810 to CJE; R43HG006078 to Lucigen Corporation), the Auburn University Department of Chemistry and Biochemistry, and the College of Science and Mathematics. Thanks go to Charles Ellis in Electrical Engineering for clean room use, as well as Tony Overfelt and Michael Miller for use of the confocal microscope in the Auburn University Research Instrumentation Facility (AURIF). Finally, the authors thank Yonnie Wu and Michael Meadows for their assistance in mass spectrometry and NMR, respectively.
Footnotes
SUPPORTING INFORMATION: Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tawfik DS, Griffiths AD. Nature Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 2.Williams R, Peisajovich SG, Miller OJ, Magdassi S, Tawfik DS, Griffiths AD. Nature Methods. 2006;3(7):545–550. doi: 10.1038/nmeth896. [DOI] [PubMed] [Google Scholar]
- 3.Kelly BT, Baret JC, Taly V, Griffiths AD. Chem Commun. 2007:1773–1788. doi: 10.1039/b616252e. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths AD, Tawfik DS. EMBO. 2003;22(1):24–35. doi: 10.1093/emboj/cdg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. PNAS. 2003;100(15):8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dresman D. Nature Methods. 2006;3(7):551–559. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 7.Easley CJ, Rocheleau JV, Head WS, Piston DW. Anal Chem. 2009;81:9086–9095. doi: 10.1021/ac9017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun S, Slaney TR, Kennedy RT. Anal Chem. 2012;84:5794–5800. doi: 10.1021/ac3011389. [DOI] [PubMed] [Google Scholar]
- 9.Abate AR, Weitz DA. Biomicrofluidics. 2011;5:014107-1–8. doi: 10.1063/1.3567093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng Y, Shin MM, Wang TY. Lab Chip. 2013;13:267–273. doi: 10.1039/c2lc40906b. [DOI] [PubMed] [Google Scholar]
- 11.Matosevic S, Paegel BM. J Am Chem Soc. 2011;133:2798–2800. doi: 10.1021/ja109137s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tadros TF. Applied Surfactants-Principles and Applications. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 13.Curran DP. Angew Chem Int Ed. 1998;37:1174–1196. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1174::AID-ANIE1174>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 14.Hildebrand JH, Cochran DRF. JACS. 1949;71:22–25. [Google Scholar]
- 15.Scott RL. JACS. 1948;70:4090–4093. doi: 10.1021/ja01192a036. [DOI] [PubMed] [Google Scholar]
- 16.Holtze C, Rowat AC, Agresti JJ, Hutchison JB, Angilé FE, Schmitz CHJ, Köster S, Duan H, Humphry KJ, Scanga RA, et al. Lab Chip. 2008;8:1632–1639. doi: 10.1039/b806706f. [DOI] [PubMed] [Google Scholar]
- 17.Piirma I. Polymeric Surfactants. CRC Press; Marcel Dekker; 1992. [Google Scholar]
- 18.Skhiri Y, Gruner P, Semin B, Brosseau Q, Pekin D, Mazutis L, Goust V, Kleinschmidt F, El Harak A, Hutchison JB, et al. Soft Matter. 2012;8:10618–10627. [Google Scholar]
- 19.Mazutis L, Araghi AF, Miller OJ, Baret JC, Frenz L, Janoshazi A, Taly V, Miller BJ, Hutchison JB, Link D, Griffiths AD, Ryckelynck M. Anal Chem. 2009;81(12):4813–4821. doi: 10.1021/ac900403z. [DOI] [PubMed] [Google Scholar]
- 20.Theberge AB, Whyte G, Huck WTS. Anal Chem. 2010;82(9):3449–3453. doi: 10.1021/ac1005316. [DOI] [PubMed] [Google Scholar]
- 21.O’Neal KL, Weber SG. J Phys Chem. 2009;113(1):149–158. doi: 10.1021/jp8084155. [DOI] [PubMed] [Google Scholar]
- 22.O’Neal KL, Geib S, Weber SG. Anal Chem. 2007;79(8):3117–3125. doi: 10.1021/ac062287+. [DOI] [PubMed] [Google Scholar]
- 23.O’Neal KL, Zhang H, Yang Y, Hong L, Lu D, Weber SG. J Chromatography A. 2010;1217(16):2287–2295. doi: 10.1016/j.chroma.2009.11.077. [DOI] [PubMed] [Google Scholar]
- 24.Matochko WL, Ng S, Jafari MR, Romaniuk J, Tang SKY, Derda R. Methods. 2012;58:18–27. doi: 10.1016/j.ymeth.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Shim J, Olguin LF, Whyte G, Scott D, Babtie A, Abell C, Huck WTS, Hollfelder F. JACS. 2009;131:15251–15256. doi: 10.1021/ja904823z. [DOI] [PubMed] [Google Scholar]
- 26.Temtem M, Casimiro T, Santos AG, Macedo AL, Cabrita EJ, Aguiar-Ricardo A. J Phys Chem B. 2007;111:1318–1326. doi: 10.1021/jp0660233. [DOI] [PubMed] [Google Scholar]
- 27.Jones GK, McGhie AR, Farrington GC. Macromolecules. 1991;24:3285–3290. [Google Scholar]
- 28.Goss K. Environ Sci Technol. 2008;42:456–458. doi: 10.1021/es702192c. [DOI] [PubMed] [Google Scholar]
- 29.Henne AL, Fox CJ. JACS. 1951;73:2323–2325. [Google Scholar]
- 30.Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U. Nat Biotechnol. 2002;20(5):473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 31.Heyduk E, Dummit B, Chang Y, Heyduk T. Anal Chem. 2008;80(13):5152–5159. doi: 10.1021/ac8004154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S, Emslie KR. Anal Chem. 2012;84:1003–1011. doi: 10.1021/ac202578x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogelstein B, Kinzler KW. PNAS. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong Q, Bhattacharya S, Kotsopoulos S, Olson J, Taly V, Griffiths AD, Link DR, Larson JW. Lab Chip. 2011;11:2167–2174. doi: 10.1039/c1lc20126c. [DOI] [PubMed] [Google Scholar]
- 35.Heyries KA, Tropini C, Vanlnsberghe M, Doolin C, Petriv OI, Singhal A, Leung K, Hughesman CB, Hansen CL. Nature Methods. 2011;8(8):649–651. doi: 10.1038/nmeth.1640. [DOI] [PubMed] [Google Scholar]
- 36.Piepenburg O, Williams CH, Stemple DL, Armes NA. PLoS Biol. 2006;4(7):e204. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Easley CJ. Bioanalysis. 2011;3(2):227–239. doi: 10.4155/bio.10.172. [DOI] [PubMed] [Google Scholar]
- 38.Shen F, Davydova EK, Du W, Kreutz JE, Piepenburg O, Ismagilov RF. Anal Chem. 2011;83:3533–3540. doi: 10.1021/ac200247e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brouzes E, Medkova M, Savenelli N, Marran D, Twardowski M, Hutchison JB, Rothberg JM, Link DR, Perrimon N, Samuels ML. Proc Natl Acad Sci USA. 2009;106:14195–14200. doi: 10.1073/pnas.0903542106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Courtois F, Olguin LF, Whyte G, Theberge AB, Huck WTS, Hollfelder F, Abell C. Anal Chem. 2009;81:3008–3016. doi: 10.1021/ac802658n. [DOI] [PubMed] [Google Scholar]
- 41.Baret JC. Lab Chip. 2012;12:422–433. doi: 10.1039/c1lc20582j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.