

Abstract
Dioxygen addition to coordinatively unsaturated [Fe(II)(OMe2N4(6-Me-DPEN))](PF6) (1) is shown to afford a complex containing a dihydroxo-bridged Fe(III)2(μ-OH)2 diamond core, [FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH)2(PF6)2•(CH3CH2CN)2 (2). The diamond core of 2 resembles the oxidized methane monooxygenase (MMOox) resting state, as well as the active site product formed following H-atom abstraction from Tyr-OH by ribonucleotide reductase (RNR). The Fe-OH bond lengths of 2 are comparable with those of the MMOHox suggesting that MMOHox contains a Fe(III)2(μ-OH)2 as opposed to Fe(III)2(μ-OH)(μ-OH2) diamond core as had been suggested. Isotopic labeling experiments with 18O2 and CD3CN indicate that the oxygen and proton of the μ-OH bridges of 2 are derived from dioxygen and acetonitrile. Deuterium incorporation (from CD3CN) suggests that an unobserved intermediate capable of abstracting a H-atom from CH3CN forms en route to 2. Given the high C–H bond dissociation energy (BDE= 97 kcal/mol) of acetonitrile, this indicates that this intermediate is a potent oxidant, possibly a high-valent iron oxo. Consistent with this, iodosylbenzene (PhIO) also reacts with 1 in CD3CN to afford the deuterated Fe(III)2(μ-OD)2 derivative of 2. Intermediates are not spectroscopically observed in either reaction (O2 and PhIO) even at low-temperatures (−80 °C), indicating that this intermediate has a very short life-time, likely due to its highly reactive nature. Hydroxo-bridged 2 was found to stoichiometrically abstract hydrogen atoms from 9,10-dihydroanthracene (C-H BDE= 76 kcal/mol) at ambient temperatures.
Introduction
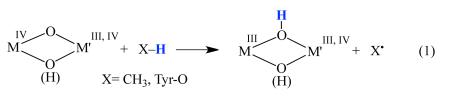
The activation of O2 is involved in many fundamental biochemical processes promoted by iron metalloenzymes. These transformations typically involve reactive iron-superoxo, -peroxo or high-valent oxo intermediates,1–12 which are, in some cases, capable of activating strong C–H bonds.1,13–15 For example, cytochrome P450 binds and subsequently activates O2 en route to an Fe(IV)-OH porphyrin radical cation intermediate (Compound I).13,15 Non-heme iron enzymes, such as α-ketoglutarate taurine dioxygenase (TauD)11,16 and halogenase SyrB2,17 activate O2 to form high-valent Fe(IV)-oxo intermediates. The reduced binuclear Fe(II)2 active site of methane monooxygenase (MMO)1,10,18–21 and ribonucleotide reductase (RNR)10,22–25 react with O2 to afford highly reactive oxidized binuclear Fe(IV)2(μ-O)2 (A) or M(III)(μ-O)(μ-OH)M'(IV) (M, M'= Mn, Fe; B) diamond cores26–28, respectively, (Scheme 1) which abstract H-atoms from either CH4 or TyrOH, respectively, to afford Fe(III)(μ-OH)(μ-O)Fe(IV) (B) or M(III)(μ-OH)2M'(III) (C) species.3,19,21,25,29,30 The highly reactive nature of these enzymatic intermediates has prompted numerous investigations aimed at establishing benchmark structural, spectroscopic, and reactivity properties of these species.1,4,10–12,16,18–28,31–36 With RNR, the introduction of an electron along with O2 affords structure B (Scheme 1), which in Chlamydia tranchomatis has been shown to contain an oxo/hydroxo bridged Fe(III)Mn(IV) dimer based on Mn and Fe K-edge EXAFS and DFT calculations.25 It's likely that the diiron RNRs have an analogous Fe(III)Fe(IV) core B that converts to core C upon reaction with Tyr-OH substrate (eqn (1)). The significantly stronger C-H bond of CH4 requires the higher valent core A (Scheme 1) of methane monooxygenase (MMO) to abstract a H-atom. It has been debated as to whether the structure of the MMO oxidized resting state (MMOHox) contains a bis-hydroxo bridged Fe(III)(μ-OH)2Fe(III) (C) or a hydroxo/aquo bridged Fe(III)(μ-OH)(μ-OH2)Fe(III) (D) core.21,37–39 One would expect the metal ion Lewis acidity of Fe(III) and Fe(IV) to disfavor protonation of a hydroxide especially if it is bridging. Water has been observed to bridge between two less Lewis acidic Fe(II) ions, however.40
Scheme 1.

 |
Many of the insights regarding the reactivity structures and properties of these oxidized bimetallic diamond cores have been provided by synthetic analogues.4,12,31–36 With synthetic complexes, solubility in non-aqueous solvents with low freezing points and small molecule crystallography can help provide mechanistic details, such as the probable identity and spectroscopic properties of reaction intermediates18,41–45 or the location of and transfer of key protons.46 Research efforts in our group have recently focused on the O2 reactivity of biologically-relevant first-row transition metal complexes.47,48 Herein we describe the reactivity of a new coordinatively unsaturated Fe(II) complex with O2 to afford a dihydroxo-bridged binuclear Fe(III) complex. Spectroscopic evidence has been obtained which suggests that an unobserved intermediate formed during this reaction abstracts a hydrogen atom from CH3CN solvent.
Experimental Section
General Methods
All manipulations were performed using Schlenk line techniques or under a N2 atmosphere in a glovebox. The highest purity reagents and solvents available were purchased and used without further purification unless otherwise noted. CH3OH and CH3CN (CaH2) were dried and distilled prior to use. Et2O was purified using solvent purification columns housed in a custom stainless steel cabinet and dispensed by a stainless steel schlenk-line (GlassContour). CD3CN and CD2Cl2 were purchased from Cambridge Isotope Laboratories, Inc. CD3CN was either dried over CaH2, vacuum transferred and stored in a dry box, or obtained from sealed ampules (Cambridge Isotopes (99.8%)) containing volumes small enough for a single experiment. All solvents were rigorously degasses prior to use. 1H NMR spectra were recorded on either a Bruker AV 301 or Bruker AV 300 FT NMR spectrometer at ambient temperature and were referenced to residual deuterated solvent. Infrared spectra were recorded on NaCl salt plates as Nujol mulls with a Perkin Elmer 1720 FT-IR spectrometer. Electrospray-ionization mass spectra were obtained on a Bruker Esquire Liquid Chromatograph-Ion Trap mass spectrometer. Gas chromatography – mass spectrometry data was recorded using an HP 5971A gas chromatograph interfaced with a quadrupole mass spectrometer equipped with a 7673A autosampler. Electronic absorption spectra were recorded on a Varian Cary 50 spectrophotometer equipped with a fiber optic cable connected to a “dip” ATR probe (C-technologies). A custom-built two-neck solution sample holder equipped with a threaded glass connector was sized specifically to fit the “dip” probe. Cyclic voltammograms were recorded in CH3CN (100 mM (nBu4N)(PF6) supporting electrolyte) on a PAR 263A potentiostat with a glassy carbon working electrode, Ag/AgNO3 reference electrode, and a platinum auxiliary electrode. EPR spectra were recorded on a Varian CW-EPR spectrometer at 15 K equipped with an Oxford helium cryostat. Solution magnetic moments were calculated by the Evans method,49 with temperature correction made in the manner described by Van Geet.50 Pascal's constants were used to correct for diamagnetic contributions to the experimental magnetic moment. X-ray crystallography data was recorded on a Bruker APEX II single crystal X-ray diffractometer with Mo-radiation. Elemental analyses were performed by Atlantic Microlabs, Norcross, GA. Experiments involving iodosylbenzene were conducted using a 2 mM stock solution in CH3OH. N,N-bis(6-methyl-2-pyridilmethyl)ethane-1,2-diamine (6-Me-DPEN) was prepared according to literature procedures.48
Synthesis of [FeII(OMe2N4(6-Me-DPEN))](PF6) (1)
FeCl2 (0.094 g, 0.74 mmol), N,N-bis(6-methyl-2-pyridilmethyl)ethane-1,2-diamine (0.20 g, 0.74 mmol), 3-hydroxy-3-methyl-2-butanone (0.09 g, 0.86 mmol), NaOCH3 (0.40 g, 0.74 mmol), and NaPF6 (0.12 g, 0.74 mmol) were combined in CH3OH (10 mL) and allowed to stir under an inert atmosphere in a glovebox at room temperature for two days. All volatiles were then removed in vacuo to yield a crude yellow solid, which was recrystallized from CH3CN/Et2O at 258 K to yield the title compound as a bright yellow solid in 47 % yield (0.20 g, 0.35 mmol). Electronic absorption spectrum: λmax (nm) (ε (M−1cm−1)) (CH3CN): 412 (250). Redox potential (CH3CN vs. Fc+/0, 298 K): Epa = +230 mV, Epc = +10 mV, ΔE = 220 mV. Magnetic moment (299 K, CD3OD solution): 4.71 B.M. ESI-MS: Expected m/z for [C21H29N4OFe]+ = 409.3, found m/z = 409.2. Elemental analysis results were obtained with the BPh4− salt of 1, which was synthesized by the same procedure described above except that NaBPh4 was used instead of NaPF6. Elemental analysis for C45H49N4BOFe; Calculated C, 72.06; H, 7.16; N, 6.86. Found C, 71.11; H, 7.60; N, 6.41.
Synthesis of [FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH)2(PF6)2•(CH3CH2CN)2 (2)
Complex 1 (0.20 g, 0.36 mmol) was dissolved in CH3CN (5 mL) under an inert atmosphere in a glovebox at room temperature. The resulting solution was placed into a gas tight syringe, removed from the glovebox, and injected into a custom-made two-neck vial equipped with a septum cap that had previously been purged with argon and contained a stir bar. Dioxygen, from a pressurized tank (Praxair, 99.9999%, <0.5 ppm H2O) and run through a column of anhydrous CaSO4 (Drierite), was gently bubbled into the stirring solution for approximately five minutes. The reaction mixture was allowed to continue stirring at room temperature for two hours, during which the solution had turned from yellow to dark orange. All volatiles were then removed in vacuo to yield a dark orange solid, which was recrystallized from CH3CN/Et2O (1/6 vol/vol) at 253 K to afford the title compound in 96 % yield (0.19 g, 0.17 mmol). X-ray quality crystals of 2 were grown from a concentrated CH3CH2CN solution at −80 °C, as well as from a concentrated CH3CN solution at room temperature. FT-IR (NaCl plates, Nujol mull, 298 K): ν(O-H) = 3610 cm−1; ν(O-D) = 2730 cm−1. Elemental analysis for C42H58N8O4F12P2Fe2; Calculated C, 44.50; H, 5.69; N, 10.61. Found C, 45.84; H, 5.33; N, 10.40.
Characterization of 2 in CH3CN solution
Electronic absorption spectrum: λmax (nm) (ε (M−1cm−1)): 310 (610), 375 (br, 440) (extinction coefficients calculated per Fe(III)). Redox potential (CH3CN vs. Fc+/0, 298 K): Epa = +235 mV, Epc = +26 mV. Magnetic moment (300 K, CD3CN solution): 5.82 B.M/Fe(III). ESI-MS: Expected m/z for [C21H30N4O2Fe]+ = 426.3, found m/z = 426.2. Expected m/z for [C21H29DN4O2Fe]+ = 427.3, found m/z = 427.2. Expected m/z for [C21H30N4O18OFe]+ = 428.3, found m/z = 428.2. EPR spectrum (1:1 CH3CN/toluene glass, 14 K): g = 1.96, 4.07, 10.2.
Synthesis and Isolation of Deuterium Labeled [FeIII(OMe2N4(6-Me-DPEN))]2(μ-OD)2(PF6)2•(CH3CH2CN)2 (2) for FT-IR Experiments
Method 1
A solution of 1 (0.25 g, 0.45 mmol) was prepared in dry CD3CN (30 mL; Cambridge Isotopes (99.8%)) under an inert atmosphere in a glovebox. The resulting solution was placed into a gas tight syringe, removed from the glovebox, and injected into a custom-made two-neck vial equipped with a septum cap that had previously been purged with argon and contained a stir bar. Dioxygen, derived from a pressurized tank (Praxair, 99.9999%, <0.5 ppm H2O) and run through a column of anhydrous CaSO4 (Drierite), was gently bubbled into the stirring solution for approximately five minutes. The reaction was allowed to continue to stir at room temperature for 30 minutes, after which all volatiles were removed in vacuo to afford 2 as a dark orange solid. The resulting solid was redissolved in a minimal amount of CH3CH2CN and crystallized at −80 °C. Crystalline samples of 2 were harvested from the mixture via filtration through a fine fritted filter, briefly dried in vacuo at room temperature, and subsequently used for FT-IR experiments.
Method 2
A solution of 2 (0.090 g, 0.16 mmol) was prepared in dry CH3CN (10 mL) under aerobic conditions at room temperature. D2O (0.1 mL) was added to this solution, and the resulting reaction mixture was allowed to gently stir at room temperature overnight. All volatiles were then removed in vacuo to afford a dark orange solid, which was left under vacuum for 72 hours. The solid was then redissolved in a minimal amount of CH3CH2CN, crystallized at −80 °C, harvested via filtration, and subsequently used for FT-IR experiments.
Analysis of Products From Reactions Between Complex 2 and 9,10-dihydroanthracene in CH3CN
In a typical reaction, 2 was dissolved in CH3CN (3–4 mL, ~0.5 mM solution of 2) at room temperature under an inert atmosphere in a glovebox. The resulting solution was placed into a gas tight syringe, removed from the glovebox, and injected into a custom-made two-neck vial containing a stir bar and equipped with a septum cap and threaded dip-probe feed-through adaptor that had previously been purged with Ar. 9,10-dihydroanthracene (0.5 equivalents per Fe(III)) was then added to the solution in a similar fashion from a concentrated stock solution that had been prepared in CH3CN in a glovebox. Each reaction mixture (the reaction was repeated three times and shown to be reproducible in all three cases) was allowed to stir under anaerobic conditions for 10–15 minutes, and then dried in vacuo. The solid products were then washed with Et2O (3 mL) and the Et2O soluble components were subsequently analyzed by GC/MS and quantified using standard calibration curves.
X-Ray Crystallography
A yellow crystal of 1 with dimensions 0.05 × 0.04 × 0.4 mm3 was mounted on a glass capillary with oil. Data was collected at −143 °C. The crystal-to-detector distance was set to 30 mm and exposure time was 60 seconds per degree for all sets of exposure. The scan width was 2.0°. Data collection was 99.4% complete to 25.0° in ϑ. A total of 56,116 partial and complete reflections were collected covering the indices, h = −21 to 21, k = −19 to 19, l = −32 to 32. 69,506 reflections were symmetry independent and the Rint = 0.125 indicated that the data was poor. Indexing and unit cell refinement indicated an orthorhombic P lattice with the space group P b c a (No. 61).
A pale red plate of 2 with dimensions 0.20 × 0.10 × 0.10 mm3 was mounted on a glass capillary with oil. Data was collected at −173 °C. The crystal-to-detector distance was set to 40 mm and exposure time was 30 seconds per degree for all sets of exposure. The scan width was 0.5°. Data collection was 99.9% complete to 25.0° in ϑ. A total of 51,770 partial and complete reflections were collected covering the indices, h = −15 to 14, k = −15 to 15, l = −17 to 17. 7,264 reflections were symmetry independent and the Rint = 0.0512 indicated that the data was good. Indexing and unit cell refinement indicated a triclinic P lattice with the space group P -1 (No. 2).
All data was integrated and scaled using SAINT, SADABS within the APEX2 software package by Bruker. Solutions by direct methods (SHELXS, SIR97) produced complete heavy atom phasing models which were consistent with each proposed structure. Scattering factors are from Waasmair and Kirfel.51 Crystal data for 1 and 2 are provided in Table 1. All non-hydrogen atoms were refined anistropically by full-matrix least-squares methods. Most hydrogen atoms were placed using a riding model, however the hydrogen atoms on the bridging hydroxide ligands in 2 were refined from the difference map. Crystallographic data is summarized in Table 1. Selected distances and angles are summarized in Table 2.
Table 1.
Crystal data for [FeII(OMe2N4(6-Me-DPEN))](PF6) (1) and [FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH)2(PF6)2 •(CH3CH2CN)2 (2).
| 1 | 2 | |
|---|---|---|
| Formula | C21H29F6FeN4OP | C48H70F12Fe2N10O4P2 |
| MW | 553.40 | 1252.78 |
| T, K | 130(2) | 100(2) |
| Unit Cella | Orthorhombic | Triclinic |
| a, Å | 15.9090(11) | 11.237(2) |
| b, Å | 17.1316(12) | 11.403(2) |
| c, Å | 17.6610(13) | 13.334(2) |
| α, deg | 90 | 70.554(9) |
| β, deg | 90 | 66.281(9) |
| γ, deg | 90 | 87.391(10) |
| V, Å3 | 4813.3(6) | 1467.3(5) |
| Z | 8 | 1 |
| d(calc), g/cm3 | 1.530 | 1.418 |
| Space group | P b c a | P −1 |
| Rb | 0.0592 | 0.0366 |
| Rwc | 0.1548 | 0.0796 |
| GOF | 0.928 | 1.007 |
Table 2.
Selected Bond Distances (Å) and Bond Angles (deg) for [FeII(OMe2N4(6-Me-DPEN))](PF6) (1) and [FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH)2(PF6)2 •(CH3CH2CN)2 (2).
| 1 | 2 | |
|---|---|---|
|
| ||
| Fe(1)-O(1) | 1.925(3) | 1.8721(13) |
| Fe(1) -N(1) | 2.086(4) | 2.1121(15) |
| Fe(1) -N(2) | 2.273(5) | 2.2556(15) |
| Fe(1) -N(3) | 2.139(4) | 2.2144(16) |
| Fe(1) -N(4) | 2.121(4) | N/A |
| Fe(1)–O(2) | N/A | 1.9715(13) |
| Fe(1')–O(2) | N/A | 2.0037(14) |
| Fe(1)•••Fe(1')% | N/A | 3.172 |
| O(1)- Fe -N(1) | 78.97(16) | 78.34(6) |
| O(1)- Fe -N(2) | 154.82(15) | 148.61(5) |
| O(1)- Fe -N(3) | 117.97(16) | 98.17(6) |
| O(1)- Fe -N(4) | 116.24(16) | N/A |
| N(1)- Fe -N(3) | 116.03(17) | 107.26(6) |
| N(1)- Fe -N(4) | 122.64(17) | N/A |
| N(3)- Fe -N(4) | 104.41(15) | N/A |
| O(1)– Fe–O(2) | N/A | 106.24(5) |
| O(1)– Fe –O(2') | N/A | 104.88(6) |
| N(1)– Fe –O(2) | N/A | 163.94(6) |
| Fe(1)–O(2)–Fe(1') | N/A | 105.88(6) |
| O(2)-Fe(1)-O(2#) | N/A | 74.12(6) |
Fe(1) and Fe(1') and O(2) and O(2') are equivalent in 2 and related by a crystallographically imposed inversion center, ie only one half of the dimer is crystallographically independent.
Results and Discussion
Synthesis and Structure of Coordinatively Unsaturated Fe(II) Alkoxide Complex
The alkoxide-ligated complex [FeII(OMe2N4(6-Me-DPEN))](PF6) (1) was synthesized via an Fe(II)-templated Schiff-base condensation between 6-Me-DPEN48 and 3-hydroxy-3-methyl-2-butanone in CH3OH under an inert atmosphere at ambient temperature. X-ray quality crystals of 1 were obtained via vapor diffusion of Et2O into a saturated CH3CN solution at room temperature. As shown in the ORTEP diagram of Figure 1, the Fe(II) ion of 1 is found in a distorted trigonal bipyramidal coordination environment (τ = 0.61)52 comprised of an alkoxide oxygen (O(1)), imine nitrogen (N(1)), tertiary amine (N(2)), and two pyridine nitrogens (N(3) and N(4)) (Figure 1). Coordinatively unsaturated 1 is high-spin (S = 2, μeff = 4.71 B.M. in CD3OD) and exhibits a quasi-reverisble FeIII/II redox couple in CH3CN (under nitrogen) with E1/2 = +120 mV vs. Fc+/0 (ΔE = 220 mV, Figure S-1). The room temperature electronic absorption spectrum of 1 in CH3CN contains a single absorption band with λmax = 412 nm (ε = 250 M−1cm−1, Figure S-2).
Figure 1.

ORTEP diagram (50% probability) of [FeII(OMe2N4(6-Me-DPEN))]+ (1) with hydrogen atoms and counterion omitted for clarity.
Reactivity with Dioxygen to Afford Dihydroxo-Bridged Iron(III,III)(μ-OH)2 Diamond Core
If excess amounts of dry O2, derived from a pressurized tank (Praxair, 99.9999%, <0.5 ppm H2O) and dried via a column of anhydrous CaSO4 (Drierite), is bubbled into a CH3CN solution of 1 for ~2–3 minutes at ambient temperature the solution changes from yellow to dark orange, and a broad band at λmax = 310 nm (Figure S-3) grows in in the electronic absorption spectrum, and the broad band at 412 nm (Figure S-2) disappears. Low temperature reactions were also performed at either −40 °C in CH3CN or −80 °C in CH3CH2CN, however no transient intermediates were observed under these conditions. X-ray quality crystals of the orange product obtained from this reaction, [FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH)2(PF6)2 •(CH3CH2CN)2 (2), were obtained in high yield (96 %) via vapor diffusion of Et2O into a concentrated CH3CH2CN solution at −80 °C (Figure S-4). As shown in the ORTEP diagram of Figure 2, both Fe(III) ions of 2 are located in a distorted octahedral environment consisting of an alkoxide oxygen (O(1)), two hydroxide bridging ligands (O(2) and O(2')), an imine nitrogen (N(1)), a tertiary amine (N(2)), and a pyridine nitrogen (N(3)) (Figure 2). The location and refinement of a hydrogen atom on each of the bridging oxygen atoms (O(2)) in the X-ray structure, along with the presence of two PF6− counterions per diiron complex, conclusively identified the bridging ligands of 2 as hydroxides. Hydrogen-bonding interactions ((O(2)-H⋯N(21)) = 2.200 Å; O(2)-H-N(21)= 171.29°) between the bridging hydroxide ligands and co-crystallized CH3CH2CN nitrogens (Figure S-4) further verified this assignment.
Figure 2.

ORTEP diagram (50% probability) of the Fe(III)(μ-OH)2Fe'(III) diamond core and Fe coordination sphere of dicationic{[FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH) 22+ (2). The two halves of the molecule are related by a crystallographic inversion center. An ORTEP diagram of the entire dicationic complex is shown in Figure S-4.
The hydroxide ligands in 2 are asymmetrically bridging with Fe(1)-O(2) and Fe(1)-O(2') bond lengths of 1.9715(13) Å and 2.0037(14) Å, respectively (Table 2). These distances are comparable to other structurally characterized Fe(III)2(μ-OH)2 and Fe(III)2 (μ-OH)2 (μ-O2CR) complexes (1.88–2.08 Å).35,36,53–64 The Fe(1)⋯Fe(1') distance in 2 (3.172 Å) is, on the other hand, longer than the reported Fe(III)2(μ-OH)2 core range (3.07–3.12 Å), while the Fe(1)-O(2)-Fe(1') bridging angle 105.88(6)° is at the larger end of the previously reported range (92.3°–105.8°).53–63 This likely results from steric congestion around each of the two Fe(III) ions, as is further indicated by the dissociation of a pyridine arm away from each metal ion in both halves of the dimer (Figure S-4).
Isotopic Labeling Experiments
Isotopic labeling studies conducted with 18O2 confirmed that the two bridging hydroxide ligands in 2 are indeed derived from O2 (Scheme 2; Figure S-5–Figure S-7). As shown by the Fe-isotope distribution pattern in the ESI mass spectra of Figure 3a, the μ-hydroxo bridge cleaves in the mass spectrometer to afford a monomeric monocationic Fe-hydroxo species with a parent ion m/z peak at 426.2 (Figure S-5), which corresponds to half of the {[FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH)2}2+ dimer 2, [FeIII(OMe2N4(6-Me-DPEN))(OH)]+, and is 17 mass units greater than that (m/z= 409.2) of 1 (Figure S-6). The parent ion peak (at m/z= 426.2; Figure 3a) shifts by two mass units (to m/z= 428.2; Figure 3c) upon the incorporation of 18O from 18O-labeled dioxygen (Figure S-7). In order to determine the source of the hydrogen atoms found on the hydroxide bridging ligands in 2, deuterium labeling studies were conducted. If O2 is added to 1 in deuterated acetonitrile (CD3CN), then a parent ion peak at m/z= 427.2 (Figure 3b), which is shifted by one mass unit (Figure S-8) relative to that observed at m/z= 426.2 (Figure 3a) in the CH3CN reaction (Figure S-5), is observed consistent with the incorporation of deuterium. Again, the Fe-isotope distribution pattern indicates that the bis-hydroxo bridged diiron complex cleaves in the mass spectrometer to afford a monomeric [FeIII(OMe2N4(6-Me-DPEN))(OD)]+. Although one can not rule out some sort of impurity in the CD3CN as the source of deuterium, it is unlikely that D2O from “wet” CD3CN is responsible, given that the 18O-label in the spectrum of Figure 3c is not “washed out” via exchange with water (H216O) contamination in the CH3CN. It is also unlikely that D2O, as opposed to H2O, would be a contaminant in the CD3CN. Deuterium labeled 2 was also obtained from a reaction between 1 and O2 in CD2Cl2. Additional evidence to support the incorporation of deuterium from CD3CN is shown by the vibrational spectrum (Figure S-9) of crystalline 2 (synthesized in CH3CN), which displays a sharp feature at 3610 cm−1 that shifts to 2730 cm−1 when 2 is synthesized in CD3CN. The energies and observed isotopic shift (νO-H/νO-D = 1.32 vs. theoretical = 1.35) for these vibrational features are consistent with O-H and O-D stretches, respectively.
Scheme 2.

Figure 3.

ESI-MS of the cleaved monomeric product observed when dicationic {[FeIII(OMe2N4(6-Me-DPEN))]2(μ-OH)22+ (2), obtained from (a) the reaction between 1 and 16O2 in CH3CN, (b) the reaction between 1 and 16O2 in CD3CN, and (c) the reaction between 1 and 18O2 in CH3CN, is placed in the mass spectrometer.
Implications Regarding the Involvement of a Reactive Intermediate
Deuterium incorporation (from CD3CN) suggests that an unobserved intermediate capable of abstracting a H-atom from CH3CN forms in the dioxygen reaction (Scheme 2) en route to 2. Given the high C–H bond dissociation energy (BDE= 97 kcal/mol) of acetonitrile, this indicates that this intermediate is a potent oxidant, possibly a high-valent iron oxo. Consistent with this, iodosylbenzene (PhIO) also reacts with 1 in CD3CN (Scheme 3) to afford the deuterated Fe(III)2(μ-OD)2 derivative of 2 (93–96 % yield), as verified by FT-IR (νO-D = 2727 cm−1; Figure S-11) and ESI-MS (m/z= 427.2; Figure S-10). Intermediates are not spectroscopically observed in either reaction (O2 and PhIO) even at low-temperatures (−80 °C), indicating that this intermediate has a very short life-time, likely due to its highly reactive nature. Iodosylbenzene (PhIO) would be expected to afford a Fe(IV)=O upon its reaction with Fe(II) (Scheme 3).65–69 High-valent Fe(IV)=O species have been shown to be highly reactive and capable of abstracting H-atoms, especially in the presence of anionic ligands.68–73 Hydrogen atom abstraction from acetonitrile has been previously observed during the O2-promoted oxidation of [Mn(II)1cyp]1- to [Mn(III)1cyp(OH)]1- via a high-valent Mn-oxo intermediate.74 High-valent iron oxo compounds [Fe(IV)(O)BnTPEN)]2+,75 [Fe(IV)(PY5)(O)]2+,76 and aqueous Fe(IV)O2+ have also been shown to abstract hydrogen atoms from CH3CN.77
Scheme 3.

Reactivity of Dihydroxo-Bridged 2 with Substrates Containing Weaker C-H Bonds
The reactivity of the dihydroxo-bridged product 2 towards stoichiometric amounts of substrates containing relatively weak C-H bonds was explored under anaerobic conditions in CH3CN at room temperature. Under these conditions, 9,10-dihydroanthracene (C-H BDFE = 76.0 kcal·mol−1)78 was oxidized to anthracene in 23(3) % yield (Figures S-12, S-13). Complex 2 was unreactive, however, with organic compounds containing stronger C-H bonds, such as toluene and cyclohexane (C-H BDFE = 87 kcal·mol−1 and 99 kcal·mol−1, respectively).78 This reactivity is similar to other Fe(III)-OH species, such as lipoxygenase79,80 and [FeIII(PY5)(OH)]2+,76 which have been found to abstract hydrogen atoms from substrates containing relatively weak C-H bonds.
Conclusions
In conclusion, we have described the synthesis and characterization of a new coordinatively unsaturated Fe(II) complex (1) that reacts with O2 to form an Fe(III)2(μ-OH)2 diamond core (2) in high yield. Isotopic labeling studies suggest that an unobserved intermediate formed during the conversion of 1 to 2 is capable of oxidizing CH3CN, presumeably via H• atom transfer. Iodosylbenzene (PhIO, 1.0 eq) also oxidizes 1 to afford 2 in high yield in CH3CN. Although low temperature intermediates were not observed during reactions between 1 and either O2 or PhIO, an unobserved high-valent Fe(IV)=O intermediate is proposed as a common intermediate in both of these reactions that is capable of, and responsible for, the observed solvent oxidation. Reactivity studies have shown that hydroxo-bridged 2 is itself also capable of oxidizing the relatively weak C-H bonds of DHA at room temperature. Efforts towards further exploring this reactivity, as well as comparing the properties and reactivity of thiolate-ligated analogues, are currently underway.
Supplementary Material
ACKNOWLEDGMENT
NIH funding (#RO1GM45881-20) is gratefully acknowledged.
Footnotes
Supporting Information Cyclic Voltammogram, electronic absorption spectrum, and ESI-MS data for 1, and ESI-MS data for 2, and isotopic (18O and D) derivatives thereof, derived from O2 and PhIO in CH3CN or CD3CN. More detailed ORTEP diagram of 2, along with crystallographic tables of both 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Tinberg CE, Lippard SJ. Acc. Chem. Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kovaleva EG, Lipscomb JD. Science. 2007;316:453–456. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Feig AL, Lippard SJ. Chem. Rev. 1994;94:759–805. [Google Scholar]
- (4).Costas M, Mehn MP, Jensen MP, Que LJ. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- (5).Cho J, Jeon S, Wilson SA, Liu LV, Kang EA, Braymer JJ, Lim MH, Hedman B, Hodgson KO, Valentine JS, Solomon EI, Nam W. Nature Chem. 2011;478:502–505. doi: 10.1038/nature10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr. Acc. Chem. Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Acc. Chem. Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Que L., Jr. Acc. Chem. Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- (9).Kovacs JA. Science. 2003;299:1024–1025. doi: 10.1126/science.1081792. [DOI] [PubMed] [Google Scholar]
- (10).Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem. Rev. 2000;100:233–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- (11).Price JC, Barr EW, Tirupati B, Bollinger JM, Jr., Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- (12).Que L, Tolman WB. Nature. 2008;455:333–340. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- (13).Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- (14).Borovik AS. Chem. Soc. Rev. 2011;40:1870–1874. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sono M, Roach MP, Coulter ED, Dawson JH. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- (16).Elkins JM, Ryle MJ, Clifton IJ, Hotopp JCD, Lloyd JS, Burzlaff NI, Baldwin JE, Hausinger RP, Roach PL. Biochemistry. 2002;41:5185–5192. doi: 10.1021/bi016014e. [DOI] [PubMed] [Google Scholar]
- (17).Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr. Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Xue G, Fiedler AT, Martinho M, Munck E, Que L., Jr. Proc. Natl. Acad. Sci, USA. 2008;105:20615–20620. [Google Scholar]
- (19).Valentine AM, Stahl SS, Lippard SJ. J. Am. Chem. Soc. 1999;121:3876–3887. [Google Scholar]
- (20).Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J. Am. Chem. Soc. 1995;117:10174–10185. [Google Scholar]
- (21).Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew. Chem., Int. Ed. Engl. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- (22).Bollinger JM, Jr., Tong WH, Ravi N, Huyuh BH, Edmondson ED, Stubbe J. J. Am. Chem. Soc. 1994;116:8015–8023. [Google Scholar]
- (23).Bollinger JM, Jr., Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- (24).Cotruvo JA, Jr., Stich TA, Britt RS, Stubbe J. J. Am. Chem. Soc. 2013;135:4027–4039. doi: 10.1021/ja312457t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Younker JM, Krest CM, Jiang W, Krebs C, Bollinger JM, Jr, Green MT. J. Am. Chem. Soc. 2008;130:15022–15027. doi: 10.1021/ja804365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xue G, Geng C, Ye S, Fiedler AT, Neese F, Que L., Jr. Inorg. Chem. 2013;52:3976–3984. doi: 10.1021/ic3027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lee S-K, Nesheim JC, Lipscomb JD. J. Biol. Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- (28).Que LJ, Tolman WB. Angew. Chem. Int. Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- (29).Brunold TC. Proc. Natl. Acad. Sci, USA. 2007;104:20641–20642. doi: 10.1073/pnas.0710734105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tshuva EY, Lippard SJ. Chem. Rev. 2004;104:987–1012. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- (31).Decker A, Solomon EI. Angew. Chem lnl. Ed. EngI. 2005;44:2252–2255. doi: 10.1002/anie.200462182. [DOI] [PubMed] [Google Scholar]
- (32).Xue G, Wang D, De Hont R, Fiedler AT, Shan X, Munck E, Que L., Jr. Proc. Natl. Acad. Sci, USA. 2007;104:20173–20178. doi: 10.1073/pnas.0708516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- (34).Shook RL, Peterson SM, Greaves J, Moore C, Rheingold AL, Borovik AS. J. Am. Chem. Soc. 2011;133:5810–5817. doi: 10.1021/ja106564a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lee D, Lippard SJ. J. Am. Chem. Soc. 1998;120:12153–12154. [Google Scholar]
- (36).Lee D, Lippard SJ. J. Am. Chem. Soc. 2001;123:4611–4612. doi: 10.1021/ja0100930. [DOI] [PubMed] [Google Scholar]
- (37).Tinberg CE, Lippard SJ. Biochemistry. 2010;49:7902–7912. doi: 10.1021/bi1009375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Elango N, Radhakrishnan R, Forland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem. Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- (40).Hagen KS, Lachicotte R. J. Am. Chem. Soc. 1992;114:8741–8742. [Google Scholar]
- (41).Kodera M, Kawahara Y, Hitomi Y, Nomura T, Ogura T, Kobayashi Y. J. Am. Chem. Soc. 2012;134:13236–13239. doi: 10.1021/ja306089q. [DOI] [PubMed] [Google Scholar]
- (42).Kim K, Lippard SL. J. Am. Chem. Soc. 1996;118:4914–4915. [Google Scholar]
- (43).Dong Y, Menage S, Brennan BA, Elgren TE, Jang HG, Pearce LL, Que LJ. J. Am. Chem. Soc. 1993;115:1851–1859. [Google Scholar]
- (44).Shook RL, Gunderson WA, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2008;130:8888–8889. doi: 10.1021/ja802775e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).MacBeth CE, Golombek AP, Young VG, Jr., Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- (46).Shearer J, Scarrow RC, Kovacs JA. J. Am. Chem. Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]
- (47).Theisen RM, Shearer J, Kaminsky W, Kovacs JA. Inorg. Chem. 2004;43:7682–7690. doi: 10.1021/ic0491884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Coggins MK, Sun X, Kwak Y, Solomon EI, Rybak-Akimova EV, Kovacs JA. J. Am. Chem. Soc. 2013;135:5631–5640. doi: 10.1021/ja311166u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Evans DA. J. Chem. Soc. 1959:2005. [Google Scholar]
- (50).Van Geet AL. Anal. Chem. 1968;40:2227–2229. [Google Scholar]
- (51).Waasmaier D, Kirfel A. Acta Crystallogr. A. 1995;51:416. [Google Scholar]
- (52).Addison AW, Rao TN, Reedijk J. J. Chem. Soc. Dalton Trans. 1984:1349–1356. [Google Scholar]
- (53).Matsumoto K, Egami H, Oguma T, Katsuki T. Chem. Comm. 2012;48:5823–5825. doi: 10.1039/c2cc18090a. [DOI] [PubMed] [Google Scholar]
- (54).Bruijnincx PCA, Buurmans ILC, Huang Y, Juhász G, Viciano-Chumillas M, Quesada M, Reedijk J, Lutz M, Spek AL, Münck E, Bominaar EL, Gebbink RJMK. Inorg. Chem. 2011;50:9243–9255. doi: 10.1021/ic200332y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Safaei E, Sheykhi H, Weyhermüller T, Bill E. Inorg. Chim. Acta. 2011;384:69–75. [Google Scholar]
- (56).Egami H, Matsumoto K, Oguma T, Kunisu T, Katsuki T. J. Am. Chem. Soc. 2010;132:13633–13635. doi: 10.1021/ja105442m. [DOI] [PubMed] [Google Scholar]
- (57).Boudalis AK, Clemente-Juan JM, Dahan F, Psycharis V, Raptopoulou CP, Donnadieu B, Sanakis Y, Tuchagues J-P. Inorg. Chem. 2008;47:11314–11323. doi: 10.1021/ic800716r. [DOI] [PubMed] [Google Scholar]
- (58).Ghiladi M, Larsen FB, McKenzie CJ, Sotofte I, Tuchagues J-P. J. Chem. Soc., Dalton Trans. 2005:1687–1692. doi: 10.1039/b418430k. [DOI] [PubMed] [Google Scholar]
- (59).Zheng H, Zang Y, Dong Y, Young VG, Que LJ. J. Am. Chem. Soc. 1999;121:2226–2235. [Google Scholar]
- (60).Nanda KK, Dutta SK, Baitalik S, Venkatsubramanian K, Nag K. J. Chem. Soc. Dalton Trans. 1995:1239–1244. [Google Scholar]
- (61).Turowski PN, Armstrong WH, Liu S, Brown SN, Lippard SJ. Inorg. Chem. 1994;33:636–645. [Google Scholar]
- (62).Ou C-C, Lalancette RA, Potenza JA, Schugar HJ. J. Am. Chem. Soc. 1978;100:2053–2057. doi: 10.1021/ja00426a078. [DOI] [PubMed] [Google Scholar]
- (63).Thich JA, Ou CC, Powers D, Vasiliou B, Mastropaolo D, Potenza JA, Schugar HJ. J. Am. Chem. Soc. 1976;98:1425–1433. [Google Scholar]
- (64).Lee D, Pierce B, Krebs C, Hendrich MP, Huynh BH, Lippard SJ. J. Am. Chem. Soc. 2002;124:3993–4007. doi: 10.1021/ja012251t. [DOI] [PubMed] [Google Scholar]
- (65).Rohde J-U, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Munck E, Nam W, Que L., Jr. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- (66).Groves JT, Haushalter RC, Nakamura M, Nemo TE, Evans BJ. J. Am. Chem. Soc. 1981;103:2884–2886. [Google Scholar]
- (67).Ye W, Ho DM, Friedle S, Palluccio TD, Rybak-Akimova EV. Inorg. Chem. 2012;51:5006–5021. doi: 10.1021/ic202435r. [DOI] [PubMed] [Google Scholar]
- (68).Nam W, Jin SW, Lim MH, Ryu JY. Inorg. Chem. 2002;41:3647–3652. doi: 10.1021/ic011145p. [DOI] [PubMed] [Google Scholar]
- (69).Sastri CV, Park MJ, Ohta T, Jackson TA, Stubna A, Seo MS, Lee J, Kim J, Kitagawa T, Munck E, Que L, Jr., Nam W. J. Am. Chem. Soc. 2005;127:12494–12495. doi: 10.1021/ja0540573. [DOI] [PubMed] [Google Scholar]
- (70).Nam W. Acc. Chem. Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- (71).Rhode J-U, Que L., Jr. Angew. Chem lnl. Ed. 2005;44:2255–2258. doi: 10.1002/anie.200462631. [DOI] [PubMed] [Google Scholar]
- (72).Hirao H, Kumar D, Que L, Jr., Shaik S. J. Am. Chem. Soc. 2006;128:8590–8606. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- (73).Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- (74).Shirin Z, Young VG, Jr., Borovik AS. Chem. Comm. 1997:1967–1968. [Google Scholar]
- (75).Klinker EJ, Shaik S, Hirao H, Que L., Jr Angew. Chem., lntl. Ed. 2009;48:1291–1295. doi: 10.1002/anie.200804029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Goldsmith CR, Stack TDP. Inorg. Chem. 2006;45:6048–6055. doi: 10.1021/ic060621e. [DOI] [PubMed] [Google Scholar]
- (77).Pestovsky O, Bakac A. J. Am. Chem. Soc. 2004;126:13757–13764. doi: 10.1021/ja0457112. [DOI] [PubMed] [Google Scholar]
- (78).Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Neidig ML, Wecksler AT, Schenk G, Holman TR, Solomon EI. J. Am. Chem. Soc. 2007;129:7531–7537. doi: 10.1021/ja068503d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Lehnert N, Solomon EI. J. Biol. Inorg. Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.